Drug Detail:Atripla (Efavirenz, emtricitabine, and tenofovir [ ef-av-ir-enz, em-trye-sye-ta-been, and-ten-of-oh-vir ])
Drug Class: Antiviral combinations
Highlights of Prescribing Information
ATRIPLA® (efavirenz, emtricitabine, and tenofovir disoproxil fumarate) tablets, for oral use
Initial U.S. Approval: 2006
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B
See full prescribing information for complete boxed warning.
Severe acute exacerbations of hepatitis B virus (HBV) have been reported in patients coinfected with HBV and HIV-1 who have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of ATRIPLA. Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue ATRIPLA. If appropriate, initiation of anti-hepatitis B therapy may be warranted. (5.1)
Recent Major Changes
| Warnings and Precautions | |
| Nervous System Symptoms (5.6) | 10/2019 |
| Immune Reconstitution Syndrome (5.12) | 10/2019 |
Indications and Usage for Atripla
ATRIPLA is a three-drug combination of efavirenz (EFV), a non-nucleoside reverse transcriptase inhibitor, and emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF), both HIV-1 nucleoside analog reverse transcriptase inhibitors, and is indicated as a complete regimen or in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 40 kg. (1)
Atripla Dosage and Administration
- Testing: Consult Full Prescribing Information for important testing recommendations prior to initiation and during treatment with ATRIPLA. (2.1)
- Recommended dosage in adults and pediatric patients weighing at least 40 kg: One tablet once daily taken orally on an empty stomach, preferably at bedtime. (2.2)
- Renal impairment: Not recommended in patients with estimated creatinine clearance below 50 mL/min. (2.3)
- Hepatic impairment: Not recommended in patients with moderate to severe hepatic impairment. (2.4)
- Dosage adjustment with rifampin coadministration: An additional 200 mg/day of efavirenz is recommended for patients weighing 50 kg or more. (2.5)
Dosage Forms and Strengths
Tablets: 600 mg of efavirenz, 200 mg of emtricitabine, and 300 mg of tenofovir disoproxil fumarate. (3)
Contraindications
- Previously demonstrated hypersensitivity (e.g., Stevens-Johnson syndrome, erythema multiforme, or toxic skin eruptions) to efavirenz, a component of ATRIPLA. (4)
- Coadministration with voriconazole. (4)
- Coadministration with elbasvir/grazoprevir. (4)
Warnings and Precautions
- Rash: Discontinue if severe rash develops. (5.2, 6.1)
- Hepatotoxicity: Monitor liver function tests before and during treatment in patients with underlying hepatic disease, including hepatitis B or C coinfection, marked transaminase elevations, or who are taking medications associated with liver toxicity. Among reported cases of hepatic failure, a few occurred in patients with no pre-existing hepatic disease. (5.3, 6.2, 8.7)
- Risk of adverse reactions or loss of virologic response due to drug interactions: Consult full prescribing information prior to and during treatment for important potential drug interactions. Consider alternatives to ATRIPLA in patients taking other medications with a known risk of Torsade de Pointes or in patients at higher risk of Torsade de Pointes. (5.4)
- Serious psychiatric symptoms: Immediate medical evaluation is recommended. (5.5, 6.1)
- Nervous system symptoms (NSS): NSS are frequent, usually begin 1−2 days after initiating therapy, and resolve in 2−4 weeks. Dosing at bedtime may improve tolerability. NSS are not predictive of onset of psychiatric symptoms. (2.2, 5.6)
- New onset or worsening renal impairment: Can include acute renal failure and Fanconi syndrome. Prior to initiation and during use of ATRIPLA, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. Avoid administering ATRIPLA with concurrent or recent use of nephrotoxic drugs. (5.7)
- Embryo fetal toxicity: Fetal harm may occur when administered to a pregnant woman during the first trimester. Avoid pregnancy while receiving ATRIPLA and for 12 weeks after discontinuation. (5.8, 8.1)
- Decreases in bone mineral density (BMD): Consider assessment of BMD in patients with a history of pathological fracture or other risk factors for osteoporosis or bone loss. (5.9)
- Convulsions: Use caution in patients with a history of seizures. (5.10)
- Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. (5.11)
- Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.12)
- Redistribution/accumulation of body fat: Observed in patients receiving antiretroviral therapy. (5.13)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence greater than or equal to 10%) observed in an active-controlled clinical trial of EFV, FTC, and TDF are diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Consult Full Prescribing Information prior to and during treatment for important potential drug interactions. (4, 5.4, 7)
- HIV-1 protease inhibitors: Coadministration of ATRIPLA with either lopinavir/ritonavir or darunavir and ritonavir increases tenofovir concentrations. Monitor for evidence of tenofovir toxicity. Coadministration of ATRIPLA with either atazanavir or atazanavir and ritonavir is not recommended. (7.3)
Use In Specific Populations
- Pregnancy: Avoid pregnancy while receiving ATRIPLA and for 12 weeks after discontinuation. (5.8, 8.3)
- Lactation: Breastfeeding is not recommended. (8.2)
- Females and Males of Reproductive Potential: Pregnancy testing and contraception are recommended. (8.3)
- Pediatrics: The incidence of rash was higher than in adults. (5.2, 6.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2021
Full Prescribing Information
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B
Severe acute exacerbations of hepatitis B virus (HBV) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), which are components of ATRIPLA.
Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue ATRIPLA. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)].
1. Indications and Usage for Atripla
ATRIPLA® is indicated as a complete regimen or in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 40 kg.
2. Atripla Dosage and Administration
2.1 Testing Prior to Initiation and During Treatment with ATRIPLA
Prior to or when initiating ATRIPLA, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to initiation and during use of ATRIPLA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.7)].
Monitor hepatic function prior to and during treatment with ATRIPLA [see Warnings and Precautions (5.3)].
Perform pregnancy testing before initiation of ATRIPLA in adolescents and adults of childbearing potential [see Warnings and Precautions (5.8), Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage for Adults and Pediatric Patients Weighing at Least 40 kg
ATRIPLA is a three-drug fixed-dose combination product containing 600 mg of efavirenz (EFV), 200 mg of emtricitabine (FTC), and 300 mg of tenofovir disoproxil fumarate (TDF). The recommended dosage of ATRIPLA in adults and pediatric patients weighing at least 40 kg is one tablet once daily taken orally on an empty stomach. Dosing at bedtime may improve the tolerability of nervous system symptoms [see Clinical Pharmacology (12.3)].
2.3 Not Recommended in Patients with Moderate or Severe Renal Impairment
ATRIPLA is not recommended in patients with moderate or severe renal impairment (estimated creatinine clearance below 50 mL/min) [see Warnings and Precautions (5.7), Use in Specific Populations (8.6)].
3. Dosage Forms and Strengths
ATRIPLA tablets are pink, capsule shaped, film coated, debossed with "123" on one side, and plain faced on the other side. Each tablet contains 600 mg of efavirenz, 200 mg of emtricitabine, and 300 mg of tenofovir disoproxil fumarate (equivalent to 245 mg of tenofovir disoproxil).
4. Contraindications
- ATRIPLA is contraindicated in patients with previously demonstrated clinically significant hypersensitivity (e.g., Stevens-Johnson syndrome, erythema multiforme, or toxic skin eruptions) to efavirenz, a component of ATRIPLA [see Warnings and Precautions (5.2)].
- ATRIPLA is contraindicated to be coadministered with voriconazole or elbasvir/grazoprevir [see Drug Interactions (7.3) and Clinical Pharmacology (12.3)].
5. Warnings and Precautions
5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
All patients should be tested for the presence of chronic HBV before or when initiating antiretroviral therapy [see Dosage and Administration (2.1)]. Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in patients who are coinfected with HBV and HIV-1 and have discontinued FTC or TDF, two of the components of ATRIPLA. Patients who are coinfected with HIV-1 and HBV should be closely monitored, with both clinical and laboratory follow-up for at least several months after stopping treatment with ATRIPLA. If appropriate, initiation of anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since posttreatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
5.2 Rash
In controlled clinical trials, 26% (266/1,008) of adult subjects treated with 600 mg EFV experienced new-onset skin rash compared with 17% (111/635) of those treated in control groups. Rash associated with blistering, moist desquamation, or ulceration occurred in 0.9% (9/1,008) of subjects treated with EFV. The incidence of Grade 4 rash (e.g., erythema multiforme, Stevens-Johnson syndrome) in adult subjects treated with EFV in all trials and expanded access was 0.1%. Rashes are usually mild-to-moderate maculopapular skin eruptions that occur within the first 2 weeks of initiating therapy with EFV (median time to onset of rash in adults was 11 days) and, in most subjects continuing therapy with EFV, rash resolves within 1 month (median duration, 16 days). The discontinuation rate for rash in adult clinical trials was 1.7% (17/1,008). ATRIPLA can be reinitiated in patients interrupting therapy because of rash. ATRIPLA should be discontinued in patients developing severe rash associated with blistering, desquamation, mucosal involvement, or fever. Appropriate antihistamines and/or corticosteroids may improve the tolerability and hasten the resolution of rash. For patients who have had a life-threatening cutaneous reaction (e.g., Stevens-Johnson syndrome), alternative therapy should be considered [see Contraindications (4)].
Experience with EFV in subjects who discontinued other antiretroviral agents of the NNRTI class is limited. Nineteen subjects who discontinued nevirapine because of rash have been treated with EFV. Nine of these subjects developed mild-to-moderate rash while receiving therapy with EFV, and two of these subjects discontinued because of rash.
Rash was reported in 59 of 182 pediatric subjects (32%) treated with EFV [see Adverse Reactions (6.1)]. Two pediatric subjects experienced Grade 3 rash (confluent rash with fever, generalized rash), and four subjects had Grade 4 rash (erythema multiforme). The median time to onset of rash in pediatric subjects was 28 days (range 3−1,642 days). Prophylaxis with appropriate antihistamines before initiating therapy with ATRIPLA in pediatric patients should be considered.
5.3 Hepatotoxicity
Postmarketing cases of hepatitis, including fulminant hepatitis progressing to liver failure requiring transplantation or resulting in death, have been reported in patients treated with EFV, a component of ATRIPLA. Reports have included patients with underlying hepatic disease, including coinfection with hepatitis B or C, and patients without pre-existing hepatic disease or other identifiable risk factors [see Warnings and Precautions (5.1)].
ATRIPLA is not recommended for patients with moderate or severe hepatic impairment. Careful monitoring is recommended for patients with mild hepatic impairment receiving ATRIPLA [see Adverse Reactions (6.2) and Use in Specific Populations (8.7)].
Monitoring of liver enzymes before and during treatment is recommended for all patients [see Dosage and Administration (2.1)]. Consider discontinuing ATRIPLA in patients with persistent elevations of serum transaminases to greater than five times the upper limit of the normal range.
Discontinue ATRIPLA if elevation of serum transaminases is accompanied by clinical signs or symptoms of hepatitis or hepatic decompensation [see Adverse Reactions (6.1)].
5.4 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of ATRIPLA and other drugs may result in potentially significant drug interactions [see Contraindications (4) and Drug Interactions (7.3)], some of which may lead to:
- Loss of therapeutic effect of concomitant drug or ATRIPLA and possible development of resistance.
- Possible clinically significant adverse reaction from greater exposures of ATRIPLA or concomitant drug.
QTc prolongation has been observed with the use of EFV [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)]. Consider alternatives to ATRIPLA when coadministered with a drug with a known risk of Torsade de Pointes or when administered to patients at higher risk of Torsade de Pointes.
See Table 3 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during ATRIPLA therapy and review concomitant medications during ATRIPLA therapy [see Dosage and Administration (2.5), Contraindications (4), and Drug Interactions (7)].
5.5 Psychiatric Symptoms
Serious psychiatric adverse experiences have been reported in patients treated with EFV, a component of ATRIPLA. In controlled trials of 1,008 subjects treated with regimens containing EFV for a mean of 2.1 years and 635 subjects treated with control regimens for a mean of 1.5 years, the frequency (regardless of causality) of specific serious psychiatric events among subjects who received EFV or control regimens, respectively, were: severe depression (2.4%, 0.9%), suicidal ideation (0.7%, 0.3%), nonfatal suicide attempts (0.5%, 0%), aggressive behavior (0.4%, 0.5%), paranoid reactions (0.4%, 0.3%), and manic reactions (0.2%, 0.3%). When psychiatric symptoms similar to those noted above were combined and evaluated as a group in a multifactorial analysis of data from Study AI266006 (006, NCT00002410), a Phase 3 randomized, open-label trial of EFV-containing regimens versus controls in 1,266 subjects (median follow-up 180 weeks, 102 weeks, and 76 weeks for subjects treated with EFV + zidovudine + lamivudine, EFV + indinavir, and indinavir + zidovudine + lamivudine, respectively), treatment with EFV was associated with an increase in the occurrence of these selected psychiatric symptoms. Other factors associated with an increase in the occurrence of these psychiatric symptoms were history of injection drug use, psychiatric history, and receipt of psychiatric medication at trial entry; similar associations were observed in both the EFV and control treatment groups. In Study 006, onset of new serious psychiatric symptoms occurred throughout the trial for both EFV-treated and control-treated subjects. One percent of EFV-treated subjects discontinued or interrupted treatment because of one or more of these selected psychiatric symptoms. There have also been occasional postmarketing reports of death by suicide, delusions, and psychosis-like behavior, although a causal relationship to the use of EFV cannot be determined from these reports. Postmarketing cases of catatonia have also been reported and may be associated with increased EFV exposure. Patients with serious psychiatric adverse experiences should seek immediate medical evaluation to assess the possibility that the symptoms may be related to the use of EFV, and if so, to determine whether the risks of continued therapy outweigh the benefits [see Adverse Reactions (6)].
5.6 Nervous System Symptoms
Fifty-three percent (531/1,008) of subjects receiving EFV in controlled trials reported central nervous system symptoms (any grade, regardless of causality) compared to 25% (156/635) of subjects receiving control regimens. These symptoms included dizziness (28.1% of the 1,008 subjects), insomnia (16.3%), impaired concentration (8.3%), somnolence (7.0%), abnormal dreams (6.2%), and hallucinations (1.2%). Other reported symptoms were euphoria, confusion, agitation, amnesia, stupor, abnormal thinking, and depersonalization. The majority of these symptoms were mild to moderate (50.7%); symptoms were severe in 2.0% of subjects. Overall, 2.1% of subjects discontinued therapy as a result. These symptoms usually begin during the first or second day of therapy and generally resolve after the first 2–4 weeks of therapy. After 4 weeks of therapy, the prevalence of nervous system symptoms of at least moderate severity ranged from 5% to 9% in subjects treated with regimens containing EFV and from 3% to 5% in subjects treated with a control regimen. Patients should be informed that these common symptoms were likely to improve with continued therapy and were not predictive of subsequent onset of the less frequent psychiatric symptoms [see Warnings and Precautions (5.5)]. Dosing at bedtime may improve the tolerability of these nervous system symptoms [see Dosage and Administration (2.2)].
Analysis of long-term data from Study 006 showed that, beyond 24 weeks of therapy, the incidences of new-onset nervous system symptoms among EFV-treated subjects were generally similar to those in the indinavir-containing control arm.
Late-onset neurotoxicity, including ataxia and encephalopathy (impaired consciousness, confusion, psychomotor slowing, psychosis, delirium), may occur months to years after beginning EFV therapy. Some events of late-onset neurotoxicity have occurred in patients with CYP2B6 genetic polymorphisms which are associated with increased EFV levels despite standard dosing of EFV. Patients presenting with signs and symptoms of serious neurologic adverse experiences should be evaluated promptly to assess the possibility that these events may be related to EFV use, and whether discontinuation of ATRIPLA is warranted.
Patients receiving ATRIPLA should be alerted to the potential for additive central nervous system effects when ATRIPLA is used concomitantly with alcohol or psychoactive drugs.
Patients who experience central nervous system symptoms such as dizziness, impaired concentration, and/or drowsiness should avoid potentially hazardous tasks such as driving or operating machinery.
5.7 New Onset or Worsening Renal Impairment
Emtricitabine and tenofovir are principally eliminated by the kidney; however, EFV is not. Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphatemia), has been reported with the use of TDF, a component of ATRIPLA [see Adverse Reactions (6.2)].
Prior to initiation and during use of ATRIPLA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. ATRIPLA is not recommended in patients with moderate or severe renal impairment (estimated creatinine clearance below 50 mL/min).
ATRIPLA should be avoided with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple non-steroidal anti-inflammatory drugs [NSAIDs]) [see Drug Interactions (7.2)]. Cases of acute renal failure after initiation of high-dose or multiple NSAIDs have been reported in HIV-infected patients with risk factors for renal dysfunction who appeared stable on TDF. Some patients required hospitalization and renal replacement therapy. Alternatives to NSAIDs should be considered, if needed, in patients at risk for renal dysfunction.
Persistent or worsening bone pain, pain in extremities, fractures, and/or muscular pain or weakness may be manifestations of proximal renal tubulopathy and should prompt an evaluation of renal function in patients at risk of renal dysfunction.
Discontinue ATRIPLA in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
5.8 Embryo-Fetal Toxicity
Efavirenz may cause fetal harm when administered during the first trimester of pregnancy. Advise adults and adolescents of childbearing potential who are receiving ATRIPLA to avoid pregnancy while receiving ATRIPLA and for 12 weeks after discontinuation [see Dosage and Administration (2.1), Use in Specific Populations (8.1, 8.3)].
5.10 Convulsions
Convulsions have been observed in adult and pediatric patients receiving EFV, generally in the presence of known medical history of seizures. Caution must be taken in any patient with a history of seizures.
Patients who are receiving concomitant anticonvulsant medications primarily metabolized by the liver, such as phenytoin and phenobarbital, may require periodic monitoring of plasma levels [see Drug Interactions (7.3)].
5.11 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including TDF and FTC, components of ATRIPLA, alone or in combination with other antiretrovirals. Treatment with ATRIPLA should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.12 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including the components of ATRIPLA. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.13 Fat Redistribution
Redistribution/accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance," has been observed in patients receiving antiretroviral therapy, including EFV. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbations of Hepatitis B in Patients Coinfected with HIV-1 and HBV [see Warnings and Precautions (5.1)].
- Rash [see Warnings and Precautions (5.2)].
- Hepatotoxicity [see Warnings and Precautions (5.3)].
- Psychiatric Symptoms [see Warnings and Precautions (5.5)].
- Nervous System Symptoms [see Warnings and Precautions (5.6)].
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.7)].
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.8)].
- Bone Loss and Mineralization Defects [see Warnings and Precautions (5.9)].
- Convulsions [see Warnings and Precautions (5.10)].
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.11)].
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.12)].
- Fat Redistribution [see Warnings and Precautions (5.13)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials in Adult Subjects
Study 934 was an open-label active-controlled trial in which 511 antiretroviral-naïve subjects received either FTC + TDF administered in combination with EFV (N=257) or zidovudine (AZT)/lamivudine (3TC) administered in combination with EFV (N=254).
The most common adverse reactions (incidence greater than or equal to 10%, any severity) occurring in Study 934 include diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. Adverse reactions observed in Study 934 were generally consistent with those seen in previous trials of the individual components (Table 1).
| FTC+TDF+EFV† | AZT/3TC+EFV | |
|---|---|---|
| N=257 | N=254 | |
|
||
| Fatigue | 9% | 8% |
| Depression | 9% | 7% |
| Nausea | 9% | 7% |
| Diarrhea | 9% | 5% |
| Dizziness | 8% | 7% |
| Upper respiratory tract infections | 8% | 5% |
| Sinusitis | 8% | 4% |
| Rash Event‡ | 7% | 9% |
| Headache | 6% | 5% |
| Insomnia | 5% | 7% |
| Anxiety | 5% | 4% |
| Nasopharyngitis | 5% | 3% |
| Vomiting | 2% | 5% |
In Study 073, subjects with stable, virologic suppression on antiretroviral therapy and no history of virologic failure were randomized to receive ATRIPLA or to stay on their baseline regimen. The adverse reactions observed in Study 073 were generally consistent with those seen in Study 934 and those seen with the individual components of ATRIPLA when each was administered in combination with other antiretroviral agents.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of EFV, FTC, or TDF. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
7. Drug Interactions
7.1 Efavirenz
Efavirenz has been shown in vivo to induce CYP3A and CYP2B6. Other compounds that are substrates of CYP3A or CYP2B6 may have decreased plasma concentrations when coadministered with EFV.
Drugs that induce CYP3A activity (e.g., phenobarbital, rifampin, rifabutin) would be expected to increase the clearance of EFV, resulting in lowered plasma concentrations [see Dosage and Administration (2.2)].
There is limited information available on the potential for a pharmacodynamic interaction between EFV and drugs that prolong the QTc interval. QTc prolongation has been observed with the use of EFV [see Clinical Pharmacology (12.2)]. Consider alternatives to ATRIPLA when coadministered with a drug with a known risk of Torsade de Pointes.
7.2 Drugs Affecting Renal Function
FTC and tenofovir are primarily eliminated by the kidneys [see Clinical Pharmacology (12.3)]. Coadministration of ATRIPLA with drugs that are eliminated by active tubular secretion may increase concentrations of FTC, tenofovir, and/or the coadministered drug. Some examples include, but are not limited to, acyclovir, adefovir dipivoxil, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.7)]. Drugs that decrease renal function may increase concentrations of FTC and/or tenofovir.
7.3 Established and Potentially Significant Interactions
Other important drug interaction information for ATRIPLA is summarized in Table 3. The drug interactions described are based on trials conducted with either ATRIPLA, the components of ATRIPLA (EFV, FTC, or TDF) as individual agents, or are potential drug interactions [see Clinical Pharmacology (12.3)].
| Concomitant Drug Class: Drug Name | Effect | Clinical Comment |
|---|---|---|
|
||
| HIV antiviral agents | ||
| Protease inhibitor:
atazanavir | ↓ atazanavir ↑ tenofovir | Coadministration of atazanavir with ATRIPLA is not recommended. The combined effect of EFV plus TDF on atazanavir plasma concentrations is not known. There are insufficient data to support dosing recommendations for atazanavir or atazanavir/ritonavir in combination with ATRIPLA. |
| Protease inhibitor: fosamprenavir calcium | ↓ amprenavir | Fosamprenavir (unboosted): Appropriate doses of fosamprenavir and ATRIPLA with respect to safety and efficacy have not been established. Fosamprenavir/ritonavir: An additional 100 mg/day (300 mg total) of ritonavir is recommended when ATRIPLA is administered with fosamprenavir/ritonavir once daily. No change in the ritonavir dose is required when ATRIPLA is administered with fosamprenavir plus ritonavir twice daily. |
| Protease inhibitor: indinavir | ↓ indinavir | The optimal dose of indinavir, when given in combination with EFV, is not known. Increasing the indinavir dose to 1000 mg every 8 hours does not compensate for the increased indinavir metabolism due to EFV. |
| Protease inhibitor: darunavir/ritonavir | ↑ tenofovir | Monitor patients receiving ATRIPLA concomitantly with ritonavir-boosted darunavir for TDF-associated adverse reactions. Discontinue ATRIPLA in patients who develop TDF-associated adverse reactions. |
| lopinavir/ritonavir | ↓ lopinavir ↑ tenofovir | Do not use once daily administration of lopinavir/ritonavir. Dose increase of lopinavir/ritonavir is recommended for all patients when coadministered with EFV. Refer to the Full Prescribing Information for lopinavir/ritonavir for guidance on coadministration with EFV- or tenofovir-containing regimens, such as ATRIPLA. Patients should be monitored for tenofovir-associated adverse reactions. Discontinue ATRIPLA in patients who develop TDF-associated adverse reactions. |
| Protease inhibitor: ritonavir | ↑ ritonavir ↑ efavirenz | When ritonavir 500 mg every 12 hours was coadministered with EFV 600 mg once daily, the combination was associated with a higher frequency of adverse clinical experiences (e.g., dizziness, nausea, paresthesia) and laboratory abnormalities (elevated liver enzymes). Monitoring of liver enzymes is recommended when ATRIPLA is used in combination with ritonavir. |
| Protease inhibitor: saquinavir | ↓ saquinavir | Appropriate doses of the combination of EFV and saquinavir/ritonavir with respect to safety and efficacy have not been established. |
| CCR5 co-receptor antagonist: maraviroc | ↓ maraviroc | Refer to the full prescribing information for maraviroc for guidance on coadministration with ATRIPLA. |
| NRTI: didanosine | ↑ didanosine | Patients receiving ATRIPLA and didanosine should be monitored closely for didanosine-associated adverse reactions. Discontinue didanosine in patients who develop didanosine-associated adverse reactions. Higher didanosine concentrations could potentiate didanosine-associated adverse reactions, including pancreatitis, and neuropathy. Suppression of CD4+ cell counts has been observed in patients receiving TDF with didanosine 400 mg daily. In patients weighing greater than 60 kg, reduce the didanosine dose to 250 mg when it is coadministered with ATRIPLA. In patients weighing less than 60 kg, reduce the didanosine dose to 200 mg when it is coadministered with ATRIPLA. When coadministered, ATRIPLA and Videx EC may be taken under fasted conditions or with a light meal (less than 400 kcal, 20% fat). |
| NNRTI: Other NNRTIs | ↑ or ↓ efavirenz and/or NNRTI | Combining two NNRTIs has not been shown to be beneficial. ATRIPLA contains EFV and should not be coadministered with other NNRTIs. |
| Integrase strand transfer inhibitor: raltegravir | ↓ raltegravir | The clinical significance of this interaction has not been directly assessed. |
| Hepatitis C antiviral agents | ||
| boceprevir | ↓ boceprevir | Plasma trough concentrations of boceprevir were decreased when boceprevir was coadministered with EFV, which may result in loss of therapeutic effect. The combination should be avoided. |
| elbasvir/grazoprevir | ↓ elbasvir ↓ grazoprevir | Coadministration of ATRIPLA with elbasvir/grazoprevir is contraindicated [see Contraindications (4)] because it may lead to loss of virologic response to elbasvir/grazoprevir. |
| glecaprevir/pibrentasvir | ↓ glecaprevir ↓ pibrentasvir | Coadministration of ATRIPLA is not recommended because it may lead to reduced therapeutic effect of glecaprevir/pibrentasvir. |
| ledipasvir/sofosbuvir | ↑ tenofovir | Patients receiving ATRIPLA and HARVONI® (ledipasvir/sofosbuvir) concomitantly should be monitored for adverse reactions associated with TDF. |
| simeprevir | ↓ simeprevir ↔ efavirenz | Concomitant administration of simeprevir with EFV is not recommended because it may result in loss of therapeutic effect of simeprevir. |
| sofosbuvir/velpatasvir sofosbuvir/velpatasvir/voxilaprevir | ↑ tenofovir ↓ velpatasvir ↓ voxilaprevir | Coadministration of EFV-containing regimens and EPCLUSA® (sofosbuvir/velpatasvir) or VOSEVI® (sofosbuvir/velpatasvir/voxilaprevir) is not recommended. |
| Other agents | ||
| Anticoagulant: warfarin | ↑ or ↓ warfarin | Plasma concentrations and effects potentially increased or decreased by EFV. |
| Anticonvulsants: carbamazepine | ↓ carbamazepine ↓ efavirenz | There are insufficient data to make a dose recommendation for ATRIPLA. Alternative anticonvulsant treatment should be used. |
| phenytoin phenobarbital | ↓ anticonvulsant ↓ efavirenz | Potential for reduction in anticonvulsant and/or EFV plasma levels; periodic monitoring of anticonvulsant plasma levels should be conducted. |
| Antidepressants: bupropion | ↓ bupropion | The effect of EFV on bupropion exposure is thought to be due to the induction of bupropion metabolism. Increases in bupropion dosage should be guided by clinical response, but the maximum recommended dose of bupropion should not be exceeded. |
| sertraline | ↓ sertraline | Increases in sertraline dose should be guided by clinical response. |
| Antifungals: | ||
| itraconazole | ↓ itraconazole ↓ hydroxy-itraconazole | Since no dose recommendation for itraconazole can be made, alternative antifungal treatment should be considered. |
| ketoconazole | ↓ ketoconazole | Drug interaction trials with ATRIPLA and ketoconazole have not been conducted. Efavirenz has the potential to decrease plasma concentrations of ketoconazole. |
| posaconazole | ↓ posaconazole | Avoid concomitant use unless the benefit outweighs the risks. |
| voriconazole | ↓ voriconazole ↑ efavirenz | Coadministration of ATRIPLA with voriconazole is contraindicated [see Contraindications (4)] because it may lead to reduced therapeutic effect of voriconazole and increased risk of EFV-associated adverse reactions |
| Anti-infective: clarithromycin | ↓ clarithromycin ↑ 14-OH metabolite | Consider alternatives to macrolide antibiotics because of the risk of QT interval prolongation. |
| Antimycobacterial: rifabutin | ↓ rifabutin | Increase daily dose of rifabutin by 50%. Consider doubling the rifabutin dose in regimens where rifabutin is given 2 or 3 times a week. |
| rifampin | ↓ efavirenz | If ATRIPLA is coadministered with rifampin to patients weighing 50 kg or more, an additional 200 mg/day of EFV is recommended. |
| Antimalarials: artemether/lumefantrine | ↓ artemether ↓ dihydroartemisinin ↓ lumefantrine | Consider alternatives to artemether/lumefantrine because of the risk of QT interval prolongation [see Warnings and Precautions (5.4)]. |
| atovaquone/proguanil | ↓ atovaquone ↓ proguanil | Concomitant administration of atovaquone/proguanil with ATRIPLA is not recommended. |
| Calcium channel blockers: diltiazem | ↓ diltiazem ↓ desacetyl diltiazem ↓ N-monodes-methyl diltiazem | Diltiazem dose adjustments should be guided by clinical response (refer to the full prescribing information for diltiazem). No dose adjustment of ATRIPLA is necessary when administered with diltiazem. |
| Others e.g., felodipine nicardipine nifedipine verapamil | ↓ calcium channel blocker | No data are available on the potential interactions of EFV with other calcium channel blockers that are substrates of CYP3A. The potential exists for reduction in plasma concentrations of the calcium channel blocker. Dose adjustments should be guided by clinical response (refer to the full prescribing information for the calcium channel blocker). |
| HMG-CoA reductase inhibitors: atorvastatin pravastatin simvastatin | ↓ atorvastatin ↓ pravastatin ↓ simvastatin | Plasma concentrations of atorvastatin, pravastatin, and simvastatin decreased with EFV. Consult the Full Prescribing Information for the HMG-CoA reductase inhibitor for guidance on individualizing the dose. |
| Hormonal contraceptives: Oral: ethinyl estradiol/norgestimate | ↓ active metabolites of norgestimate | A reliable method of barrier contraception must be used in addition to hormonal contraceptives. Efavirenz had no effect on ethinyl estradiol concentrations, but progestin levels (norelgestromin and levonorgestrel) were markedly decreased. No effect of ethinyl estradiol/norgestimate on EFV plasma concentrations was observed. |
| Implant: etonogestrel | ↓ etonogestrel | A reliable method of barrier contraception must be used in addition to hormonal contraceptives. Decreased exposure of etonogestrel may be expected. There have been postmarketing reports of contraceptive failure with etonogestrel in EFV-exposed patients. |
| Immunosuppressants: cyclosporine, tacrolimus, sirolimus, and others metabolized by CYP3A | ↓ immuno-suppressant | Decreased exposure of the immunosuppressant may be expected due to CYP3A induction by EFV. These immunosuppressants are not anticipated to affect exposure of EFV. Dose adjustments of the immunosuppressant may be required. Close monitoring of immunosuppressant concentrations for at least 2 weeks (until stable concentrations are reached) is recommended when starting or stopping treatment with ATRIPLA. |
| Narcotic analgesic: methadone | ↓ methadone | Coadministration of EFV in HIV-1 infected individuals with a history of injection drug use resulted in signs of opiate withdrawal. Methadone dose was increased by a mean of 22% to alleviate withdrawal symptoms. Patients should be monitored for signs of withdrawal and their methadone dose increased as required to alleviate withdrawal symptoms. |
7.4 Efavirenz Assay Interference
Cannabinoid Test Interaction: Efavirenz does not bind to cannabinoid receptors. False-positive urine cannabinoid test results have been reported with some screening assays in uninfected and HIV-infected subjects receiving EFV. Confirmation of positive screening tests for cannabinoids by a more specific method is recommended.
8. Use In Specific Populations
8.4 Pediatric Use
The effectiveness and safety of ATRIPLA as a complete regimen for the treatment of HIV-1 infection was established in pediatric patients with body weight greater than or equal to 40 kg [see Dosage and Administration (2.2)]. Use of ATRIPLA in this age group is supported by adequate and well-controlled studies of ATRIPLA in adults with HIV-1 infection and data from pediatric studies of the individual components of ATRIPLA (EFV, FTC, and TDF).
ATRIPLA should only be administered to pediatric patients with a body weight greater than or equal to 40 kg. Because ATRIPLA is a fixed-dose combination tablet, the dose of ATRIPLA cannot be adjusted for patients of lower weight [see Warnings and Precautions (5.2, 5.9), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical trials of EFV, FTC, or TDF did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for elderly patients should be cautious, keeping in mind the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Because ATRIPLA is a fixed-dose combination, and cannot be dose adjusted, it is not recommended in patients with moderate or severe renal impairment (estimated creatinine clearance below 50 mL/min) [see Dosage and Administration (2.3), Warnings and Precautions (5.7)].
8.7 Hepatic Impairment
ATRIPLA is not recommended for patients with moderate or severe hepatic impairment because there are insufficient data to determine an appropriate dose. Patients with mild hepatic impairment may be treated with ATRIPLA at the approved dose. Because of the extensive cytochrome P450-mediated metabolism of EFV and limited clinical experience in patients with hepatic impairment, caution should be exercised in administering ATRIPLA to these patients [see Dosage and Administration (2.4), Warnings and Precautions (5.3), and Clinical Pharmacology (12.3)].
10. Overdosage
If overdose occurs, the patient should be monitored for evidence of toxicity, and standard supportive treatment applied as necessary. Administration of activated charcoal may be used to aid removal of unabsorbed EFV. Hemodialysis can remove both FTC and TDF (refer to detailed information below) but is unlikely to significantly remove EFV from the blood.
11. Atripla Description
ATRIPLA is a fixed-dose combination tablet containing EFV, FTC, and TDF. EFV is a non-nucleoside reverse transcriptase inhibitor (NNRTI). FTC is a synthetic nucleoside analog of cytidine. TDF, which is converted in vivo to tenofovir, is an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate.
ATRIPLA tablets are for oral administration. Each tablet contains 600 mg of EFV, 200 mg of FTC, and 300 mg of TDF (equivalent to 245 mg of tenofovir disoproxil) as active ingredients. The tablets include the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The tablets are film coated with a coating material containing black iron oxide, polyethylene glycol, polyvinyl alcohol, red iron oxide, talc, and titanium dioxide.
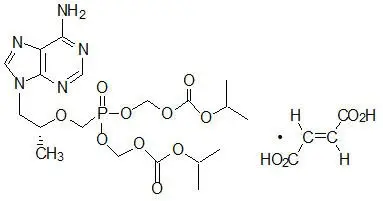
12. Atripla - Clinical Pharmacology
12.1 Mechanism of Action
ATRIPLA is a fixed-dose combination of antiviral drugs EFV, FTC, and TDF [see Microbiology (12.4)].
12.3 Pharmacokinetics
Specific Populations
Assessment of Drug Interactions
The drug interaction trials described were conducted with either ATRIPLA or the components of ATRIPLA (EFV, FTC, or TDF) as individual agents.
Efavirenz: The steady-state pharmacokinetics of EFV and tenofovir were unaffected when EFV and TDF were administered together versus each agent dosed alone. Specific drug interaction trials have not been performed with EFV and NRTIs other than tenofovir, lamivudine, and zidovudine. Clinically significant interactions would not be expected based on NRTIs elimination pathways.
Efavirenz has been shown in vivo to cause hepatic enzyme induction, thus increasing the biotransformation of some drugs metabolized by CYP3A and CYP2B6. In vitro studies have shown that EFV inhibited CYP isozymes 2C9 and 2C19 with Ki values (8.5–17 µM) in the range of observed EFV plasma concentrations. In in vitro studies, EFV did not inhibit CYP2E1 and inhibited CYP2D6 and CYP1A2 (Ki values 82–160 µM) only at concentrations well above those achieved clinically. Coadministration of EFV with drugs primarily metabolized by CYP2C9, CYP2C19, CYP3A or CYP2B6 isozymes may result in altered plasma concentrations of the coadministered drug. Drugs which induce CYP3A and CYP2B6 activity would be expected to increase the clearance of EFV resulting in lowered plasma concentrations.
Drug interaction trials were performed with EFV and other drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interaction. There was no clinically significant interaction observed between EFV and zidovudine, lamivudine, azithromycin, fluconazole, lorazepam, cetirizine, or paroxetine. Single doses of famotidine or an aluminum and magnesium antacid with simethicone had no effects on EFV exposures. The effects of coadministration of EFV on Cmax, AUC, and Cmin are summarized in Table 4 (effect of other drugs on EFV) and Table 5 (effect of EFV on other drugs) see [Drug Interactions (7)].
| Mean % Change of EFV Pharmacokinetic Parameters* (90% CI) | ||||||
|---|---|---|---|---|---|---|
| Coadministered Drug | Dose of Coadministered Drug (mg) | EFV Dose (mg) | N | Cmax | AUC | Cmin |
| NA = not available | ||||||
|
||||||
| Lopinavir/ritonavir | 400/100 mg q12h × 9 days | 600 mg qd × 9 days | 11, 12† | ↔ | ↓ 16 (↓ 38 to ↑ 15) | ↓ 16 (↓ 42 to ↑ 20) |
| Nelfinavir | 750 mg q8h × 7 days | 600 mg qd × 7 days | 10 | ↓ 12 (↓ 32 to ↑13)‡ | ↓ 12 (↓ 35 to ↑ 18)‡ | ↓ 21 (↓ 53 to ↑ 33) |
| Ritonavir | 500 mg q12h × 8 days | 600 mg qd × 10 days | 9 | ↑ 14 (↑ 4 to ↑ 26) | ↑ 21 (↑ 10 to ↑ 34) | ↑ 25 (↑ 7 to ↑ 46)‡ |
| Boceprevir | 800 mg tid × 6 days | 600 mg qd × 16 days | NA | ↑11 (↑ 2 to ↑ 20) | ↑ 20 (↑ 15 to ↑ 26) | NA |
| Rifabutin | 300 mg qd × 14 days | 600 mg qd × 14 days | 11 | ↔ | ↔ | ↓ 12 (↓ 24 to ↑ 1) |
| Rifampin | 600 mg × 7 days | 600 mg qd × 7 days | 12 | ↓ 20 (↓ 11 to ↓ 28) | ↓ 26 (↓ 15 to ↓ 36) | ↓ 32 (↓ 15 to ↓ 46) |
| Artemether/lumefantrine | Artemether 20 mg/lumefantrine 120 mg tablets (6 4-tablet doses over 3 days) | 600 mg qd × 26 days | 12 | ↔ | ↓17 | NA |
| Simvastatin | 40 mg qd × 4 days | 600 mg qd × 15 days | 14 | ↓ 12 (↓ 28 to ↑ 8) | ↔ | ↓ 12 (↓ 25 to ↑ 3) |
| Carbamazepine | 200 mg qd × 3 days, 200 mg bid × 3 days, then 400 mg qd × 15 days | 600 mg qd × 35 days | 14 | ↓ 21 (↓ 15 to ↓ 26) | ↓ 36 (↓ 32 to ↓ 40) | ↓ 47 (↓ 41 to ↓ 53) |
| Diltiazem | 240 mg × 14 days | 600 mg qd × 28 days | 12 | ↑ 16 (↑ 6 to ↑ 26) | ↑ 11 (↑ 5 to ↑ 18) | ↑ 13 (↑ 1 to ↑ 26) |
| Voriconazole | 400 mg po q12h × 1 day then 200 mg po q12h × 8 days | 400 mg qd × 9 days | NA | ↑ 38§ | ↑ 44§ | NA |
| 300 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↓ 14¶
(↓ 7 to ↓ 21) | ↔¶ | NA | |
| 400 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↔¶ | ↑ 17¶
(↑ 6 to ↑ 29) | NA | |
No effect on the pharmacokinetic parameters of EFV was observed with the following coadministered drugs: indinavir, saquinavir soft gelatin capsule, simeprevir, ledipasvir/sofosbuvir, sofosbuvir, clarithromycin, itraconazole, atorvastatin, pravastatin, or sertraline.
| Mean % Change of Coadministered Drug Pharmacokinetic Parameters* (90% CI) | ||||||
|---|---|---|---|---|---|---|
| Coadministered Drug | Dose of Coadministered Drug (mg) | EFV Dose (mg) | N | Cmax | AUC | Cmin |
| NA = not available | ||||||
|
||||||
| Atazanavir | 400 mg qd with a light meal d 1–20 | 600 mg qd with a light meal d 7–20 | 27 | ↓ 59 (↓ 49 to ↓ 67) | ↓ 74 (↓ 68 to ↓78) | ↓ 93 (↓ 90 to ↓ 95) |
| 400 mg qd d 1–6, then 300 mg qd d 7–20 with ritonavir 100 mg qd and a light meal | 600 mg qd 2 h after atazanavir and ritonavir d 7–20 | 13 | ↑ 14†
(↓ 17 to ↑ 58) | ↑ 39†
(↑ 2 to ↑ 88) | ↑ 48†
(↑ 24 to ↑ 76) |
|
| 300 mg qd/ritonavir 100 mg qd d 1–10 (pm), then 400 mg qd/ritonavir 100 mg qd d 11–24 (pm) (simultaneous with EFV) | 600 mg qd with a light snack d 11–24 (pm) | 14 | ↑ 17 (↑ 8 to ↑ 27) | ↔ | ↓ 42 (↓ 31 to ↓ 51) |
|
| Indinavir | 1000 mg q8h × 10 days | 600 mg qd × 10 days | 20 | |||
| After morning dose | ↔‡ | ↓ 33‡
(↓ 26 to ↓ 39) | ↓ 39‡
(↓ 24 to ↓ 51) |
|||
| After afternoon dose | ↔‡ | ↓ 37‡
(↓ 26 to ↓ 46) | ↓ 52‡
(↓ 47 to ↓ 57) |
|||
| After evening dose | ↓ 29‡
(↓ 11 to ↓ 43) | ↓ 46‡
(↓ 37 to ↓ 54) | ↓ 57‡
(↓ 50 to ↓ 63) |
|||
| Lopinavir/ritonavir | 400/100 mg q12h × 9 days | 600 mg qd × 9 days | 11, 7§ | ↔¶ | ↓ 19¶
(↓ 36 to ↑ 3) | ↓ 39¶
(↓ 3 to ↓ 62) |
| Nelfinavir | 750 mg q8h × 7 days | 600 mg qd × 7 days | 10 | ↑ 21 (↑ 10 to ↑ 33) | ↑ 20 (↑ 8 to ↑ 34) | ↔ |
| Metabolite AG-1402 | ↓ 40 (↓ 30 to ↓ 48) | ↓ 37 (↓ 25 to ↓ 48) | ↓ 43 (↓ 21 to ↓ 59) |
|||
| Ritonavir | 500 mg q12h × 8 days | 600 mg qd × 10 days | 11 | |||
| After AM dose | ↑ 24 (↑ 12 to ↑ 38) | ↑ 18 (↑ 6 to ↑ 33) | ↑ 42 (↑ 9 to ↑ 86)# |
|||
| After PM dose | ↔ | ↔ | ↑ 24 (↑ 3 to ↑ 50)# |
|||
| Saquinavir SGCÞ | 1200 mg q8h × 10 days | 600 mg qd × 10 days | 12 | ↓ 50 (↓ 28 to ↓ 66) | ↓ 62 (↓ 45 to ↓ 74) | ↓ 56 (↓ 16 to ↓ 77)# |
| Maraviroc | 100 mg bid | 600 mg qd | 12 | ↓ 51 (↓ 37 to ↓ 62) | ↓ 45 (↓ 38 to ↓ 51) | ↓ 45 (↓ 28 to ↓ 57) |
| Raltegravir | 400 mg single dose | 600 mg qd | 9 | ↓ 36 (↓ 2 to ↓ 59) | ↓ 36 (↓ 20 to ↓ 48) | ↓ 21 (↓ 51 to ↑ 28) |
| Boceprevir | 800 mg tid × 6 days | 600 mg qd × 16 days | NA | ↓ 8 (↓ 22 to ↑ 8) | ↓ 19 (↓ 11 to ↓ 25) | ↓ 44 (↓ 26 to ↓ 58) |
| Simeprevir | 150 mg qd × 14 days | 600 mg qd × 14 days | 23 | ↓ 51 (↓ 46 to ↓ 56) | ↓ 71 (↓ 67 to ↓ 74) | ↓ 91 (↓ 88 to ↓ 92) |
| Ledipasvir/sofosbuvirß | 90/400 mg qd × 14 days | 600 mg qd × 14 days | 15 | |||
| Ledipasvir | ↓ 34 (↓ 25 to ↓ 41) | ↓ 34 (↓ 25 to ↓ 41) | ↓ 34 (↓ 24 to ↓ 43) |
|||
| Sofosbuvir | ↔ | ↔ | NA | |||
| GS-331007à | ↔ | ↔ | ↔ | |||
| Sofosbuvirè | 400 mg qd single dose | 600 mg qd × 14 days | 16 | ↓ 19 (↓ 40 to ↑ 10) | ↔ | NA |
| GS-331007à | ↓ 23 (↓ 16 to ↓ 30) | ↓ 16 (↓ 24 to ↓ 8) | NA | |||
| Sofosbuvir/velpatasvirð | 400/100 mg qd × 14 days | 600 mg qd × 14 days | 14 | |||
| Sofosbuvir | ↑ 38 (↑ 14 to ↑ 67) | ↔ | NA | |||
| GS-331007à | ↓ 14 (↓ 20 to ↓ 7) | ↔ | ↔ | |||
| Velpatasvir | ↓ 47 (↓ 57 to ↓ 36) | ↓ 53 (↓ 61 to ↓ 43) | ↓ 57 (↓ 64 to ↓ 48) |
|||
| Clarithromycin | 500 mg q12h × 7 days | 400 mg qd × 7 days | 11 | ↓ 26 (↓ 15 to ↓ 35) | ↓ 39 (↓ 30 to ↓ 46) | ↓ 53 (↓ 42 to ↓ 63) |
| 14-OH metabolite | ↑ 49 (↑ 32 to ↑ 69) | ↑ 34 (↑ 18 to ↑ 53) | ↑ 26 (↑ 9 to ↑ 45) |
|||
| Itraconazole | 200 mg q12h × 28 days | 600 mg qd × 14 days | 18 | ↓ 37 (↓ 20 to ↓ 51) | ↓ 39 (↓ 21 to ↓ 53) | ↓ 44 (↓ 27 to ↓ 58) |
| Hydroxy-itraconazole | ↓ 35 (↓ 12 to ↓ 52) | ↓ 37 (↓ 14 to ↓ 55) | ↓ 43 (↓ 18 to ↓ 60) |
|||
| Posaconazole | 400 mg (oral suspension) bid × 10 and 20 days | 400 mg qd × 10 and 20 days | 11 | ↓ 45 (↓ 34 to ↓ 53) | ↓ 50 (↓ 40 to ↓ 57) | NA |
| Rifabutin | 300 mg qd × 14 days | 600 mg qd × 14 days | 9 | ↓ 32 (↓ 15 to ↓ 46) | ↓ 38 (↓ 28 to ↓ 47) | ↓ 45 (↓ 31 to ↓ 56) |
| Artemether/lumefantrine | Artemether 20 mg/lumefantrine 120 mg tablets (6 4-tablet doses over 3 days) | 600 mg qd × 26 days | 12 | |||
| Artemether | ↓ 21 | ↓ 51 | NA | |||
| dihydroartemisinin | ↓ 38 | ↓ 46 | NA | |||
| lumefantrine | ↔ | ↓ 21 | NA | |||
| Atorvastatin | 10 mg qd × 4 days | 600 mg qd × 15 days | 14 | ↓ 14 (↓ 1 to ↓ 26) | ↓ 43 (↓ 34 to ↓ 50) | ↓ 69 (↓ 49 to ↓ 81) |
| Total active (including metabolites) | ↓ 15 (↓ 2 to ↓ 26) | ↓ 32 (↓ 21 to ↓ 41) | ↓ 48 (↓ 23 to ↓ 64) |
|||
| Pravastatin | 40 mg qd × 4 days | 600 mg qd × 15 days | 13 | ↓ 32 (↓ 59 to ↑ 12) | ↓ 44 (↓ 26 to ↓ 57) | ↓ 19 (↓ 0 to ↓ 35) |
| Simvastatin | 40 mg qd × 4 days | 600 mg qd × 15 days | 14 | ↓ 72 (↓ 63 to ↓ 79) | ↓ 68 (↓ 62 to ↓ 73) | ↓ 45 (↓ 20 to ↓ 62) |
| Total active (including metabolites) | ↓ 68 (↓ 55 to ↓ 78) | ↓ 60 (↓ 52 to ↓ 68) | NAø | |||
| Carbamazepine | 200 mg qd × 3 days, 200 mg bid × 3 days, then 400 mg qd × 29 days | 600 mg qd × 14 days | 12 | ↓ 20 (↓ 15 to ↓ 24) | ↓ 27 (↓ 20 to ↓ 33) | ↓ 35 (↓ 24 to ↓ 44) |
| Epoxide metabolite | ↔ | ↔ | ↓ 13 (↓ 30 to ↑ 7) |
|||
| Diltiazem | 240 mg × 21 days | 600 mg qd × 14 days | 13 | ↓ 60 (↓ 50 to ↓ 68) | ↓ 69 (↓ 55 to ↓ 79) | ↓ 63 (↓ 44 to ↓ 75) |
| Desacetyl diltiazem | ↓ 64 (↓ 57 to ↓ 69) | ↓ 75 (↓ 59 to ↓ 84) | ↓ 62 (↓ 44 to ↓ 75) |
|||
| N-monodesmethyl diltiazem | ↓ 28 (↓ 7 to ↓ 44) | ↓ 37 (↓ 17 to ↓ 52) | ↓ 37 (↓ 17 to ↓ 52) |
|||
| Ethinyl estradiol/ norgestimate | 0.035 mg/0.25 mg × 14 days | 600 mg qd × 14 days | ||||
| Ethinyl estradiol | 21 | ↔ | ↔ | ↔ | ||
| Norelgestromin | 21 | ↓ 46 (↓ 39 to ↓ 52) | ↓ 64 (↓ 62 to ↓ 67) | ↓ 82 (↓ 79 to ↓ 85) |
||
| Levonorgestrel | 6 | ↓ 80 (↓ 77 to ↓ 83) | ↓ 83 (↓ 79 to ↓ 87) | ↓ 86 (↓ 80 to ↓ 90) |
||
| Methadone | Stable maintenance 35–100 mg daily | 600 mg qd × 14–21 days | 11 | ↓ 45 (↓ 25 to ↓ 59) | ↓ 52 (↓ 33 to ↓ 66) | NA |
| Bupropion | 150 mg single dose (sustained-release) | 600 mg qd × 14 days | 13 | ↓ 34 (↓ 21 to ↓ 47) | ↓ 55 (↓ 48 to ↓ 62) | NA |
| Hydroxybupropion | ↑ 50 (↑ 20 to ↑ 80) | ↔ | NA | |||
| Sertraline | 50 mg qd × 14 days | 600 mg qd × 14 days | 13 | ↓ 29 (↓ 15 to ↓ 40) | ↓ 39 (↓ 27 to ↓ 50) | ↓ 46 (↓ 31 to ↓ 58) |
| Voriconazole | 400 mg po q12h × 1 day then 200 mg po q12h × 8 days | 400 mg qd × 9 days | NA | ↓ 61ý | ↓ 77ý | NA |
| 300 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↓ 36£
(↓ 21 to ↓ 49) | ↓ 55£
(↓ 45 to ↓ 62) | NA | |
| 400 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↑ 23£
(↓ 1 to ↑ 53) | ↓ 7£
(↓ 23 to ↑ 13) | NA | |
13. Nonclinical Toxicology
14. Clinical Studies
Clinical Study 934 (NCT00112047) supports the use of ATRIPLA tablets in antiretroviral treatment-naïve HIV-1 infected patients.
Clinical Study 073 (NCT00365612) provides clinical experience in subjects with stable, virologic suppression and no history of virologic failure who switched from their current regimen to ATRIPLA.
In antiretroviral treatment-experienced patients, the use of ATRIPLA tablets may be considered for patients with HIV-1 strains that are expected to be susceptible to the components of ATRIPLA as assessed by treatment history or by genotypic or phenotypic testing [see Microbiology (12.4)].
16. How is Atripla supplied
ATRIPLA tablets are pink, capsule shaped, film coated, debossed with "123" on one side and plain faced on the other side. Each bottle contains 30 tablets (NDC 15584-0101-1) and silica gel desiccant, and is closed with a child-resistant closure.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: October/2019 | |||
| Patient Information
ATRIPLA® (uh TRIP luh) (efavirenz, emtricitabine, and tenofovir disoproxil fumarate) tablets |
||||
| What is the most important information I should know about ATRIPLA? ATRIPLA can cause serious side effects, including:
|
||||
| What is ATRIPLA?
ATRIPLA is a prescription medicine that contains efavirenz, emtricitabine, and tenofovir disoproxil fumarate combined in 1 tablet. ATRIPLA is used alone as a complete regimen, or in combination with other anti-HIV-1 medicines to treat people with HIV-1 infection who weigh at least 88 lbs (40 kg). It is not known if ATRIPLA is safe and effective for use in children with HIV-1 infection who weigh less than 88 lbs (40 kg). |
||||
| Who should not take ATRIPLA? Do not take ATRIPLA if you:
|
||||
Before taking ATRIPLA, tell your healthcare provider about all of your medical conditions, including if you:
Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine. ATRIPLA and some medicines may interact with each other causing serious side effects. You can ask your healthcare provider or pharmacist for a list of medicines that interact with ATRIPLA. Do not start a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take ATRIPLA with other medicines. |
||||
How should I take ATRIPLA?
|
||||
What should I avoid while taking ATRIPLA?
|
||||
| What are the possible side effects of ATRIPLA? ATRIPLA may cause serious side effects, including:
|
||||
|
|
|||
| If you have dizziness, trouble concentrating or sleepiness, do not drive a car, use machinery, or do anything that needs you to be alert. | ||||
|
||||
|
|
|||
|
||||
|
|
|||
| These are not all the possible side effects of ATRIPLA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||
How should I store ATRIPLA?
|
||||
| General information about the safe and effective use of ATRIPLA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ATRIPLA for a condition for which it was not prescribed. Do not give ATRIPLA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ATRIPLA that is written for health professionals. |
||||
| What are the ingredients of ATRIPLA? Active Ingredients: efavirenz, emtricitabine, and tenofovir disoproxil fumarate Inactive Ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium sterate, microcrystalline cellulose, and sodium lauryl sulfate. The film coating contains black iron oxide, polyethylene glycol, polyvinyl alcohol, red iron oxide, talc, and titanium dioxide. Manufactured and distributed by: Gilead Sciences, Inc. Foster City, CA 94404 For more information go to www.ATRIPLA.com or call 1-800-445-3235. ATRIPLA, EMTRIVA, TRUVADA, and VIREAD are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners. © 2019 Gilead Sciences, LLC. All rights reserved. 21937-GS-020 |
||||

| ATRIPLA
efavirenz, emtricitabine, and tenofovir disoproxil fumarate tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - Gilead Sciences, LLC (780297904) |
