Drug Detail:Enhertu (Fam-trastuzumab deruxtecan [ fam-tras-tooz-ue-mab-der-ux-tee-kan ])
Drug Class: HER2 inhibitors
Highlights of Prescribing Information
ENHERTU® (fam-trastuzumab deruxtecan-nxki) for injection, for intravenous use
Initial U.S. Approval: 2019
WARNING: INTERSTITIAL LUNG DISEASE and EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning.
- Interstitial lung disease (ILD) and pneumonitis, including fatal cases, have been reported with ENHERTU. Monitor for and promptly investigate signs and symptoms including cough, dyspnea, fever, and other new or worsening respiratory symptoms. Permanently discontinue ENHERTU in all patients with Grade 2 or higher ILD/pneumonitis. Advise patients of the risk and to immediately report symptoms. (2.3, 5.1)
- Exposure to ENHERTU during pregnancy can cause embryo-fetal harm. Advise patients of these risks and the need for effective contraception. (5.4, 8.1, 8.3)
Recent Major Changes
| Indications and Usage (1.1) | 05/2022 |
| Indications and Usage (1.2) | 11/2022 |
| Indications and Usage (1.3) | 08/2022 |
| Dosage and Administration (2.1) | 08/2022 |
| Dosage and Administration (2.2) | 05/2022 |
| Dosage and Administration (2.2) | 08/2022 |
| Dosage and Administration (2.3) | 08/2022 |
| Warnings and Precautions (5.1, 5.2, 5.3) | 08/2022 |
Indications and Usage for Enhertu
ENHERTU is a HER2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of:
- adult patients with unresectable or metastatic HER2-positive breast cancer who have received a prior anti-HER2-based regimen either:
- in the metastatic setting, or
- in the neoadjuvant or adjuvant setting and have developed disease recurrence during or within six months of completing therapy. (1.1)
- adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer, as determined by an FDA-approved test, who have received a prior chemotherapy in the metastatic setting or developed disease recurrence during or within 6 months of completing adjuvant chemotherapy. (1.2)
- adult patients with unresectable or metastatic non-small cell lung cancer (NSCLC) whose tumors have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have received a prior systemic therapy. (1.3)
This indication is approved under accelerated approval based on objective response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. (1.3, 14.3) - adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen. (1.4)
Enhertu Dosage and Administration
- Do not substitute ENHERTU for or with trastuzumab or ado-trastuzumab emtansine. (2.2, 2.4)
- For intravenous infusion only. Do not administer as an intravenous push or bolus. DO NOT use Sodium Chloride Injection, USP. (2.4)
- Premedicate for prevention of chemotherapy-induced nausea and vomiting. (2.2)
- The recommended dosage of ENHERTU for breast cancer is 5.4 mg/kg given as an intravenous infusion once every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity. (2.2, 2.3)
- The recommended dosage of ENHERTU for lung cancer is 5.4 mg/kg given as an intravenous infusion once every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity. (2.2, 2.3)
- The recommended dosage of ENHERTU for gastric cancer is 6.4 mg/kg given as an intravenous infusion once every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity. (2.2, 2.3)
- Management of adverse reactions (ILD, neutropenia, thrombocytopenia, or left ventricular dysfunction) may require temporary interruption, dose reduction, or discontinuation of ENHERTU. (2.3)
Dosage Forms and Strengths
For injection: 100 mg lyophilized powder in a single-dose vial (3)
Contraindications
None. (4)
Warnings and Precautions
- Neutropenia: Monitor complete blood counts prior to initiation of ENHERTU and prior to each dose, and as clinically indicated. Manage through treatment interruption or dose reduction. (2.3, 5.2)
- Left Ventricular Dysfunction: Assess LVEF prior to initiation of ENHERTU and at regular intervals during treatment as clinically indicated. Manage through treatment interruption or discontinuation. Permanently discontinue ENHERTU in patients with symptomatic congestive heart failure (CHF). (2.3, 5.3)
Adverse Reactions/Side Effects
The most common adverse reactions (≥20%), including laboratory abnormalities, in patients with:
- metastatic breast cancer and HER2-mutant NSCLC were nausea, decreased white blood cell count, decreased hemoglobin, decreased neutrophil count, decreased lymphocyte count, fatigue, decreased platelet count, increased aspartate aminotransferase, vomiting, increased alanine aminotransferase, alopecia, increased blood alkaline phosphatase, constipation, musculoskeletal pain, decreased appetite, hypokalemia, diarrhea, and respiratory infection. (6.1)
- gastric cancer were decreased hemoglobin, decreased white blood cell count, decreased neutrophil count, decreased lymphocyte count, decreased platelet count, nausea, decreased appetite, increased aspartate aminotransferase, fatigue, increased blood alkaline phosphatase, increased alanine aminotransferase, diarrhea, hypokalemia, vomiting, constipation, increased blood bilirubin, pyrexia, and alopecia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Daiichi Sankyo, Inc. at 1-877-437-7763 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
- Females and Males of Reproductive Potential: Verify pregnancy status of females prior to initiation of ENHERTU. (8.3)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2022
Full Prescribing Information
WARNING: INTERSTITIAL LUNG DISEASE and EMBRYO-FETAL TOXICITY
- Interstitial Lung Disease (ILD) and pneumonitis, including fatal cases, have been reported with ENHERTU. Monitor for and promptly investigate signs and symptoms including cough, dyspnea, fever, and other new or worsening respiratory symptoms. Permanently discontinue ENHERTU in all patients with Grade 2 or higher ILD/pneumonitis. Advise patients of the risk and the need to immediately report symptoms [see Dosage and Administration (2.3), Warnings and Precautions (5.1)].
- Embryo-Fetal Toxicity: Exposure to ENHERTU during pregnancy can cause embryo-fetal harm. Advise patients of these risks and the need for effective contraception [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
1. Indications and Usage for Enhertu
1.1 HER2-Positive Metastatic Breast Cancer
ENHERTU is indicated for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received a prior anti-HER2-based regimen either:
- in the metastatic setting, or
- in the neoadjuvant or adjuvant setting and have developed disease recurrence during or within six months of completing therapy.
1.2 HER2-Low Metastatic Breast Cancer
ENHERTU is indicated for the treatment of adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer, as determined by an FDA-approved test, who have received a prior chemotherapy in the metastatic setting or developed disease recurrence during or within 6 months of completing adjuvant chemotherapy [see Dosage and Administration (2.1)].
1.3 Unresectable or Metastatic HER2-Mutant Non-Small Cell Lung Cancer
ENHERTU is indicated for the treatment of adult patients with unresectable or metastatic non-small cell lung cancer (NSCLC) whose tumors have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have received a prior systemic therapy.
This indication is approved under accelerated approval based on objective response rate and duration of response [see Clinical Studies (14.3)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
2. Enhertu Dosage and Administration
2.2 Recommended Dosage and Schedules
Do not substitute ENHERTU for or with trastuzumab or ado-trastuzumab emtansine.
Slow or interrupt the infusion rate if the patient develops infusion-related symptoms.
Permanently discontinue ENHERTU in case of severe infusion reactions.
2.3 Dosage Modifications
Management of adverse reactions may require temporary interruption, dose reduction, or treatment discontinuation of ENHERTU as described in Tables 1 and 2.
Do not re-escalate the ENHERTU dose after a dose reduction is made.
If a planned dose is delayed or missed, administer as soon as possible; do not wait until the next planned cycle. Adjust the schedule of administration to maintain a 3-week interval between doses. Administer the infusion at the dose and rate the patient tolerated in the most recent infusion.
| Dose Reduction Schedule | Breast Cancer and NSCLC | Gastric Cancer |
|---|---|---|
| Recommended starting dose | 5.4 mg/kg | 6.4 mg/kg |
| First dose reduction | 4.4 mg/kg | 5.4 mg/kg |
| Second dose reduction | 3.2 mg/kg | 4.4 mg/kg |
| Requirement for further dose reduction | Discontinue treatment. | Discontinue treatment. |
| Adverse Reaction | Severity | Treatment Modification | |
|---|---|---|---|
| Toxicity grades are in accordance with National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0 (NCI CTCAE v.5.0). | |||
| Interstitial Lung Disease (ILD)/ Pneumonitis [see Warnings and Precautions (5.1)] | Asymptomatic ILD/pneumonitis (Grade 1) | Interrupt ENHERTU until resolved to Grade 0, then:
|
|
| Symptomatic ILD/pneumonitis (Grade 2 or greater) |
|
||
| Neutropenia [see Warnings and Precautions (5.2)] | Grade 3 (less than 1.0 to 0.5 × 109/L) |
|
|
| Grade 4 (less than 0.5 × 109/L) |
|
||
| Febrile Neutropenia [see Warnings and Precautions (5.2)] | Absolute neutrophil count of less than 1.0 × 109/L and temperature greater than 38.3°C or a sustained temperature of 38°C or greater for more than one hour |
|
|
| Thrombocytopenia [see Adverse Reactions (6.1)] | Grade 3 (platelets less than 50 to 25 × 109/L) |
|
|
| Grade 4 (platelets less than 25 × 109/L) |
|
||
| Left Ventricular Dysfunction [see Warnings and Precautions (5.3)] | LVEF greater than 45% and absolute decrease from baseline is 10% to 20% |
|
|
| LVEF 40% to 45% | And absolute decrease from baseline is less than 10% |
|
|
| And absolute decrease from baseline is 10% to 20% |
|
||
| LVEF less than 40% or absolute decrease from baseline is greater than 20% |
|
||
| Symptomatic congestive heart failure (CHF) |
|
||
2.4 Preparation and Administration
In order to prevent medication errors, check the vial labels to ensure that the drug being prepared and administered is ENHERTU (fam-trastuzumab deruxtecan-nxki) and not trastuzumab or ado-trastuzumab emtansine.
Reconstitute and further dilute ENHERTU prior to intravenous infusion. Use appropriate aseptic technique.
ENHERTU (fam-trastuzumab deruxtecan-nxki) is a hazardous drug. Follow applicable special handling and disposal procedures.1
3. Dosage Forms and Strengths
For injection: 100 mg of fam-trastuzumab deruxtecan-nxki as a white to yellowish white lyophilized powder in a single-dose vial for reconstitution and further dilution
5. Warnings and Precautions
5.1 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD), including pneumonitis, can occur in patients treated with ENHERTU [see Adverse Reactions (6.1)]. A higher incidence of Grade 1 and 2 ILD/pneumonitis has been observed in patients with moderate renal impairment.
Advise patients to immediately report cough, dyspnea, fever, and/or any new or worsening respiratory symptoms. Monitor patients for signs and symptoms of ILD. Promptly investigate evidence of ILD. Evaluate patients with suspected ILD by radiographic imaging. Consider consultation with a pulmonologist. For asymptomatic (Grade 1) ILD, consider corticosteroid treatment (e.g., ≥0.5 mg/kg/day prednisolone or equivalent). Withhold ENHERTU until recovery [see Dosage and Administration (2.3)]. In cases of symptomatic ILD (Grade 2 or greater), promptly initiate systemic corticosteroid treatment (e.g., ≥1 mg/kg/day prednisolone or equivalent) and continue for at least 14 days followed by gradual taper for at least 4 weeks. Permanently discontinue ENHERTU in patients who are diagnosed with symptomatic (Grade 2 or greater) ILD [see Dosage and Administration (2.3)].
5.2 Neutropenia
Severe neutropenia, including febrile neutropenia, can occur in patients treated with ENHERTU.
Monitor complete blood counts prior to initiation of ENHERTU and prior to each dose, and as clinically indicated. Based on the severity of neutropenia, ENHERTU may require dose interruption or reduction [see Dosage and Administration (2.3)].
5.3 Left Ventricular Dysfunction
Patients treated with ENHERTU may be at increased risk of developing left ventricular dysfunction. Left ventricular ejection fraction (LVEF) decrease has been observed with anti-HER2 therapies, including ENHERTU.
Assess LVEF prior to initiation of ENHERTU and at regular intervals during treatment as clinically indicated. Manage LVEF decrease through treatment interruption. Permanently discontinue ENHERTU if LVEF of less than 40% or absolute decrease from baseline of greater than 20% is confirmed. Permanently discontinue ENHERTU in patients with symptomatic congestive heart failure (CHF) [see Dosage and Administration (2.3)].
Treatment with ENHERTU has not been studied in patients with a history of clinically significant cardiac disease or LVEF less than 50% prior to initiation of treatment.
5.4 Embryo-Fetal Toxicity
Based on its mechanism of action, ENHERTU can cause fetal harm when administered to a pregnant woman. In postmarketing reports, use of a HER2-directed antibody during pregnancy resulted in cases of oligohydramnios manifesting as fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death. Based on its mechanism of action, the topoisomerase inhibitor component of ENHERTU, DXd, can also cause embryo-fetal harm when administered to a pregnant woman because it is genotoxic and targets actively dividing cells [see Use in Specific Populations (8.1), Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. Advise patients of the potential risks to a fetus.
Verify the pregnancy status of females of reproductive potential prior to the initiation of ENHERTU. Advise females of reproductive potential to use effective contraception during treatment and for 7 months after the last dose of ENHERTU. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ENHERTU and for 4 months after the last dose of ENHERTU [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.1)]
- Neutropenia [see Warnings and Precautions (5.2)]
- Left Ventricular Dysfunction [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
HER2-Positive Metastatic Breast Cancer
DESTINY-Breast03
The safety of ENHERTU was evaluated in 257 patients with unresectable or metastatic HER2-positive breast cancer who received at least one dose of ENHERTU 5.4 mg/kg in DESTINY-Breast03 [see Clinical Studies (14.1)]. ENHERTU was administered by intravenous infusion once every three weeks. The median duration of treatment was 14 months (range: 0.7 to 30) for patients who received ENHERTU and 7 months (range: 0.7 to 25) for patients who received ado-trastuzumab emtansine.
Serious adverse reactions occurred in 19% of patients receiving ENHERTU. Serious adverse reactions in >1% of patients who received ENHERTU were vomiting, interstitial lung disease, pneumonia, pyrexia, and urinary tract infection. Fatalities due to adverse reactions occurred in 0.8% of patients including COVID-19 and sudden death (one patient each).
ENHERTU was permanently discontinued in 14% of patients, of which ILD/pneumonitis accounted for 8%. Dose interruptions due to adverse reactions occurred in 44% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose interruption were neutropenia, leukopenia, anemia, thrombocytopenia, pneumonia, nausea, fatigue, and ILD/pneumonitis. Dose reductions occurred in 21% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose reduction were nausea, neutropenia, and fatigue.
The most common (≥20%) adverse reactions, including laboratory abnormalities, were nausea, decreased white blood cell count, decreased neutrophil count, increased aspartate aminotransferase, decreased hemoglobin, decreased lymphocyte count, increased alanine aminotransferase, decreased platelet count, fatigue, vomiting, increased blood alkaline phosphatase, alopecia, hypokalemia, constipation, musculoskeletal pain, diarrhea, decreased appetite, headache, respiratory infection, abdominal pain, increased blood bilirubin, and stomatitis.
Tables 3 and 4 summarize common adverse reactions and laboratory abnormalities observed in DESTINY-Breast03.
| Adverse Reactions | ENHERTU 5.4 mg/kg N=257 | Ado-trastuzumab emtansine 3.6 mg/kg N=261 |
||
|---|---|---|---|---|
| All Grades % | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
| Events were graded using NCI CTCAE version 5.0. | ||||
|
||||
| Gastrointestinal Disorders | ||||
| Nausea | 76 | 7 | 30 | 0.4 |
| Vomiting | 49 | 1.6 | 10 | 0.8 |
| Constipation | 34 | 0 | 20 | 0 |
| Diarrhea | 29 | 1.2 | 7 | 0.4 |
| Abdominal pain* | 21 | 0.8 | 8 | 0.4 |
| Stomatitis† | 20 | 0.8 | 5 | 0 |
| Dyspepsia | 11 | 0 | 6 | 0 |
| General Disorders and Administration Site Conditions | ||||
| Fatigue‡ | 49 | 6 | 35 | 0.8 |
| Blood and Lymphatic System Disorders | ||||
| Anemia§ | 33 | 7 | 17 | 6 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Alopecia¶ | 37 | 0.4 | 3.1 | 0 |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Musculoskeletal pain# | 31 | 1.2 | 25 | 0.4 |
| Metabolism and Nutrition Disorders | ||||
| Decreased appetite | 29 | 1.6 | 17 | 0.4 |
| Investigations | ||||
| Decreased weight | 17 | 1.2 | 6 | 0.4 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Respiratory infectionÞ | 22 | 0.8 | 12 | 1.1 |
| Epistaxis | 11 | 0 | 16 | 0.4 |
| Cough | 11 | 0.4 | 10 | 0 |
| Interstitial lung diseaseß | 11 | 0.8 | 1.9 | 0 |
| Nervous System Disorders | ||||
| Headacheà | 22 | 0.4 | 16 | 0 |
| Peripheral neuropathyè | 13 | 0.4 | 14 | 0.4 |
| Dizziness | 13 | 0.4 | 8 | 0 |
Other clinically relevant adverse reactions reported in less than 10% of patients in the ENHERTU-treated group were:
- Respiratory, Thoracic and Mediastinal Disorders: dyspnea (8%)
- Skin and Subcutaneous Tissue Disorders: pruritus (8%) and skin hyperpigmentation (6%) [including skin hyperpigmentation, skin discoloration, and pigmentation disorder]
- Nervous System Disorders: dysgeusia (6%)
- Metabolism and Nutrition Disorders: dehydration (4.3%)
- Eye Disorders: blurred vision (3.5%)
- Cardiac Disorders: asymptomatic left ventricular ejection fraction decrease (2.7%) [see Warnings and Precautions (5.3)]
- Injury, Poisoning and Procedural Complications: infusion-related reactions (2.3%) [including hypersensitivity and infusion-related reactions]
- Blood and Lymphatic System Disorders: febrile neutropenia (0.8%)
| Laboratory Parameter | ENHERTU 5.4 mg/kg N=257 | Ado-trastuzumab emtansine 3.6 mg/kg N=261 |
||
|---|---|---|---|---|
| All Grades % | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
| Percentages were calculated using patients with worsening laboratory values from baseline and the number of patients with both baseline and post-treatment measurements as the denominator. | ||||
| Frequencies were based on NCI CTCAE v.5.0 grade-derived laboratory abnormalities. | ||||
| Hematology | ||||
| Decreased white blood cell count | 74 | 8 | 24 | 0.8 |
| Decreased neutrophil count | 70 | 18 | 30 | 2.3 |
| Decreased hemoglobin | 64 | 7 | 38 | 6 |
| Decreased lymphocyte count | 55 | 14 | 23 | 3.9 |
| Decreased platelet count | 52 | 7 | 79 | 24 |
| Chemistry | ||||
| Increased aspartate aminotransferase | 67 | 0.8 | 83 | 5 |
| Increased alanine aminotransferase | 53 | 1.6 | 67 | 6 |
| Increased blood alkaline phosphatase | 49 | 0.8 | 46 | 0.8 |
| Hypokalemia | 35 | 4.7 | 39 | 1.5 |
| Increased blood bilirubin | 20 | 0 | 14 | 0 |
| Increased blood creatinine | 16 | 0.8 | 8 | 0.4 |
DESTINY-Breast01 and Study DS8201-A-J101
The safety of ENHERTU was evaluated in a pooled analysis of 234 patients with unresectable or metastatic HER2-positive breast cancer who received at least one dose of ENHERTU 5.4 mg/kg in DESTINY-Breast01 and Study DS8201-A-J101 (NCT02564900) [see Clinical Studies (14.1)]. ENHERTU was administered by intravenous infusion once every three weeks. The median duration of treatment was 7 months (range: 0.7 to 31).
In the pooled 234 patients, the median age was 56 years (range: 28-96), 74% of patients were <65 years, 99.6% of patients were female, and the majority were White (51%) or Asian (42%). Patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 (58%) or 1 (42%) at baseline. Ninety-four percent had visceral disease, 31% had bone metastases, and 13% had brain metastases.
Serious adverse reactions occurred in 20% of patients receiving ENHERTU. Serious adverse reactions in >1% of patients who received ENHERTU were interstitial lung disease, pneumonia, vomiting, nausea, cellulitis, hypokalemia, and intestinal obstruction. Fatalities due to adverse reactions occurred in 4.3% of patients including interstitial lung disease (2.6%), and the following events occurred in one patient each (0.4%): acute hepatic failure/acute kidney injury, general physical health deterioration, pneumonia, and hemorrhagic shock.
ENHERTU was permanently discontinued in 9% of patients, of which ILD accounted for 6%. Dose interruptions due to adverse reactions occurred in 33% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose interruption were neutropenia, anemia, thrombocytopenia, leukopenia, upper respiratory tract infection, fatigue, nausea, and ILD. Dose reductions occurred in 18% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose reduction were fatigue, nausea, and neutropenia.
The most common (≥20%) adverse reactions, including laboratory abnormalities, were nausea, decreased white blood cell count, decreased hemoglobin, decreased neutrophil count, fatigue, vomiting, alopecia, increased aspartate aminotransferase, increased alanine aminotransferase, decreased platelet count, constipation, decreased appetite, diarrhea, hypokalemia, and cough.
Tables 5 and 6 summarize common adverse reactions and laboratory abnormalities observed in ENHERTU-treated patients in DESTINY-Breast01 and Study DS8201-A-J101.
| Adverse Reactions | ENHERTU 5.4 mg/kg N=234 |
|
|---|---|---|
| All Grades % | Grades 3 or 4 % |
|
| Events were graded using NCI CTCAE version 4.03. | ||
|
||
| Gastrointestinal Disorders | ||
| Nausea | 79 | 7 |
| Vomiting | 47 | 3.8 |
| Constipation | 35 | 0.9 |
| Diarrhea | 29 | 1.7 |
| Abdominal pain* | 19 | 1.3 |
| Stomatitis† | 14 | 0.9 |
| Dyspepsia | 12 | 0 |
| General Disorders and Administration Site Conditions | ||
| Fatigue‡ | 59 | 6 |
| Skin and Subcutaneous Tissue Disorders | ||
| Alopecia | 46 | 0.4§ |
| Rash¶ | 10 | 0 |
| Metabolism and Nutrition Disorders | ||
| Decreased appetite | 32 | 1.3 |
| Blood and Lymphatic System Disorders | ||
| Anemia# | 31 | 7 |
| Respiratory, Thoracic and Mediastinal Disorders | ||
| Cough | 20 | 0 |
| Dyspnea | 13 | 1.3 |
| Epistaxis | 13 | 0 |
| Interstitial lung diseaseÞ | 9 | 2.6ß |
| Nervous System Disorders | ||
| Headacheà | 19 | 0 |
| Dizziness | 10 | 0 |
| Infections and Infestations | ||
| Upper respiratory tract infectionè | 15 | 0 |
| Eye Disorders | ||
| Dry eye | 11 | 0.4ð |
Other clinically relevant adverse reactions reported in less than 10% of patients were:
- Injury, Poisoning and Procedural Complications: infusion-related reactions (2.6%)
- Blood and Lymphatic System Disorders: febrile neutropenia (1.7%)
| Laboratory Parameter | ENHERTU 5.4 mg/kg N=234 |
|
|---|---|---|
| All Grades % | Grades 3 or 4 % |
|
| Percentages were calculated using patients with worsening laboratory values from baseline and the number of patients with both baseline and post-treatment measurements as the denominator. | ||
| Frequencies were based on NCI CTCAE v.4.03 grade-derived laboratory abnormalities. | ||
| Hematology | ||
| Decreased white blood cell count | 70 | 7 |
| Decreased hemoglobin | 70 | 7 |
| Decreased neutrophil count | 62 | 16 |
| Decreased platelet count | 37 | 3.4 |
| Chemistry | ||
| Increased aspartate aminotransferase | 41 | 0.9 |
| Increased alanine aminotransferase | 38 | 0.4 |
| Hypokalemia | 26 | 3 |
HER2-Low Metastatic Breast Cancer
The safety of ENHERTU was evaluated in 371 patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer who received ENHERTU 5.4 mg/kg in DESTINY-Breast04 [see Clinical Studies (14.2)]. ENHERTU was administered by intravenous infusion once every three weeks. The median duration of treatment was 8 months (range: 0.2 to 33) for patients who received ENHERTU.
Serious adverse reactions occurred in 28% of patients receiving ENHERTU. Serious adverse reactions in >1% of patients who received ENHERTU were ILD/pneumonitis, pneumonia, dyspnea, musculoskeletal pain, sepsis, anemia, febrile neutropenia, hypercalcemia, nausea, pyrexia, and vomiting. Fatalities due to adverse reactions occurred in 4.0% of patients including ILD/pneumonitis (3 patients); sepsis (2 patients); and ischemic colitis, disseminated intravascular coagulation, dyspnea, febrile neutropenia, general physical health deterioration, pleural effusion, and respiratory failure (1 patient each).
ENHERTU was permanently discontinued in 16% of patients, of which ILD/pneumonitis accounted for 8%. Dose interruptions due to adverse reactions occurred in 39% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose interruption were neutropenia, fatigue, anemia, leukopenia, COVID-19, ILD/pneumonitis, increased transaminases, and hyperbilirubinemia. Dose reductions occurred in 23% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose reduction were fatigue, nausea, thrombocytopenia, and neutropenia.
The most common (≥20%) adverse reactions, including laboratory abnormalities, were nausea, decreased white blood cell count, decreased hemoglobin, decreased neutrophil count, decreased lymphocyte count, fatigue, decreased platelet count, alopecia, vomiting, increased aspartate aminotransferase, increased alanine aminotransferase, constipation, increased blood alkaline phosphatase, decreased appetite, musculoskeletal pain, diarrhea, and hypokalemia.
Tables 7 and 8 summarize common adverse reactions and laboratory abnormalities observed in DESTINY-Breast04.
| Adverse Reactions | ENHERTU 5.4 mg/kg | Chemotherapy | ||
|---|---|---|---|---|
| N=371 | N=172 | |||
| All Grades % | Grades 3 or 4 % | All Grades % | Grades 3 or 4 % |
|
| Events were graded using NCI CTCAE version 5.0. | ||||
|
||||
| Gastrointestinal Disorders | ||||
| Nausea | 76 | 4.6 | 30 | 0 |
| Vomiting | 40 | 1.6 | 13 | 0 |
| Constipation | 34 | 0.8 | 22 | 0 |
| Diarrhea | 27 | 1.3 | 22 | 1.7 |
| Abdominal pain* | 18 | 0.5 | 13 | 0 |
| Stomatitis† | 13 | 0.3 | 12 | 0.6 |
| General Disorders and Administration Site Conditions | ||||
| Fatigue‡ | 54 | 9 | 48 | 4.7 |
| Pyrexia | 12 | 0.3 | 13 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Alopecia | 40 | 0 | 33 | 0 |
| Rash§ | 13 | 0 | 23 | 4.7 |
| Blood and Lymphatic System Disorders | ||||
| Anemia¶ | 39 | 10 | 27 | 5 |
| Metabolism and Nutrition Disorders | ||||
| Decreased appetite | 32 | 2.4 | 19 | 1.2 |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Musculoskeletal pain# | 32 | 1.3 | 31 | 0.6 |
| Investigations | ||||
| Decreased weight | 16 | 0.3 | 8 | 0 |
| Vascular Disorders | ||||
| HemorrhageÞ | 16 | 0 | 3.5 | 0 |
| Nervous System Disorders | ||||
| Headacheß | 15 | 0.3 | 6 | 0 |
| Peripheral neuropathyà | 13 | 0 | 29 | 5 |
| Dizzinessè | 11 | 0.5 | 6 | 0 |
| Infections and Infestations | ||||
| Upper respiratory tract infectionð | 14 | 0.3 | 5 | 0 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Interstitial lung diseaseø | 12 | 1.3 | 0.6 | 0 |
| Dyspnea | 10 | 1.3 | 9 | 1.2 |
Other clinically relevant adverse reactions reported in less than 10% of patients treated with ENHERTU:
- Nervous System Disorders: dysgeusia (10%)
- Respiratory, Thoracic and Mediastinal Disorders: cough (10%)
- Gastrointestinal Disorders: abdominal distension (5%), gastritis (2.7%), flatulence (2.4%)
- Eye Disorders: blurred vision (4.9%) [including blurred vision and visual impairment]
- Skin and Subcutaneous Tissue Disorders: pruritus (3.2%) and skin hyperpigmentation (2.7%) [including skin hyperpigmentation, skin discoloration, and pigmentation disorder]
- Metabolism and Nutrition Disorders: dehydration (1.9%)
- Blood and Lymphatic System Disorders: febrile neutropenia (1.1%)
- Injury, Poisoning and Procedural Complications: infusion-related reactions (0.5%) [including injection site reaction and chills]
| Laboratory Parameter | ENHERTU 5.4 mg/kg | Chemotherapy | ||
|---|---|---|---|---|
| N=371 | N=172 | |||
| All Grades % | Grades 3 or 4 % | All Grades % | Grades 3 or 4 % |
|
| Percentages were calculated using patients with worsening laboratory values from baseline and the number of patients with both baseline and post-treatment measurements as the denominator. | ||||
| Frequencies were based on NCI CTCAE v.5.0 grade-derived laboratory abnormalities. | ||||
| Hematology | ||||
| Decreased white blood cell count | 70 | 9 | 78 | 25 |
| Decreased hemoglobin | 64 | 8 | 53 | 6 |
| Decreased neutrophil count | 64 | 14 | 73 | 38 |
| Decreased lymphocyte count | 55 | 18 | 40 | 11 |
| Decreased platelet count | 44 | 6 | 21 | 0.6 |
| Chemistry | ||||
| Increased aspartate aminotransferase | 38 | 2.2 | 38 | 4.1 |
| Increased alanine aminotransferase | 36 | 0.8 | 38 | 4.1 |
| Increased blood alkaline phosphatase | 34 | 0.3 | 24 | 0 |
| Hypokalemia | 25 | 3.3 | 17 | 1.2 |
| Increased blood bilirubin | 16 | 2.7 | 15 | 0.6 |
| Increased blood creatinine | 15 | 1.1 | 9 | 0.6 |
Unresectable or Metastatic HER2-Mutant NSCLC
DESTINY-Lung02 evaluated two dose levels (5.4 mg/kg [n=101] and 6.4 mg/kg [n=50]); however, only the results for the recommended dose of 5.4 mg/kg intravenously every 3 weeks are described below due to increased toxicity observed with the higher dose in patients with NSCLC, including ILD/pneumonitis.
The safety of ENHERTU was evaluated in 101 patients in DESTINY-Lung02 [see Clinical Studies (14.3)]. Patients received ENHERTU 5.4 mg/kg intravenously once every three weeks until disease progression or unacceptable toxicity. Nineteen percent of patients were exposed for greater than 6 months. The median age was 59 years (range 30 to 83); 64% were female; 23% were White, 64% were Asian, and 14% were other races.
Serious adverse reactions occurred in 30% of patients receiving ENHERTU. Serious adverse reactions in >1% of patients who received ENHERTU were ILD/pneumonitis, thrombocytopenia, dyspnea, nausea, pleural effusion, and increased troponin I. Fatality occurred in 1 patient with suspected ILD/pneumonitis (1%).
ENHERTU was permanently discontinued due to an adverse reaction in 8% of patients. Adverse reactions which resulted in permanent discontinuation of ENHERTU were ILD/pneumonitis, diarrhea, hypokalemia, hypomagnesemia, myocarditis, and vomiting. Dose interruptions of ENHERTU due to adverse reactions occurred in 23% of patients. Adverse reactions which required dose interruption (>2%) included neutropenia and ILD/pneumonitis. Dose reductions due to an adverse reaction occurred in 11% of patients.
The most common (≥20%) adverse reactions, including laboratory abnormalities, were nausea, decreased white blood cell count, decreased hemoglobin, decreased neutrophil count, decreased lymphocyte count, decreased platelet count, decreased albumin, increased aspartate aminotransferase, increased alanine aminotransferase, fatigue, constipation, decreased appetite, vomiting, increased alkaline phosphatase, and alopecia.
Tables 9 and 10 summarize common adverse reactions and laboratory abnormalities observed in DESTINY-Lung02.
| Adverse Reactions | ENHERTU 5.4 mg/kg N=101 |
|
|---|---|---|
| All Grades % | Grades 3 or 4 % |
|
| Events were graded using NCI CTCAE version 5.0. | ||
|
||
| Gastrointestinal Disorders | ||
| Nausea | 61 | 3.0 |
| Constipation | 31 | 1.0 |
| Vomiting* | 26 | 2.0 |
| Diarrhea | 19 | 1.0 |
| Stomatitis† | 12 | 0 |
| Blood and Lymphatic System Disorders | ||
| Anemia | 34 | 10 |
| General Disorders and Administration Site Conditions | ||
| Fatigue‡ | 32 | 4.0 |
| Metabolism and Nutrition Disorders | ||
| Decreased appetite | 30 | 1.0 |
| Skin and Subcutaneous Tissue Disorders | ||
| Alopecia | 21 | 0 |
| Musculoskeletal and Connective Tissue Disorders | ||
| Musculoskeletal pain§ | 15 | 1.0 |
Other clinically relevant adverse reactions reported in less than 10% of patients were:
- Respiratory, Thoracic and Mediastinal Disorders: interstitial lung disease (6%) [including interstitial lung disease that was adjudicated as ILD including pneumonitis, interstitial lung disease, pulmonary toxicity, and respiratory failure], dyspnea (5%), and epistaxis (3%)
- Gastrointestinal Disorders: abdominal pain (9%) [including abdominal discomfort, abdominal pain, and upper abdominal pain]
- Skin and Subcutaneous Disorders: rash (3%) [including rash and maculo-papular rash]
- Infections and Infestations: upper respiratory tract infection (4%) [including upper respiratory tract infection, pharyngitis, and laryngitis]
- Nervous System Disorders: headache (4%) [including headache and migraine]
| Laboratory Parameter | ENHERTU 5.4 mg/kg N=101* |
|
|---|---|---|
| All Grades†
% | Grades 3 or 4†
% |
|
|
||
| Hematology‡ | ||
| Decreased white blood cell count | 60 | 4.0 |
| Decreased hemoglobin | 58 | 10 |
| Decreased neutrophil count | 52 | 12 |
| Decreased lymphocyte count | 43 | 16 |
| Decreased platelet count | 40 | 4.0 |
| Chemistry | ||
| Decreased albumin | 39 | 0 |
| Increased aspartate aminotransferase | 35 | 1.0 |
| Increased alanine aminotransferase | 34 | 2.0 |
| Increased alkaline phosphatase | 22 | 0 |
| Hypokalemia | 17 | 2.0 |
Locally Advanced or Metastatic Gastric Cancer
The safety of ENHERTU was evaluated in 187 patients with locally advanced or metastatic HER2-positive gastric or GEJ adenocarcinoma in DESTINY-Gastric01 [see Clinical Studies (14.4)]. Patients intravenously received at least one dose of either ENHERTU (N=125) 6.4 mg/kg once every three weeks or either irinotecan (N=55) 150 mg/m2 biweekly or paclitaxel (N=7) 80 mg/m2 weekly for 3 weeks. The median duration of treatment was 4.6 months (range: 0.7 to 22.3) in the ENHERTU group and 2.8 months (range: 0.5 to 13.1) in the irinotecan/paclitaxel group.
Serious adverse reactions occurred in 44% of patients receiving ENHERTU 6.4 mg/kg. Serious adverse reactions in >2% of patients who received ENHERTU were decreased appetite, ILD, anemia, dehydration, pneumonia, cholestatic jaundice, pyrexia, and tumor hemorrhage. Fatalities due to adverse reactions occurred in 2.4% of patients: disseminated intravascular coagulation, large intestine perforation, and pneumonia occurred in one patient each (0.8%).
ENHERTU was permanently discontinued in 15% of patients, of which ILD accounted for 6%. Dose interruptions due to adverse reactions occurred in 62% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose interruption were neutropenia, anemia, decreased appetite, leukopenia, fatigue, thrombocytopenia, ILD, pneumonia, lymphopenia, upper respiratory tract infection, diarrhea, and hypokalemia. Dose reductions occurred in 32% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose reduction were neutropenia, decreased appetite, fatigue, nausea, and febrile neutropenia.
The most common (≥20%) adverse reactions, including laboratory abnormalities, were decreased hemoglobin, decreased white blood cell count, decreased neutrophil count, decreased lymphocyte count, decreased platelet count, nausea, decreased appetite, increased aspartate aminotransferase, fatigue, increased blood alkaline phosphatase, increased alanine aminotransferase, diarrhea, hypokalemia, vomiting, constipation, increased blood bilirubin, pyrexia, and alopecia.
Tables 11 and 12 summarize adverse reactions and laboratory abnormalities observed in patients receiving ENHERTU 6.4 mg/kg in DESTINY-Gastric01.
| ENHERTU 6.4 mg/kg N=125 | Irinotecan or Paclitaxel N=62 |
|||
|---|---|---|---|---|
| Adverse Reactions | All Grades % | Grades 3 or 4 % | All Grades % | Grades 3 or 4 % |
| Events were graded using NCI CTCAE version 4.03. | ||||
|
||||
| Gastrointestinal Disorders | ||||
| Nausea | 63 | 4.8 | 47 | 1.6 |
| Diarrhea | 32 | 2.4 | 32 | 1.6 |
| Vomiting | 26 | 0 | 8 | 0 |
| Constipation | 24 | 0 | 23 | 0 |
| Abdominal pain* | 14 | 0.8 | 15 | 3.2 |
| Stomatitis† | 11 | 1.6 | 4.8 | 0 |
| Metabolism and Nutrition Disorders | ||||
| Decreased appetite | 60 | 17 | 45 | 13 |
| Dehydration | 6 | 2.4 | 3.2 | 1.6 |
| Blood and Lymphatic System Disorders | ||||
| Anemia‡ | 58 | 38 | 31 | 23 |
| Febrile neutropenia | 4.8 | 4.8 | 3.2 | 3.2 |
| General Disorders and Administration Site Conditions | ||||
| Fatigue§ | 55 | 9 | 44 | 4.8 |
| Pyrexia | 24 | 0 | 16 | 0 |
| Peripheral edema | 10 | 0 | 0 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Alopecia | 22 | 0 | 15 | 0 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Interstitial lung disease¶ | 10 | 2.4 | 0 | 0 |
| Hepatobiliary Disorders | ||||
| Abnormal hepatic function | 8 | 3.2 | 1.6 | 1.6 |
Other clinically relevant adverse reactions reported in less than 10% of patients were:
- Cardiac Disorders: asymptomatic left ventricular ejection fraction decrease (8%) [see Warnings and Precautions (5.3)]
- Infections and Infestations: pneumonia (6%)
- Injury, Poisoning and Procedural Complications: infusion-related reactions (1.6%)
| Laboratory Parameter | ENHERTU 6.4 mg/kg N=125 | Irinotecan or Paclitaxel N=62 |
||
|---|---|---|---|---|
| All Grades % | Grades 3 or 4 % | All Grades % | Grades 3 or 4 % |
|
| Percentages were calculated using patients with worsening laboratory values from baseline and the number of patients with both baseline and post-treatment measurements as the denominator. | ||||
| Frequencies were based on NCI CTCAE v.4.03 grade-derived laboratory abnormalities. | ||||
| Hematology | ||||
| Decreased hemoglobin | 75 | 38 | 55 | 23 |
| Decreased white blood cell count | 74 | 29 | 53 | 13 |
| Decreased neutrophil count | 72 | 51 | 45 | 23 |
| Decreased lymphocyte count | 70 | 28 | 53 | 12 |
| Decreased platelet count | 68 | 12 | 12 | 5 |
| Chemistry | ||||
| Increased aspartate aminotransferase | 58 | 9 | 32 | 8 |
| Increased blood alkaline phosphatase | 54 | 8 | 34 | 10 |
| Increased alanine aminotransferase | 47 | 9 | 17 | 1.7 |
| Hypokalemia | 30 | 4.8 | 18 | 8 |
| Increased blood bilirubin | 24 | 7 | 5 | 3.4 |
8. Use In Specific Populations
8.4 Pediatric Use
Safety and effectiveness of ENHERTU have not been established in pediatric patients.
8.5 Geriatric Use
Of the 883 patients with breast cancer treated with ENHERTU 5.4 mg/kg, 22% were 65 years or older and 3.6% were 75 years or older. No overall differences in efficacy within clinical studies were observed between patients ≥65 years of age compared to younger patients. There was a higher incidence of Grade 3-4 adverse reactions observed in patients aged 65 years or older (60%) as compared to younger patients (48%).
Of the 101 patients with unresectable or metastatic HER2-mutant NSCLC treated with ENHERTU 5.4 mg/kg, 40% were 65 years or older and 8% were 75 years or older. No overall differences in efficacy or safety were observed between patients ≥65 years of age compared to younger patients.
Of the 125 patients with locally advanced or metastatic HER2-positive gastric or GEJ adenocarcinoma treated with ENHERTU 6.4 mg/kg in DESTINY-Gastric01, 56% were 65 years or older and 14% were 75 years or older. No overall differences in efficacy or safety were observed between patients ≥65 years of age compared to younger patients.
8.6 Renal Impairment
No dose adjustment of ENHERTU is required in patients with mild (creatinine clearance [CLcr] ≥60 and <90 mL/min) or moderate (CLcr ≥30 and <60 mL/min) renal impairment [see Clinical Pharmacology (12.3)]. A higher incidence of Grade 1 and 2 ILD/pneumonitis has been observed in patients with moderate renal impairment [see Warnings and Precautions (5.1)]. Monitor patients with moderate renal impairment more frequently. The recommended dosage of ENHERTU has not been established for patients with severe renal impairment (CLcr <30 mL/min) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment of ENHERTU is required in patients with mild (total bilirubin ≤ULN and any AST >ULN or total bilirubin >1 to 1.5 times ULN and any AST) or moderate (total bilirubin >1.5 to 3 times ULN and any AST) hepatic impairment. In patients with moderate hepatic impairment, due to potentially increased exposure, closely monitor for increased toxicities related to the topoisomerase inhibitor, DXd [see Dosage and Administration (2.3)]. The recommended dosage of ENHERTU has not been established for patients with severe hepatic impairment (total bilirubin >3 times ULN and any AST) [see Clinical Pharmacology (12.3)].
11. Enhertu Description
Fam-trastuzumab deruxtecan-nxki is a HER2-directed antibody and topoisomerase inhibitor conjugate. Fam-trastuzumab deruxtecan-nxki is an antibody-drug conjugate (ADC) composed of three components: 1) a humanized anti-HER2 IgG1 monoclonal antibody (mAb), covalently linked to 2) a topoisomerase inhibitor, via 3) a tetrapeptide-based cleavable linker. Deruxtecan is composed of a protease-cleavable maleimide tetrapeptide linker and the topoisomerase inhibitor, DXd, which is an exatecan derivative.
The antibody is produced in Chinese hamster ovary cells by recombinant DNA technology, and the topoisomerase inhibitor and linker are produced by chemical synthesis. Approximately 8 molecules of deruxtecan are attached to each antibody molecule. Fam-trastuzumab deruxtecan-nxki has the following structure:
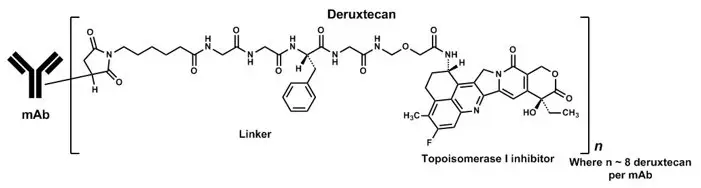
ENHERTU (fam-trastuzumab deruxtecan-nxki) is a sterile, white to yellowish white, preservative-free lyophilized powder in single-dose vials. Each vial delivers 100 mg of fam-trastuzumab deruxtecan-nxki, L-histidine (4.45 mg), L-histidine hydrochloride monohydrate (20.2 mg), polysorbate 80 (1.5 mg), and sucrose (450 mg). Following reconstitution with 5 mL of Sterile Water for Injection, USP, the resulting concentration of fam-trastuzumab deruxtecan-nxki is 20 mg/mL with a pH of 5.5. The resulting solution is administered by intravenous infusion following dilution.
12. Enhertu - Clinical Pharmacology
12.1 Mechanism of Action
Fam-trastuzumab deruxtecan-nxki is a HER2-directed antibody-drug conjugate. The antibody is a humanized anti-HER2 IgG1. The small molecule, DXd, is a topoisomerase I inhibitor attached to the antibody by a cleavable linker. Following binding to HER2 on tumor cells, fam-trastuzumab deruxtecan-nxki undergoes internalization and intracellular linker cleavage by lysosomal enzymes. Upon release, the membrane-permeable DXd causes DNA damage and apoptotic cell death.
12.3 Pharmacokinetics
The pharmacokinetics of fam-trastuzumab deruxtecan-nxki was evaluated in patients with cancer. Following a single dose, exposures (Cmax and AUC) of fam-trastuzumab deruxtecan-nxki and released topoisomerase inhibitor (DXd) increased proportionally over a dose range of 3.2 mg/kg to 8 mg/kg (approximately 0.6 to 1.5 times the recommended dose in breast cancer and NSCLC and 0.5 to 1.25 times the recommended dose in gastric cancer).
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of ENHERTU or of other anti-HER2 products.
During the median 14-month treatment period in HER2-positive breast cancer patients in DESTINY-Breast03 with a median ADA sampling period of 13 months, treatment-emergent ADA (or anti-fam-trastuzumab deruxtecan-nxki antibodies) developed in 1.6% (4/256) of patients who received ENHERTU. The incidence of treatment-emergent neutralizing antibodies against fam-trastuzumab deruxtecan-nxki was 0.4% (1/256).
During the median 7-month treatment period in HER2-positive breast cancer patients in DESTINY-Breast01 with a median ADA sampling period of 9 months, treatment-emergent ADA developed in 1.2% (3/249) of patients who received ENHERTU. The incidence of treatment-emergent neutralizing antibodies against fam-trastuzumab deruxtecan-nxki was 0% (0/249).
During the median 8-month treatment period in HER2-low breast cancer patients in DESTINY-Breast04 with a median ADA sampling period of 8 months, treatment-emergent ADA developed in 2.0% (7/357) of patients who received ENHERTU. The incidence of treatment-emergent neutralizing antibodies against fam-trastuzumab deruxtecan-nxki was 0% (0/357).
During the median 3.5-month treatment period in HER2-mutant NSCLC patients in DESTINY-Lung02 with median ADA sampling period of 2.2 months, treatment-emergent ADA developed in 0.7% (1/143) of patients who received ENHERTU. The incidence of treatment-emergent neutralizing antibodies against fam-trastuzumab deruxtecan-nxki was 0% (0/249).
During the median 4.6-month treatment period in HER2-positive gastric or GEJ adenocarcinoma patients in DESTINY-Gastric01 with a median ADA sampling period of 4.6 months, treatment-emergent ADA developed in 7.3% (9/123) of patients who received ENHERTU. The incidence of treatment-emergent neutralizing antibodies against fam-trastuzumab deruxtecan-nxki was 0% (0/123).
Due to the limited number of patients who tested positive for ADA, the effect of treatment-emergent ADAs and treatment-emergent neutralizing antibodies on the pharmacokinetics, pharmacodynamics, safety and/or effectiveness of fam-trastuzumab deruxtecan-nxki is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with fam-trastuzumab deruxtecan-nxki.
The topoisomerase inhibitor component of fam-trastuzumab deruxtecan-nxki, DXd, was clastogenic in both an in vivo rat bone marrow micronucleus assay and an in vitro Chinese hamster lung chromosome aberration assay and was not mutagenic in an in vitro bacterial reverse mutation assay.
Fertility studies have not been conducted with fam-trastuzumab deruxtecan-nxki. In a six-week repeat-dose toxicity study in rats, intravenous administration of fam-trastuzumab deruxtecan-nxki resulted in spermatid retention at 20 mg/kg and 60 mg/kg (approximately 4 and 9 times the human recommended dose of 5.4 mg/kg based on AUC, respectively). Decreased testes and epididymides weights, tubular atrophy/degeneration in testes, and reduced sperm count in epididymides were observed at a dose of 197 mg/kg (19 times the human recommended dose of 5.4 mg/kg based on AUC). In a three-month repeat-dose toxicity study in monkeys, intravenous administration of fam-trastuzumab deruxtecan-nxki resulted in decreased numbers of round spermatids in the testes at seminiferous tubule stages V to VI at ≥30 mg/kg (≥7 times the human recommended dose of 5.4 mg/kg based on AUC). Evidence of reversibility was observed in monkeys by the end of a three-month recovery period.
14. Clinical Studies
14.1 HER2-Positive Metastatic Breast Cancer
DESTINY-Breast03
The efficacy of ENHERTU was evaluated in study DESTINY-Breast03 (NCT03529110), a multicenter, open-label, randomized trial that enrolled 524 patients with HER2-positive, unresectable and/or metastatic breast cancer who received prior trastuzumab and taxane therapy for metastatic disease or developed disease recurrence during or within 6 months of completing adjuvant therapy. HER2 expression was based on archival tissue tested at a central laboratory prior to enrollment with HER2 positivity defined as HER2 IHC 3+ or ISH positive. Patients were excluded for a history of ILD/pneumonitis requiring treatment with steroids, ILD/pneumonitis at screening, or clinically significant cardiac disease. Patients were also excluded for untreated and symptomatic brain metastases, ECOG performance status >1, or prior treatment with an anti-HER2 antibody-drug conjugate in the metastatic setting.
Patients were randomized 1:1 to receive either ENHERTU 5.4 mg/kg (N=261) or ado-trastuzumab emtansine 3.6 mg/kg (N=263) by intravenous infusion every 3 weeks until unacceptable toxicity or disease progression. Randomization was stratified by hormone receptor status, prior treatment with pertuzumab, and visceral versus non-visceral disease. Tumor imaging was obtained every 6 weeks and CT/MRI of the brain was mandatory for all patients at baseline. The major efficacy outcomes were progression-free survival (PFS) as assessed by blinded independent central review (BICR) based on Response Evaluation Criteria in Solid Tumors (RECIST) v.1.1 and overall survival (OS). Confirmed objective response rate (ORR) was an additional outcome measure.
The median age was 54 years (range: 20-83); 80% were <65 years and 99.6% were female. The majority of patients were Asian (60%), White (27%) and Black (3.6%). Eleven percent (11%) of patients were of Hispanic/Latino ethnicity. Patients had an ECOG performance status of 0 (63%) or 1 (37%) at baseline. Seventy-three percent had visceral disease, 16% had brain metastases at baseline, 52% were hormone receptor positive (HR+) and 48% of patients had received one line of prior systemic therapy in the metastatic setting. The percentage of patients who had not received prior treatment for metastatic disease was 10%.
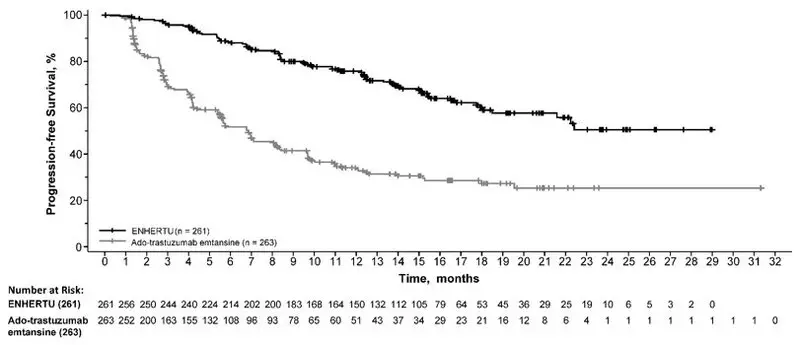
Efficacy results are summarized in Table 13 and Figure 1. At the time of the PFS analysis, 16% of patients had died and overall survival (OS) was immature.
| Efficacy Parameter | ENHERTU 5.4 mg/kg | Ado-trastuzumab emtansine 3.6 mg/kg |
|---|---|---|
| CI = confidence interval; NR = not reached; NE=not estimable | ||
|
||
| Progression-Free Survival (PFS) per BICR | ||
| N | 261 | 263 |
| Number of events (%) | 87 (33.3) | 158 (60.1) |
| Median, months (95% CI) | NR (18.5, NE) | 6.8 (5.6, 8.2) |
| Hazard ratio (95% CI) | 0.28 (0.22, 0.37) | |
| p-value | p< 0.0001 | |
| Confirmed Objective Response Rate (ORR) per BICR* | ||
| N | 248 | 241 |
| n (%) | 205 (82.7) | 87 (36.1) |
| 95% CI | (77.4, 87.2) | (30.0, 42.5) |
| Complete Response n (%) | 39 (15.7) | 20 (8.3) |
| Partial Response n (%) | 166 (66.9) | 67 (27.8) |
| Figure 1: Kaplan-Meier Plot of Progression-Free Survival per BICR (Intent-to-Treat Analysis Set) |
|---|
 |
DESTINY-Breast01
The efficacy of ENHERTU was evaluated in study DESTINY-Breast01 (NCT03248492), a multicenter, single-arm, trial that enrolled 184 female patients with HER2-positive, unresectable and/or metastatic breast cancer who had received two or more prior anti-HER2 therapies. Patients were excluded for a history of treated ILD or current ILD at screening. Patients were also excluded for history of clinically significant cardiac disease, active brain metastases, and ECOG performance status >1. HER2 expression was based on archival tissue tested at a central laboratory prior to enrollment with HER2 positivity defined as HER2 IHC 3+ or ISH positive.
Patients received ENHERTU 5.4 mg/kg by intravenous infusion every 3 weeks until unacceptable toxicity or disease progression. Tumor imaging was obtained every 6 weeks and CT/MRI of the brain was mandatory for patients with brain metastases at baseline. The major efficacy outcomes were confirmed objective response rate (ORR) assessed by independent central review (ICR) using RECIST v1.1 and duration of response (DOR).
The median age was 55 years (range: 28-96); 76% of patients were <65 years. All 184 patients were female, and the majority were White (55%) or Asian (38%). Patients had an ECOG performance status of 0 (55%) or 1 (44%) at baseline. Ninety-two percent had visceral disease, 29% had bone metastases, and 13% had brain metastases. Fifty-three percent were HR+. Sum of diameters of target lesions were <5 cm in 42%, and ≥5 cm in 50% (not evaluable by central review in 8% of patients).
The median number of prior cancer regimens in the locally advanced/metastatic setting was 5 (range: 2-17).
All patients received prior trastuzumab, ado-trastuzumab emtansine, and 66% had prior pertuzumab.
Efficacy results are summarized in Table 14.
| Efficacy Parameter | DESTINY-Breast01 N=184 |
|---|---|
| ORR 95% CI calculated using Clopper-Pearson method | |
|
|
| Confirmed Objective Response Rate (95% CI) | 60.3% (52.9, 67.4) |
| Complete Response | 4.3% |
| Partial Response | 56.0% |
| Duration of Response*
Median, months (95% CI)† | 14.8 (13.8, 16.9) |
14.2 HER2-Low Metastatic Breast Cancer
The efficacy of ENHERTU was evaluated in study DESTINY-Breast04 (NCT03734029), a randomized, multicenter, open-label study that enrolled 557 adult patients with unresectable or metastatic HER2-low breast cancer. The study included 2 cohorts: 494 hormone receptor-positive (HR+) patients and 63 hormone receptor-negative (HR-) patients. HER2-low expression was defined as IHC 1+ or IHC 2+/ISH-, as determined at a central laboratory using Ventana's PATHWAY anti-HER-2/neu (4B5) Rabbit Monoclonal Primary Antibody assay. Patients must have received chemotherapy in the metastatic setting or have developed disease recurrence during or within 6 months of completing adjuvant chemotherapy. Patients who were HR+ must have received at least one endocrine therapy or be ineligible for endocrine therapy. Patients were randomized 2:1 to receive either ENHERTU 5.4 mg/kg (N=373) by intravenous infusion every 3 weeks or physician's choice of chemotherapy (N=184, eribulin, capecitabine, gemcitabine, nab paclitaxel, or paclitaxel). Randomization was stratified by HER2 IHC status of tumor samples (IHC 1+ or IHC 2+/ISH-), number of prior lines of chemotherapy in the metastatic setting (1 or 2), and HR status/prior CDK4/6i treatment (HR+ with prior CDK4/6 inhibitor treatment, HR+ without prior CDK4/6 inhibitor treatment, or HR-). Treatment was administered until disease progression, death, withdrawal of consent, or unacceptable toxicity. The study excluded patients with a history of ILD/pneumonitis requiring treatment with steroids or ILD/pneumonitis at screening and clinically significant cardiac disease. Patients were also excluded for untreated or symptomatic brain metastases or ECOG performance status >1.
The major efficacy outcome measure was PFS in patients with HR+ breast cancer assessed by BICR based on RECIST v1.1. Additional efficacy outcome measures were PFS assessed by BICR based on RECIST v1.1 in the overall population (all randomized HR+ and HR- patients), OS in HR+ patients, and OS in the overall population.
The median age was 57 years (range: 28 to 81); 24% were age 65 or older; 99.6% were female; 48% were White, 40% were Asian, and 2% were Black or African American; 3.8% of patients were of Hispanic/Latino ethnicity. Patients had an ECOG performance status of 0 (55%) or 1 (45%) at baseline; 58% were IHC 1+, 42% were IHC 2+/ISH-; 70% had liver metastases, 33% had lung metastases, and 6% had brain metastases. In the metastatic setting, patients had a median of 3 prior lines of systemic therapy (range: 1 to 9) with 58% having 1 and 41% having 2 prior chemotherapy regimens; 3.9% were early progressors (progression in the neo/adjuvant setting). In HR+ patients, the median number of prior lines of endocrine therapy was 2 (range: 0 to 9) and 70% had prior CDK4/6i treatment.
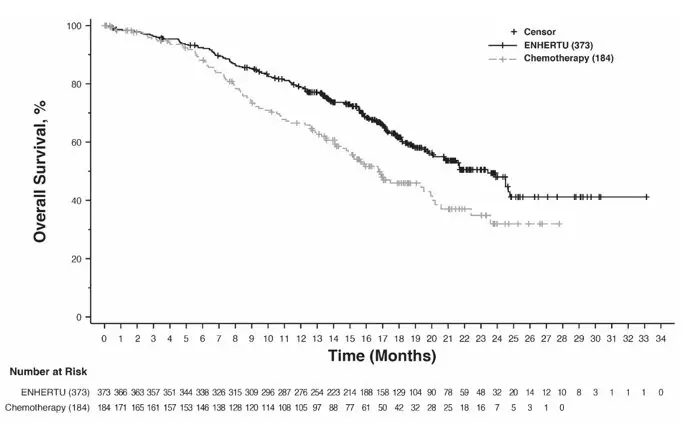
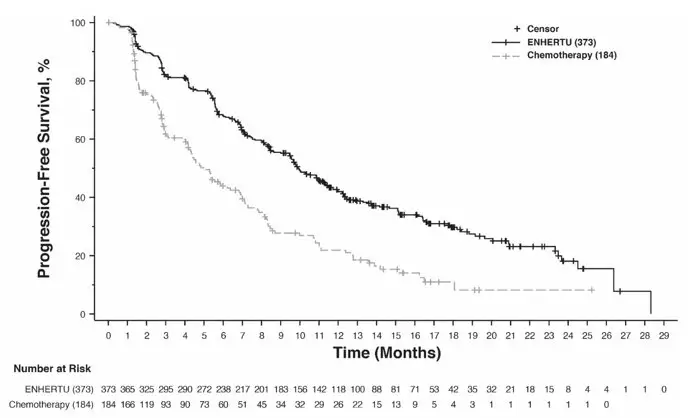
Efficacy results are summarized in Table 15 and Figures 2 and 3.
| Efficacy Parameter | HR+ Cohort | Overall Population (HR+ and HR- Cohorts) |
||
|---|---|---|---|---|
| ENHERTU (N=331) | Chemotherapy (N=163) | ENHERTU (N=373) | Chemotherapy (N=184) |
|
| CI = confidence interval | ||||
| Overall Survival | ||||
| Number of events (%) | 126 (38.1) | 73 (44.8) | 149 (39.9) | 90 (48.9) |
| Median, months (95% CI) | 23.9 (20.8, 24.8) | 17.5 (15.2, 22.4) | 23.4 (20.0, 24.8) | 16.8 (14.5, 20.0) |
| Hazard ratio (95% CI) | 0.64 (0.48, 0.86) | 0.64 (0.49, 0.84) | ||
| p-value | 0.0028 | 0.001 | ||
| Progression-Free Survival per BICR | ||||
| Number of events (%) | 211 (63.7) | 110 (67.5) | 243 (65.1) | 127 (69.0) |
| Median, months (95% CI) | 10.1 (9.5, 11.5) | 5.4 (4.4, 7.1) | 9.9 (9.0, 11.3) | 5.1 (4.2, 6.8) |
| Hazard ratio (95% CI) | 0.51 (0.40, 0.64) | 0.50 (0.40, 0.63) | ||
| p-value | <0.0001 | <0.0001 | ||
| Confirmed Objective Response Rate per BICR | ||||
| n (%) | 175 (52.9) | 27 (16.6) | 195 (52.3) | 30 (16.3) |
| 95% CI | 47.3, 58.4 | 11.2, 23.2 | 47.1, 57.4 | 11.3, 22.5 |
| Complete Response n (%) | 12 (3.6) | 1 (0.6) | 13 (3.5) | 2 (1.1) |
| Partial Response n (%) | 164 (49.5) | 26 (16.0) | 183 (49.1) | 28 (15.2) |
| Duration of Response per BICR | ||||
| Median, months (95% CI) | 10.7 (8.5, 13.7) | 6.8 (6.5, 9.9) | 10.7 (8.5, 13.2) | 6.8 (6.0, 9.9) |
| Figure 2: Kaplan-Meier Plot of Overall Survival (Overall Population) |
|---|
 |
| Figure 3: Kaplan-Meier Plot of Progression-Free Survival (Overall Population) |
|---|
 |
14.3 Unresectable or Metastatic HER2-Mutant Non-Small Cell Lung Cancer
ENHERTU was evaluated in DESTINY-Lung01 (NCT03505710) and at two dose levels in DESTINY-Lung02 (NCT04644237). Patients were prospectively selected for treatment with ENHERTU based on the presence of activating HER2 (ERBB2) mutations by local testing using tissue. Samples from DESTINY-Lung01 were retrospectively tested using Oncomine™ Dx Target Test (Life Technologies Corporation, Tissue-test) and Guardant360® CDx test (Guardant Health Inc., Plasma test). Demographic and baseline disease characteristics were similar for patients in DESTINY-Lung01 and DESTINY-Lung02, except for race (34% Asian vs 79% Asian, respectively). Response rates were consistent across dose levels. Increased rates of ILD/pneumonitis were observed at the higher dose. The approved recommended dose of 5.4 mg/kg intravenously every 3 weeks in the DESTINY-Lung02 study is described below [see Adverse Reactions (6.1)].
The efficacy of ENHERTU was evaluated in DESTINY-Lung02, a multicenter, multi-cohort, randomized, blinded, dose-optimization trial. Eligible patients were required to have unresectable or metastatic HER2-mutant non-squamous NSCLC with disease progression after one prior systemic therapy. Patients with a history of steroid dependent ILD/pneumonitis, clinically significant cardiac disease, clinically active brain metastases, and ECOG performance status >1 were excluded. Patients received ENHERTU 5.4 mg/kg by intravenous infusion every 3 weeks until disease progression or unacceptable toxicity. Tumor imaging was obtained every 6 weeks and CT/MRI of the brain was mandatory for patients with stable brain metastases at baseline.
Results from an interim efficacy analysis in a pre-specified patient cohort are described below. The major efficacy outcomes were confirmed ORR as assessed by BICR using RECIST v1.1 and DOR.
The median age was 58 years (range 30 to 78); 69% were female; 79% were Asian, 12% were White, and 10% were other races; 29% had an ECOG performance status of 0 and 71% had 1; 33% had stable brain metastases; 94% had a mutation in the ERBB2 kinase domain and 6% had a mutation in the extracellular domain. The median number of prior regimens was 2 (range: 1 to 12); 100% of patients received prior platinum therapy, 71% received prior immunotherapy, and 44% received both in combination. Fifty percent of patients were never-smokers and 50% were former smokers; 96% of patients had adenocarcinoma histology.
Efficacy results are provided in Table 16.
| Efficacy Parameter | DESTINY-Lung02 N=52 |
|---|---|
| ORR 95% CI calculated using Clopper-Pearson method | |
| NE=not estimable | |
|
|
| Confirmed Objective Response Rate (95% CI) | 57.7% (43.2, 71.3) |
| Complete Response | 1.9% |
| Partial Response | 55.8% |
| Duration of Response
Median, months (95% CI)† | 8.7 (7.1, NE) |
14.4 Locally Advanced or Metastatic Gastric Cancer
The efficacy of ENHERTU was evaluated in study DESTINY-Gastric01 (NCT03329690), a multicenter, open-label, randomized trial conducted in Japan and South Korea that enrolled 188 adult patients with HER2-positive (IHC 3+ or IHC 2+/ISH positive), locally advanced or metastatic gastric or GEJ adenocarcinoma who had progressed on at least two prior regimens including trastuzumab, a fluoropyrimidine- and a platinum-containing chemotherapy. HER2 expression was determined by a central lab on tissue obtained either before or after prior trastuzumab treatment. Patients were excluded for a history of treated or current ILD, a history of clinically significant cardiac disease, active brain metastases, or ECOG performance status >1.
Patients were randomized 2:1 to receive ENHERTU (N=126) 6.4 mg/kg intravenously every 3 weeks or physician's choice of chemotherapy: irinotecan monotherapy (N=55) 150 mg/m2 intravenously every 2 weeks or paclitaxel monotherapy (N=7) 80 mg/m2 intravenously weekly. Randomization was stratified by HER2 status (IHC 3+ or IHC 2+/ISH+), ECOG performance status (0 or 1), and region (Japan or South Korea). Tumor imaging assessments were performed at screening and every 6 weeks from the first treatment dose. Treatment was administered until unacceptable toxicity or disease progression. The major efficacy outcomes were ORR assessed by ICR according to RECIST v1.1 and OS in the intent-to-treat population. Additional efficacy outcomes were PFS and DOR.
The median age was 66 years (range 28 to 82); 76% were male; and 100% were Asian. All patients received a trastuzumab product. Patients had an ECOG performance status of either 0 (49%) or 1 (51%); 87% had gastric adenocarcinoma and 13% had GEJ adenocarcinoma; 76% were IHC 3+ and 23% were IHC 2+/ISH+; 65% had inoperable advanced cancer; 35% had postoperative recurrent cancer; 54% had liver metastases; 29% had lung metastases; 45% had three or more prior regimens in the locally advanced or metastatic setting. A total of 30% of patients were identified as HER2-positive using tissue obtained following prior treatment with a trastuzumab product.
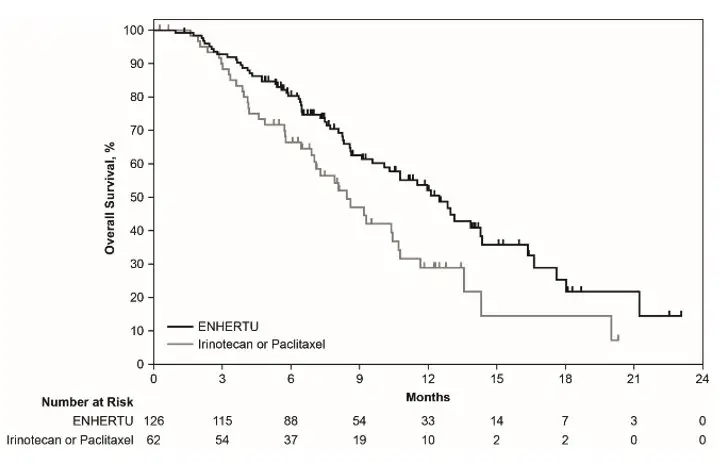
Efficacy results are summarized in Table 17, and the Kaplan-Meier curve for OS is shown in Figure 4.
| Efficacy Parameter | ENHERTU N=126 | Irinotecan or Paclitaxel N=62 |
|---|---|---|
| CI = confidence interval; NR = not reached | ||
|
||
| Overall Survival (OS)* | ||
| Median, months (95% CI)† | 12.5 (9.6, 14.3) | 8.4 (6.9,10.7) |
| Hazard ratio (95% CI)‡ | 0.59 (0.39, 0.88) | |
| p-value§ | 0.0097 | |
| Progression-Free Survival (PFS)¶ | ||
| Median, months (95% CI)† | 5.6 (4.3, 6.9) | 3.5 (2.0, 4.3) |
| Hazard ratio (95% CI)‡ | 0.47 (0.31, 0.71) | |
| Confirmed Objective Response Rate (ORR)¶ | ||
| n (%) | 51 (40.5) | 7 (11.3) |
| 95% CI# | (31.8, 49.6) | (4.7, 21.9) |
| p-valueÞ | <0.0001 | |
| Complete Response n (%) | 10 (7.9) | 0 (0.0) |
| Partial Response n (%) | 41 (32.5) | 7 (11.3) |
| Duration of Response (DOR)¶ | ||
| Median, months (95% CI)† | 11.3 (5.6, NR) | 3.9 (3.0, 4.9) |
| Figure 4: Kaplan-Meier Plot of Overall Survival |
|---|
 |
16. How is Enhertu supplied
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 08/2022 | ||
| Medication Guide
ENHERTU® (en-HER-too) (fam-trastuzumab deruxtecan-nxki) for injection |
|||
| What is the most important information I should know about ENHERTU? ENHERTU can cause serious side effects, including:
|
|||
|
| ||
Your healthcare provider will check you for these side effects during your treatment with ENHERTU. Your healthcare provider may reduce your dose, delay treatment or completely stop treatment with ENHERTU if you have severe side effects.
|
|||
| What is ENHERTU?
ENHERTU is a prescription medicine used to treat adults who have:
|
|||
Before you receive ENHERTU, tell your healthcare provider about all of your medical conditions, including if you:
|
|||
How will I receive ENHERTU?
|
|||
| What are the possible side effects of ENHERTU? ENHERTU can cause serious side effects. See "What is the most important information I should know about ENHERTU?" The most common side effects of ENHERTU, when used in people with metastatic breast cancer and HER2-mutant non-small cell lung cancer include: |
|||
|
| ||
| The most common side effects of ENHERTU, when used in people with HER2-positive gastric or GEJ adenocarcinoma, include: | |||
|
| ||
| ENHERTU may cause fertility problems in males, which may affect the ability to father children. Talk to your healthcare provider if you have concerns about fertility. These are not all of the possible side effects of ENHERTU. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
| General information about the safe and effective use of ENHERTU.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about ENHERTU that is written for healthcare professionals. |
|||
| What are the ingredients in ENHERTU?
Active Ingredient: fam-trastuzumab deruxtecan-nxki. Inactive Ingredients: L-histidine, L-histidine hydrochloride monohydrate, polysorbate 80, and sucrose. Manufactured by: Daiichi Sankyo, Inc., Basking Ridge, NJ 07920 U.S. License No. 2128 Marketed by: Daiichi Sankyo, Inc., Basking Ridge, NJ 07920 and AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850 ENHERTU® is a registered trademark of Daiichi Sankyo Company, Ltd. © 2022 Daiichi Sankyo Co., Ltd. USMG-ENH-C9-0822-r005 For more information, call 1-877-437-7763 or go to https://www.ENHERTU.com. |
|||


| ENHERTU
fam-trastuzumab deruxtecan-nxki injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Daiichi Sankyo, Inc. (068605067) |
