Drug Detail:Balversa (Erdafitinib [ er-da-fi-ti-nib ])
Drug Class: Multikinase inhibitors
Highlights of Prescribing Information
BALVERSA ® (erdafitinib) tablets, for oral use
Initial U.S. Approval: 2019
Recent Major Changes
| Warnings and Precautions ( 5.2) | 04/2022 |
Indications and Usage for Balversa
BALVERSA is a kinase inhibitor indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) that has
- susceptible FGFR3 or FGFR2 genetic alterations and
- progressed during or following at least one line of prior platinum-containing chemotherapy including within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA. ( 1, 2.1)
This indication is approved under accelerated approval based on tumor response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. ( 1, 14.1)
Balversa Dosage and Administration
- Confirm the presence of FGFR genetic alterations in tumor specimens prior to initiation of treatment with BALVERSA. ( 2.1)
- Recommended initial dosage: 8 mg orally once daily with a dose increase to 9 mg daily if criteria are met. ( 2.2)
- Swallow whole with or without food. ( 2.2)
Dosage Forms and Strengths
Tablets: 3 mg, 4 mg, and 5 mg. ( 3)
Contraindications
None. ( 4)
Warnings and Precautions
- Ocular disorders: BALVERSA can cause central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED). Perform monthly ophthalmological examinations during the first four months of treatment, every 3 months afterwards, and at any time for visual symptoms. Withhold BALVERSA when CSR/RPED occurs and permanently discontinue if it does not resolve within 4 weeks or if Grade 4 in severity. ( 2.3, 5.1)
- Hyperphosphatemia: Increases in phosphate levels are a pharmacodynamic effect of BALVERSA. Monitor for hyperphosphatemia and manage with dose modifications when required. ( 2.3, 5.2)
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to the fetus and to use effective contraception ( 5.3, 8.1, 8.3)
Adverse Reactions/Side Effects
The most common adverse reactions including laboratory abnormalities (≥20%) were phosphate increased, stomatitis, fatigue, creatinine increased, diarrhea, dry mouth, nail disorder, alanine aminotransferase increased, alkaline phosphatase increased, sodium decreased, decreased appetite, albumin decreased, dysgeusia, hemoglobin decreased, dry skin, aspartate aminotransferase increased, magnesium decreased, dry eye, alopecia, palmar-plantar erythrodysesthesia syndrome, constipation, phosphate decreased, abdominal pain, calcium increased, nausea, and musculoskeletal pain. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Products, LP. at 1-800-526-7736 (1-800-JANSSEN and www.BALVERSA.com) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Moderate CYP2C9 or strong CYP3A4 inhibitors: Consider alternative agents or monitor closely for adverse reactions. ( 7.1)
- Strong CYP2C9 or CYP3A4 inducers: Avoid concomitant use with BALVERSA. ( 7.1)
- Moderate CYP2C9 or CYP3A4 inducers: Increase BALVERSA dose up to 9 mg. ( 7.1)
- Serum phosphate level-altering agents: Avoid concomitant use with agents that can alter serum phosphate levels before the initial dose modification period. ( 2.3, 7.1)
- CYP3A4 substrates: Avoid concomitant use with sensitive CYP3A4 substrates with narrow therapeutic indices. ( 7.2)
- OCT2 substrates: Consider alternative agents or consider reducing the dose of OCT2 substrates based on tolerability. ( 7.2)
- P-gp substrates: Separate BALVERSA administration by at least 6 hours before or after administration of P-gp substrates with narrow therapeutic indices. ( 7.2)
Use In Specific Populations
- Lactation: Advise not to breastfeed. ( 8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2023
Full Prescribing Information
1. Indications and Usage for Balversa
BALVERSA is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC), that has:
- susceptible FGFR3 or FGFR2 genetic alterations, and
- progressed during or following at least one line of prior platinum-containing chemotherapy, including within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA [see Dosage and Administration (2.1) and Clinical Studies (14.1)] .
This indication is approved under accelerated approval based on tumor response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials [see Clinical Studies (14.1)] .
2. Balversa Dosage and Administration
2.1 Patient Selection
Select patients for the treatment of locally advanced or metastatic urothelial carcinoma with BALVERSA based on the presence of susceptible FGFR genetic alterations in tumor specimens as detected by an FDA-approved companion diagnostic [see Clinical Studies (14.1)] .
Information on FDA-approved tests for the detection of FGFR genetic alterations in urothelial cancer is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage and Schedule
The recommended starting dose of BALVERSA is 8 mg (two 4 mg tablets) orally once daily, with a dose increase to 9 mg (three 3 mg tablets) once daily based on serum phosphate (PO 4) levels and tolerability at 14 to 21 days [see Dosage and Administration (2.3)].
Swallow tablets whole with or without food. If vomiting occurs any time after taking BALVERSA, the next dose should be taken the next day. Treatment should continue until disease progression or unacceptable toxicity occurs.
If a dose of BALVERSA is missed, it can be taken as soon as possible on the same day. Resume the regular daily dose schedule for BALVERSA the next day. Extra tablets should not be taken to make up for the missed dose.
2.3 Dose Modifications for Adverse Reactions
The recommended dose modifications for adverse reactions are listed in Table 1.
| Dose | 1 st dose reduction | 2 nd dose reduction | 3 rd dose reduction | 4 th dose reduction | 5 th dose reduction |
|---|---|---|---|---|---|
| 9 mg ⭢
(three 3 mg tablets) | 8 mg
(two 4 mg tablets) | 6 mg
(two 3 mg tablets) | 5 mg
(one 5 mg tablet) | 4 mg
(one 4 mg tablet) | Stop |
| 8 mg ⭢
(two 4 mg tablets) | 6 mg
(two 3 mg tablets) | 5 mg
(one 5 mg tablet) | 4 mg
(one 4 mg tablet) | Stop | |
Table 2 summarizes recommendations for dose interruption, reduction, or discontinuation of BALVERSA in the management of specific adverse reactions.
| Adverse Reaction | BALVERSA Dose Modification |
|---|---|
|
|
| Hyperphosphatemia | |
| In all patients, restrict phosphate intake to 600–800 mg daily. If serum phosphate is above 7.0 mg/dL, consider adding an oral phosphate binder until serum phosphate level returns to < 5.5 mg/dL. | |
| 5.6–6.9 mg/dL (1.8–2.3 mmol/L) | Continue BALVERSA at current dose. |
| 7.0–9.0 mg/dL (2.3–2.9 mmol/L) | Withhold BALVERSA with weekly reassessments until level returns to < 5.5 mg/dL (or baseline). Then restart BALVERSA at the same dose level. A dose reduction may be implemented for hyperphosphatemia lasting > 1 week. |
| > 9.0 mg/dL (> 2.9 mmol/L) | Withhold BALVERSA with weekly reassessments until level returns to < 5.5 mg/dL (or baseline). Then may restart BALVERSA at 1 dose level lower. |
| > 10.0 mg/dL (> 3.2 mmol/L) or significant alteration in baseline renal function or Grade 3 hypercalcemia | Withhold BALVERSA with weekly reassessments until level returns to < 5.5 mg/dL (or baseline). Then may restart BALVERSA at 2 dose levels lower. |
| Central Serous Retinopathy/Retinal Pigment Epithelial Detachment (CSR/RPED) | |
| Grade 1: Asymptomatic; clinical or diagnostic observations only | Withhold until resolution. If resolves within 4 weeks, resume at the next lower dose level. Then, if no recurrence for a month, consider re-escalation. If stable for 2 consecutive eye exams but not resolved, resume at the next lower dose level. |
| Grade 2: Visual acuity 20/40 or better or ≤ 3 lines of decreased vision from baseline | Withhold until resolution. If resolves within 4 weeks, may resume at the next lower dose level. |
| Grade 3: Visual acuity worse than 20/40 or > 3 lines of decreased vision from baseline | Withhold until resolution. If resolves within 4 weeks, may resume two dose levels lower. If recurs, consider permanent discontinuation. |
| Grade 4: Visual acuity 20/200 or worse in affected eye | Permanently discontinue. |
| Other Adverse Reactions * | |
| Grade 3 | Withhold BALVERSA until resolves to Grade 1 or baseline, then may resume dose level lower. |
| Grade 4 | Permanently discontinue. |
5. Warnings and Precautions
5.1 Ocular Disorders
BALVERSA can cause ocular disorders, including central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED) resulting in visual field defect.
CSR/RPED was reported in 25% of patients treated with BALVERSA, with a median time to first onset of 50 days. Grade 3 CSR/RPED, involving central field of vision, was reported in 3% of patients. CSR/RPED resolved in 13% of patients and was ongoing in 13% of patients at the study cutoff. CSR/RPED led to dose interruptions and reductions in 9% and 14% of patients, respectively and 3% of patients discontinued BALVERSA.
Dry eye symptoms occurred in 28% of patients during treatment with BALVERSA and were Grade 3 in 6% of patients. All patients should receive dry eye prophylaxis with ocular demulcents as needed.
Perform monthly ophthalmological examinations during the first 4 months of treatment and every 3 months afterwards, and urgently at any time for visual symptoms. Ophthalmological examination should include assessment of visual acuity, slit lamp examination, fundoscopy, and optical coherence tomography.
Withhold BALVERSA when CSR occurs and permanently discontinue if it does not resolve within 4 weeks or if Grade 4 in severity. For ocular adverse reactions, follow the dose modification guidelines [see Dosage and Administration (2.3)] .
5.2 Hyperphosphatemia and Soft Tissue Mineralization
BALVERSA can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcinosis, non-uremic calciphylaxis and vascular calcification. Increases in phosphate levels are a pharmacodynamic effect of BALVERSA [see Pharmacodynamics (12.2)]. Hyperphosphatemia was reported as adverse reaction in 76% of patients treated with BALVERSA. The median onset time for any grade event of hyperphosphatemia was 20 days (range: 8–116) after initiating BALVERSA. Thirty-two percent of patients received phosphate binders during treatment with BALVERSA. Cutaneous calcinosis, non-uremic calciphylaxis and vascular calcification have been observed in 0.3% of patients treated with BALVERSA.
Monitor for hyperphosphatemia throughout treatment. In all patients, restrict phosphate intake to 600–800 mg daily. If serum phosphate is above 7.0 mg/dL, consider adding an oral phosphate binder until serum phosphate level returns to <5.5 mg/dL. Withhold, dose reduce, or permanently discontinue BALVERSA based on duration and severity of hyperphosphatemia according to Table 2 [see Dosage and Administration (2.3)].
5.3 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animal reproduction studies, BALVERSA can cause fetal harm when administered to a pregnant woman. In an embryo-fetal toxicity study, oral administration of erdafitinib to pregnant rats during the period of organogenesis caused malformations and embryo-fetal death at maternal exposures that were less than the human exposures at the maximum human recommended dose based on area under the curve (AUC). Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with BALVERSA and for one month after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with BALVERSA and for one month after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)] .
6. Adverse Reactions/Side Effects
The following serious adverse reactions are also described elsewhere in the labeling:
- Ocular Disorders [see Warning and Precautions (5.1)] .
- Hyperphosphatemia [see Warning and Precautions (5.2)] .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of BALVERSA was evaluated in the BLC2001 study that included 87 patients with locally advanced or metastatic urothelial carcinoma which had susceptible FGFR3 or FGFR2 genetic alterations, and which progressed during or following at least one line of prior chemotherapy including within 12 months of neoadjuvant or adjuvant chemotherapy [see Clinical Studies (14.1)] . Patients were treated with BALVERSA at 8 mg orally once daily; with a dose increase to 9 mg in patients with phosphate levels <5.5 mg/dL on Day 14 of Cycle 1. Median duration of treatment was 5.3 months (range: 0 to 17 months).
The most common adverse reactions (ARs) including laboratory abnormalities (≥20%) were phosphate increased, stomatitis, fatigue, creatinine increased, diarrhea, dry mouth, nail disorder, alanine aminotransferase increased, alkaline phosphatase increased, sodium decreased, decreased appetite, albumin decreased, dysgeusia, hemoglobin decreased, dry skin, aspartate aminotransferase increased, magnesium decreased, dry eye, alopecia, palmar-plantar erythrodysesthesia syndrome, constipation, phosphate decreased, abdominal pain, calcium increased, nausea, and musculoskeletal pain. The most common Grade 3 or greater ARs (>1%) were stomatitis, nail dystrophy, palmar-plantar erythrodysesthesia syndrome, paronychia, nail disorder, keratitis, and hyperphosphatemia.
An adverse reaction with a fatal outcome in 1% of patients was acute myocardial infarction.
Serious adverse reactions occurred in 41% of patients including eye disorders (10%).
Permanent discontinuation due to an adverse reaction occurred in 13% of patients. The most frequent reasons for permanent discontinuation included eye disorders (6%).
Dosage interruptions occurred in 68% of patients. The most frequent adverse reactions requiring dosage interruption included hyperphosphatemia (24%), stomatitis (17%), eye disorders (17%), and palmar-plantar erythro-dysesthesia syndrome (8%).
Dose reductions occurred in 53% of patients. The most frequent adverse reactions for dose reductions included eye disorders (23%), stomatitis (15%), hyperphosphatemia (7%), palmar-plantar erythro-dysesthesia syndrome (7%), paronychia (7%), and nail dystrophy (6%).
Table 3 presents ARs reported in ≥10% of patients treated with BALVERSA at 8 mg once daily.
| BALVERSA 8 mg daily (N=87) | ||
|---|---|---|
| Adverse Reaction | All Grades (%) | Grade 3–4 (%) |
|
||
| Any | 100 | 67 |
| Gastrointestinal disorders | 92 | 24 |
| Stomatitis | 56 | 9 |
| Diarrhea | 47 | 2 |
| Dry mouth | 45 | 0 |
| Constipation | 28 | 1 |
| Abdominal pain * | 23 | 2 |
| Nausea | 21 | 1 |
| Vomiting | 13 | 2 |
| Metabolism and nutrition disorders | 90 | 16 |
| Decreased appetite | 38 | 0 |
| General disorders and admin. site conditions | 69 | 13 |
| Fatigue † | 54 | 10 |
| Pyrexia | 14 | 1 |
| Skin and subcutaneous disorders | 75 | 16 |
| Nail disorder ‡ | 45 | 10 |
| Dry skin § | 34 | 0 |
| Palmar-plantar erythrodysesthesia | 26 | 6 |
| Alopecia | 26 | 0 |
| Nail discoloration | 11 | 0 |
| Eye disorders | 62 | 11 |
| Dry eye ¶ | 28 | 6 |
| Vision blurred | 17 | 0 |
| Lacrimation increased | 10 | 0 |
| Nervous system disorders | 57 | 5 |
| Dysgeusia | 37 | 1 |
| Infections and infestations | 56 | 20 |
| Paronychia | 17 | 3 |
| Urinary tract infection | 17 | 6 |
| Conjunctivitis | 11 | 0 |
| Respiratory, thoracic and mediastinal disorders | 40 | 7 |
| Oropharyngeal pain | 11 | 1 |
| Dyspnea # | 10 | 2 |
| Renal and urinary tract disorders | 38 | 10 |
| Hematuria | 11 | 2 |
| Musculoskeletal and connective tissue disorders | 31 | 0 |
| Musculoskeletal pain Þ | 20 | 0 |
| Arthralgia | 11 | 0 |
| Investigations | 44 | 5 |
| Weight decreased ß | 16 | 0 |
| BALVERSA 8 mg daily (N=86 *) | ||
|---|---|---|
| Laboratory Abnormality | All Grades (%) | Grade 3–4 (%) |
|
||
| Hematology | ||
| Hemoglobin decreased | 35 | 3 |
| Platelets decreased | 19 | 1 |
| Leukocytes decreased | 17 | 0 |
| Neutrophils decreased | 10 | 2 |
| Chemistry | ||
| Phosphate increased | 76 | 1 |
| Creatinine increased | 52 | 5 |
| Sodium decreased | 40 | 16 |
| Alanine aminotransferase increased | 41 | 1 |
| Alkaline phosphatase increased | 41 | 1 |
| Albumin decreased | 37 | 0 |
| Aspartate aminotransferase increased | 30 | 0 |
| Magnesium decreased | 30 | 1 |
| Phosphate decreased | 24 | 9 |
| Calcium increased | 22 | 3 |
| Potassium increased | 16 | 0 |
| Fasting glucose decreased | 10 | 0 |
7. Drug Interactions
7.1 Effect of Other Drugs on BALVERSA
Table 5 summarizes drug interactions that affect the exposure of BALVERSA or serum phosphate level and their clinical management.
| Moderate CYP2C9 or Strong CYP3A4 Inhibitors | |
| Clinical Impact |
|
| Clinical Management |
|
| Strong CYP2C9 or CYP3A4 Inducers | |
| Clinical Impact |
|
| Clinical Management |
|
| Moderate CYP2C9 or CYP3A4 Inducers | |
| Clinical Impact |
|
| Clinical Management |
|
| Serum Phosphate Level-Altering Agents | |
| Clinical Impact |
|
| Clinical Management |
|
7.2 Effect of BALVERSA on Other Drugs
Table 6 summarizes the effect of BALVERSA on other drugs and their clinical management.
| CYP3A4 Substrates | |
| Clinical Impact |
|
| Clinical Management |
|
| OCT2 Substrates | |
| Clinical Impact |
|
| Clinical Management |
|
| P-glycoprotein (P-gp) Substrates | |
| Clinical Impact |
|
| Clinical Management |
|
8. Use In Specific Populations
8.4 Pediatric Use
Safety and effectiveness of BALVERSA in pediatric patients have not been established.
In 4 and 13-week repeat-dose toxicology studies in rats and dogs, toxicities in bone and teeth were observed at an exposure less than the human exposure (AUC) at the maximum recommended human dose. Chondroid dysplasia/metaplasia were reported in multiple bones in both species, and tooth abnormalities included abnormal/irregular denting in rats and dogs and discoloration and degeneration of odontoblasts in rats.
8.5 Geriatric Use
Of the 416 patients treated with BALVERSA in clinical studies, 45% were 65 years of age or older, and 12% were 75 years of age or older. No overall differences in safety or effectiveness were observed between these patients and younger patients [see Clinical Studies (14.1)].
8.6 Renal Impairment
No dose adjustment is recommended for patients with mild to moderate renal impairment [estimated glomerular filtration rate (eGFR) 30 to 89 mL/min/1.73 m 2]. No data are available in patients with severe renal impairment [see Clinical Pharmacology (12.3)] .
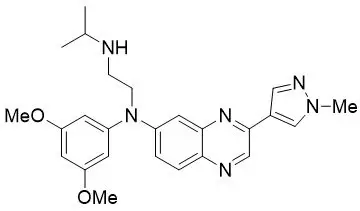
11. Balversa Description
Erdafitinib, the active ingredient in BALVERSA, is a kinase inhibitor. The chemical name is N-(3,5-dimethoxyphenyl)-N'-(1-methylethyl)-N-[3-(1-methyl-1H-pyrazol-4-yl)quinoxalin-6-yl]ethane-1,2-diamine. Erdafitinib is a yellow powder. It is practically insoluble, or insoluble to freely soluble in organic solvents, and slightly soluble to practically insoluble, or insoluble in aqueous media over a wide range of pH values. The molecular formula is C 25H 30N 6O 2 and molecular weight is 446.56.
Chemical structure of erdafitinib is as follows:
BALVERSA ® (erdafitinib) tablets are supplied as 3 mg, 4 mg or 5 mg film-coated tablets for oral administration and contains the following inactive ingredients:
Tablet Core: Croscarmellose sodium, Magnesium stearate (from vegetable source), Mannitol, Meglumine, and Microcrystalline Cellulose.
Film Coating: (Opadry amb II): Glycerol monocaprylocaprate Type I, Polyvinyl alcohol-partially hydrolyzed, Sodium lauryl sulfate, Talc, Titanium dioxide, Iron oxide yellow, Iron oxide red (for the orange and brown tablets only), Ferrosoferric oxide/iron oxide black (for the brown tablets only).
12. Balversa - Clinical Pharmacology
12.1 Mechanism of Action
Erdafitinib is a kinase inhibitor that binds to and inhibits enzymatic activity of FGFR1, FGFR2, FGFR3 and FGFR4 based on in vitro data. Erdafitinib also binds to RET, CSF1R, PDGFRA, PDGFRB, FLT4, KIT, and VEGFR2. Erdafitinib inhibited FGFR phosphorylation and signaling and decreased cell viability in cell lines expressing FGFR genetic alterations, including point mutations, amplifications, and fusions. Erdafitinib demonstrated antitumor activity in FGFR-expressing cell lines and xenograft models derived from tumor types, including bladder cancer.
12.2 Pharmacodynamics
12.3 Pharmacokinetics
Following administration of 8 mg once daily, the mean (coefficient of variation [CV%]) erdafitinib steady-state maximum observed plasma concentration (C max), area under the curve (AUC tau), and minimum observed plasma concentration (C min) were 1,399 ng/mL (51%), 29,268 ng∙h/mL (60%), and 936 ng/mL (65%), respectively.
Following single and repeat once daily dosing, erdafitinib exposure (maximum observed plasma concentration [C max] and area under the plasma concentration time curve [AUC]) increased proportionally across the dose range of 0.5 to 12 mg (0.06 to 1.3 times the maximum approved recommended dose). Steady state was achieved after 2 weeks with once daily dosing and the mean accumulation ratio was 4-fold.
Drug Interaction Studies
12.5 Pharmacogenomics
CYP2C9 activity is reduced in individuals with genetic variants, such as the CYP2C9*2 and CYP2C9*3 polymorphisms. Erdafitinib exposure was similar in subjects with CYP2C9*1/*2 and *1/*3 genotypes relative to subjects with CYP2C9*1/*1 genotype (wild type). No data are available in subjects characterized by other genotypes (e.g., *2/*2, *2/*3, *3/*3). Simulation suggested no clinically meaningful differences in erdafitinib exposure in subjects with CYP2C9*2/*2 and *2/*3 genotypes. The exposure of erdafitinib is predicted to be 50% higher in subjects with the CYP2C9*3/*3 genotype, estimated to be present in 0.4% to 3% of the population among various ethnic groups.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenicity studies have not been conducted with erdafitinib.
Erdafitinib was not mutagenic in a bacterial reverse mutation (Ames) assay and was not clastogenic in an in vitro micronucleus or an in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with erdafitinib. In the 3-month repeat-dose toxicity study, erdafitinib showed effects on female reproductive organs (necrosis of the ovarian corpora lutea) in rats at an exposure less than the human exposure (AUC) at maximum recommended human dose.
14. Clinical Studies
14.1 Urothelial Carcinoma with Susceptible FGFR Genetic Alterations
Study BLC2001 (NCT02365597) was a multicenter, open-label, single-arm study to evaluate the efficacy and safety of BALVERSA in patients with locally advanced or metastatic urothelial carcinoma (mUC). Fibroblast growth factor receptor (FGFR) mutation status for screening and enrollment of patients was determined by a clinical trial assay (CTA). The efficacy population consists of a cohort of eighty-seven patients who were enrolled in this study with disease that had progressed on or after at least one prior chemotherapy and that had at least 1 of the following genetic alterations: FGFR3 gene mutations (R248C, S249C, G370C, Y373C) or FGFR gene fusions (FGFR3-TACC3, FGFR3-BAIAP2L1, FGFR2-BICC1, FGFR2-CASP7), as determined by the CTA performed at a central laboratory. Tumor samples from 69 patients were tested retrospectively by the QIAGEN therascreen® FGFR RGQ RT-PCR Kit, which is the FDA-approved test for selection of patients with mUC for BALVERSA.
Patients received a starting dose of BALVERSA at 8 mg once daily with a dose increase to 9 mg once daily in patients whose serum phosphate levels were below the target of 5.5 mg/dL between days 14 and 17; a dose increase occurred in 41% of patients. BALVERSA was administered until disease progression or unacceptable toxicity. The major efficacy outcome measures were objective response rate (ORR) and duration of response (DoR), as determined by blinded independent review committee (BIRC) according to RECIST v1.1.
The median age was 67 years (range: 36 to 87 years), 79% were male, and 74% were Caucasian. Most patients (92%) had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Sixty-six percent of patients had visceral metastases. Eighty-four (97%) patients received at least one of cisplatin or carboplatin previously. Fifty-six percent of patients only received prior cisplatin-based regimens, 29% received only prior carboplatin-based regimens, and 10% received both cisplatin and carboplatin-based regimens. Three (3%) patients had disease progression following prior platinum-containing neoadjuvant or adjuvant therapy only. Twenty-four percent of patients had been treated with prior anti PD-L1/PD-1 therapy.
Efficacy results are summarized in Table 7 and Table 8. Overall response rate was 32.2%. Responders included patients who had previously not responded to anti PD-L1/PD-1 therapy.
| BIRC * Assessment | |
|---|---|
| Endpoint | N=87 |
| ORR = CR + PR
CI = Confidence Interval |
|
|
|
| ORR (95% CI) | 32.2% (22.4, 42.0) |
| Complete response (CR) | 2.3% |
| Partial response (PR) | 29.9% |
| Median DoR in months (95% CI) | 5.4 (4.2, 6.9) |
| BIRC * Assessment | |
|---|---|
| ORR = CR + PR
CI = Confidence Interval |
|
|
|
| FGFR3 Point Mutation | N=64 |
| ORR (95% CI) | 40.6% (28.6, 52.7) |
| FGFR3 Fusion †, ‡ | N=18 |
| ORR (95% CI) | 11.1% (0, 25.6) |
| FGFR2 Fusion ‡ | N=6 |
| ORR | 0 |
16. How is Balversa supplied
BALVERSA ® (erdafitinib) tablets are available in the strengths and packages listed below:
- 3 mg tablets: Yellow, round biconvex, film-coated, debossed with "3" on one side and "EF" on the other side.
- Bottle of 56-tablets with child resistant closure (NDC 59676-030-56).
- Bottle of 84-tablets with child resistant closure (NDC 59676-030-84).
- 4 mg tablets: Orange, round biconvex, film-coated, debossed with "4" on one side and "EF" on the other side.
- Bottle of 28-tablets with child resistant closure (NDC 59676-040-28).
- Bottle of 56-tablets with child resistant closure (NDC 59676-040-56).
- 5 mg tablets: Brown, round biconvex, film-coated, debossed with "5" on one side and "EF" on the other side.
- Bottle of 28-tablets with child resistant closure (NDC 59676-050-28).

| BALVERSA
erdafitinib tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| BALVERSA
erdafitinib tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| BALVERSA
erdafitinib tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Janssen Products LP (804684207) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Cilag AG | 483237103 | api manufacture(59676-030, 59676-040, 59676-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Johnson & Johnson Private Limited | 677603030 | analysis(59676-030, 59676-040, 59676-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Cilag SpA | 542797928 | manufacture(59676-030, 59676-040, 59676-050) , analysis(59676-030, 59676-040, 59676-050) , pack(59676-030, 59676-040, 59676-050) | |
