Drug Detail:Fabrazyme (Agalsidase beta [ a-gal-sih-daze-bay-tah ])
Drug Class: Lysosomal enzymes
Highlights of Prescribing Information
FABRAZYME® (agalsidase beta) for injection, for intravenous use
Initial U.S. Approval: 2003
Indications and Usage for Fabrazyme
Fabrazyme is a hydrolytic lysosomal neutral glycosphingolipid-specific enzyme indicated for the treatment of adult and pediatric patients 2 years of age and older with confirmed Fabry disease. (1)
Fabrazyme Dosage and Administration
- The recommended dosage is 1 mg/kg body weight given every two weeks as an intravenous infusion. (2.1)
- Ensure appropriate medical support is available when Fabrazyme is administered because of the potential for anaphylaxis and severe infusion-associated reactions. (2.1, 5.1, 5.2)
- Administer antipyretics prior to infusion. (2.1)
- See the full prescribing information for the recommended infusion rate. (2.1)
Dosage Forms and Strengths
For injection: 5 mg or 35 mg lyophilized cake or powder in a single-dose vial for reconstitution (3)
Contraindications
None. (4)
Warnings and Precautions
- Anaphylaxis and Hypersensitivity Reactions: Life-threatening anaphylactic and severe hypersensitivity reactions have occurred during Fabrazyme infusions. If severe hypersensitivity or anaphylactic reactions occur, immediately discontinue the infusion and provide necessary emergency treatment. Readministration to patients who have previously experienced severe or serious hypersensitivity reactions to Fabrazyme should be done only after careful consideration of the risks and benefits of continued treatment, and only under the direct supervision of qualified personnel with appropriate medical support measures readily available. (5.1)
- Infusion-Associated Reactions: Pretreat patients who experience infusion-associated reactions with an antipyretic and antihistamine. If an infusion-associated reaction occurs, decrease the infusion rate, temporarily stop the infusion, and consider administration of additional antipyretics, antihistamines, and/or steroids. If a severe infusion-associated reaction occurs, discontinue the infusion and initiate appropriate anaphylaxis treatment. (5.2)
Adverse Reactions/Side Effects
Most common adverse reactions (≥20%) are: upper respiratory tract infection, chills, pyrexia, headache, cough, paresthesia, fatigue, peripheral edema, dizziness, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme at 1-800-745-4447 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2023
Full Prescribing Information
1. Indications and Usage for Fabrazyme
Fabrazyme® is indicated for the treatment of adult and pediatric patients 2 years of age and older with confirmed Fabry disease.
2. Fabrazyme Dosage and Administration
2.1 Recommended Dosage
- The recommended dosage of Fabrazyme is 1 mg/kg body weight infused every two weeks as an intravenous infusion.
- Infusion rate:
- –
- The initial intravenous infusion rate is 0.25 mg/min (15 mg/hour). Slow the infusion rate in the event of infusion-associated reactions [see Warnings and Precautions (5.2)].
- –
- For patients >30 kg, after patient tolerance to the infusion is well established, increase the infusion rate in increments of 0.05 to 0.08 mg/min (increments of 3 to 5 mg/hour) with each subsequent infusion. The minimum infusion duration is 1.5 hours (based on individual patient tolerability).
- –
- For patients weighing <30 kg, the maximum infusion rate is 0.25 mg/minute (15 mg/hour).
- Because of the potential for severe infusion-associated reactions, appropriate medical support measures should be readily available when Fabrazyme is administered [see Warnings and Precautions (5.2)].
- Administer antipyretics prior to infusion of Fabrazyme [see Warnings and Precautions (5.2)].
- Rechallenge: Patients who have had a positive skin test to Fabrazyme or who have tested positive for anti-Fabrazyme IgE may be successfully rechallenged with Fabrazyme. The initial rechallenge administration should be a low dose at a lower infusion rate, e.g., one-half the therapeutic dose (0.5 mg/kg) at 1/25th of the initial standard recommended rate (0.01 mg/min). Once a patient tolerates the infusion, the dose may be increased to reach the approved dose of 1 mg/kg and the infusion rate may be increased by slowly titrating upwards (doubled every 30 minutes up to a maximum rate of 0.25 mg/minute), as tolerated [see Adverse Reactions (6.2)].
3. Dosage Forms and Strengths
For injection: 5 mg or 35 mg of agalsidase beta as a white to off-white, lyophilized cake or powder in a single-dose vial for reconstitution.
5. Warnings and Precautions
5.1 Anaphylaxis and Hypersensitivity Reactions
In clinical trials and postmarketing safety experience with Fabrazyme, approximately 1% of patients developed anaphylactic or severe hypersensitivity reactions during Fabrazyme infusion.
In clinical trials with Fabrazyme, 10 of 238 patients developed IgE antibodies or skin test reactivity specific to Fabrazyme. Two of six patients in the rechallenge study discontinued treatment with Fabrazyme prematurely due to recurrent infusion-associated reactions. Four serious infusion-associated reactions occurred in three patients during Fabrazyme infusions, including bronchospasm, urticaria, hypotension, and development of Fabrazyme-specific antibodies. Other infusion-associated reactions occurring in more than one patient during the study included rigors, hypertension, nausea, vomiting, and pruritus.
Higher incidences of hypersensitivity reactions were observed in adult patients with persistent anti-Fabrazyme antibodies and in adult patients with high antibody titer compared to that in antibody-negative adult patients [see Adverse Reactions (6.2)].
Life-threatening anaphylactic and severe hypersensitivity reactions have been observed in patients during Fabrazyme infusions. Reactions have included localized angioedema (including swelling of the face, mouth, and throat), bronchospasm, hypotension, generalized urticaria, dysphagia, rash, dyspnea, flushing, chest discomfort, pruritus, and nasal congestion. Interventions have included cardiopulmonary resuscitation, oxygen supplementation, intravenous fluids, hospitalization, and treatment with inhaled beta-adrenergic agonists, epinephrine, and intravenous corticosteroids.
If anaphylactic or severe hypersensitivity reactions occur, immediately discontinue the administration of Fabrazyme and initiate necessary emergency treatment. Because of the potential for severe hypersensitivity reactions, appropriate medical support measures should be readily available when Fabrazyme is administered.
The risks and benefits of readministering Fabrazyme following an anaphylactic or severe hypersensitivity reaction should be considered. If a decision is made to readminister the product, ensure that appropriate medical emergency support is available [see Dosage and Administration (2.1) and Adverse Reactions (6.2)].
Physicians should consider testing for IgE antibodies in patients who experienced suspected hypersensitivity reactions and consider the risks and benefits of continued treatment in patients with anti-Fabrazyme IgE antibodies. There are no marketed tests for antibodies against Fabrazyme. If testing is warranted, contact Genzyme Corporation at 1-800-745-4447.
Patients who have had a positive skin test to Fabrazyme or who have tested positive for Fabrazyme-specific IgE antibody have been rechallenged with Fabrazyme using a rechallenge protocol. Rechallenge of these patients should only occur under the direct supervision of qualified personnel with appropriate medical support measures readily available [see Dosage and Administration (2.1) and Adverse Reactions (6.2)].
5.2 Infusion-Associated Reactions
In clinical trials of Fabrazyme, 59% of patients experienced infusion-associated reactions during Fabrazyme administration, some of which were severe. Infusion-associated reactions are defined as adverse reactions occurring on the same day as the infusion. The incidence of infusion-associated reactions was higher in patients who were positive for anti-Fabrazyme antibodies than in patients who were negative for anti-Fabrazyme antibodies [see Adverse Reactions (6.2)].
Severe infusion-associated reactions experienced by more than one patient in clinical trials of Fabrazyme included chills, vomiting, hypotension, and paresthesia. Other infusion-associated reactions included pyrexia, feeling hot or cold, dyspnea, nausea, flushing, headache, fatigue, pruritus, pain in extremity, hypertension, chest pain, throat tightness, abdominal pain, dizziness, tachycardia, nasal congestion, diarrhea, edema peripheral, myalgia, urticaria, bradycardia, and somnolence [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Most patients in clinical trials were pretreated with acetaminophen. In patients experiencing infusion-associated reactions, pretreatment with an antipyretic and antihistamine is recommended. Infusion-associated reactions occurred in some patients after receiving pretreatment with antipyretics, antihistamines, and oral steroids. Infusion-associated reactions tended to decline in frequency with continued use of Fabrazyme. However, infusion-associated reactions may still occur despite extended duration of Fabrazyme treatment. If an infusion-associated reaction occurs, decrease the infusion rate, temporarily stop the infusion, and consider administrating additional antipyretics, antihistamines, and/or steroids. If severe infusion-associated reactions occur, discontinue administration of Fabrazyme immediately and initiate appropriate medical treatment. Severe reactions are generally managed with administration of antihistamines, corticosteroids, intravenous fluids, and/or oxygen, when clinically indicated. Because of the potential for severe infusion-associated reactions, ensure appropriate medical support measures are readily available when Fabrazyme is administered. Monitor closely patients who have experienced infusion-associated reactions when readministering Fabrazyme. Patients with advanced Fabry disease may have compromised cardiac function, which may predispose them to a higher risk of severe complications from infusion-associated reactions. Monitor closely patients with compromised cardiac function if Fabrazyme is administered to these patients [see Warnings and Precautions (5.1)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in labeling:
- Anaphylaxis and Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Infusion-Associated Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trial of another drug and may not reflect the rates observed in patients in clinical practice.
The data described below reflect exposure of 80 patients, ages 16 to 61 years, to 1 mg/kg Fabrazyme every two weeks in two separate double-blind, placebo-controlled clinical trials, for periods ranging from 1 to 35 months (mean 15.5 months). All 58 patients enrolled in one of the two studies continued into an open-label extension study of Fabrazyme treatment for up to 54 additional months. Patients were treated with antipyretics and antihistamines prior to the infusions.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Fabrazyme in the studies described below with the incidence of antibodies in other studies or to other agalsidase beta products may be misleading.
Patients with classic Fabry disease in Study 1, Study 2, and extension studies were tested at multiple time points for antibodies to agalsidase beta during the 55 to 58-month period. Approximately 83% (110 of 133) of adult patients receiving agalsidase beta developed antibodies; 77% (102/133) of patients developed neutralizing antibody (NAb) that inhibited in vitro agalsidase beta catalytic activity, which declined over time, and 6% (8/133) of patients developed NAb that inhibited cellular uptake. In pediatric patients with Fabry disease in Study 3 receiving the recommended dose who were 8 to <16 years of age, antibodies to agalsidase beta were detected in approximately 69% (11/16) of patients. Most patients who developed antibodies did so within the first 3 months of treatment. Antibody titers generally declined over time. Approximately 18% of adult patients who developed antibodies became antibody negative by 74 weeks (median time) from the time of seroconversion; however, none of the pediatric patients became antibody negative. Female patients generally had lower incidence of antibodies and lower antibody titers compared to male patients. In Study 5, patients with truncating GLA mutations had higher incidence of antibodies and higher antibody titers compared to patients with nontruncating GLA mutations. Patients with plasma α-galactosidase A activity ≤1.5 nmol/hr/mL had higher incidence of antibodies and higher antibody titers compared to patients with plasma α-galactosidase A activity >1.5 nmol/hr/mL.
In general, over 90% of adult and pediatric patients treated with agalsidase beta achieved and maintained normalization of plasma globotriaosylceramide (GL-3) levels irrespective of developing antibodies to agalsidase beta.
Study 4 was an open-label, rechallenge study to evaluate the safety of Fabrazyme treatment in patients who had a positive skin test to Fabrazyme or who had tested positive for Fabrazyme-specific IgE antibodies. In this study, six adult male patients, who had experienced multiple or recurrent infusion-associated reactions during previous clinical trials of Fabrazyme, were rechallenged with Fabrazyme administered as a graded infusion for up to 52 weeks of treatment. The initial two rechallenge doses of Fabrazyme were administered as a 0.5 mg/kg dose per week at an initial infusion rate of 0.01 mg/min for the first 30 minutes (1/25th the usually recommended maximum infusion rate). The infusion rate was doubled every 30 minutes thereafter, as tolerated, for the remainder of the infusion up to a maximum rate of 0.25 mg/min. If the patient tolerated the infusion, the dose was increased to 1 mg/kg every two weeks and the infusion rate was increased by slow upwards titration [see Dosage and Administration (2.1)]. Pretreatment was not permitted for at least the first 4 infusions in order to allow early recognition of acute systemic hypersensitivity reactions. Four of the six patients treated in this study received at least 26 weeks of Fabrazyme (2 patients received 26 weeks and 2 patients received 52 weeks), and two patients discontinued prematurely due to recurrent infusion-associated reactions [see Warnings and Precautions (5.1, 5.2)].
Testing for IgE antibodies was performed in approximately 60 patients in clinical trials who experienced moderate to severe infusion-associated reactions or in whom mast cell activation was suspected. Seven of these patients tested positive for Fabrazyme-specific IgE antibodies or had a positive skin test to Fabrazyme. Patients who have had a positive skin test to Fabrazyme, or who have tested positive for Fabrazyme-specific IgE antibodies in clinical trials with Fabrazyme have been rechallenged [see Dosage and Administration (2.1) and Warnings and Precautions (5.1, 5.2)]. The incidences of hypersensitivity reactions were 51% (41/80) and 60% (25/42) in adult patients with persistent anti-Fabrazyme antibodies and in adult patients with high antibody titer, respectively, compared to 30% (7/23) in antibody-negative adult patients [see Warnings and Precautions (5.1)]. The incidence of infusion-associated reactions was 76% (84/110) in antibody-positive adult patients compared to 30% (7/23) in antibody-negative adult patients. The incidence of infusion-associated reactions was 46% (5/11) in antibody positive pediatric patients compared to 20% (1/5) in antibody negative pediatric patients [see Warnings and Precautions (5.2)].
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Fabrazyme. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Cardiovascular: cardiorespiratory arrest, cardiac failure, myocardial infarction, palpitations
- Hypersensitivity reactions: anaphylaxis [see Warnings and Precautions (5.1)], localized angioedema (including auricular swelling, eye swelling, dysphagia, lip swelling, edema, pharyngeal edema, face swelling, and swollen tongue), and bronchospasm
- General: hyperhidrosis, asthenia, infusion site reaction
- Lymphatic: lymphadenopathy
- Musculoskeletal: arthralgia
- Neurologic: cerebrovascular accident, hypoesthesia, oral hypoesthesia
- Pulmonary: respiratory failure, hypoxia
- Renal: renal failure
- Vascular: leukocytoclastic vasculitis
8. Use In Specific Populations
8.4 Pediatric Use
The safety and effectiveness of Fabrazyme have been established in pediatric patients based on adequate and well-controlled studies in adults, a single-arm, open-label study in 16 pediatric patients with Fabry disease aged 8 to 16 years, and additional data in 24 patients with Fabry disease aged 2 to 7 years [see Clinical Pharmacology (12.2) and Clinical Studies (14)].
The overall safety profile of Fabrazyme was similar between the pediatric and the adult population [see Adverse Reactions (6.1) and Clinical Studies (14)].
11. Fabrazyme Description
Agalsidase beta is a recombinant human α-galactosidase A enzyme with the same amino acid sequence as the native enzyme. Purified agalsidase beta is a homodimeric glycoprotein with a molecular weight of approximately 100 kD. The mature protein is comprised of two subunits of 398 amino acids (approximately 51 kD), each of which contains three N-linked glycosylation sites. The enzyme α-galactosidase A catalyzes the hydrolysis of GL-3 and other α-galactyl-terminated neutral glycosphingolipids, such as galabiosylceramide and blood group B substances to ceramide dihexoside and galactose. The specific activity of agalsidase beta is approximately 70 U/mg (one unit is defined as the amount of activity that results in the hydrolysis of 1 µmole of a synthetic substrate, p-nitrophenyl-α-D-galactopyranoside, per minute under the assay conditions).
Agalsidase beta is produced by recombinant DNA technology in a Chinese hamster ovary mammalian cell expression system.
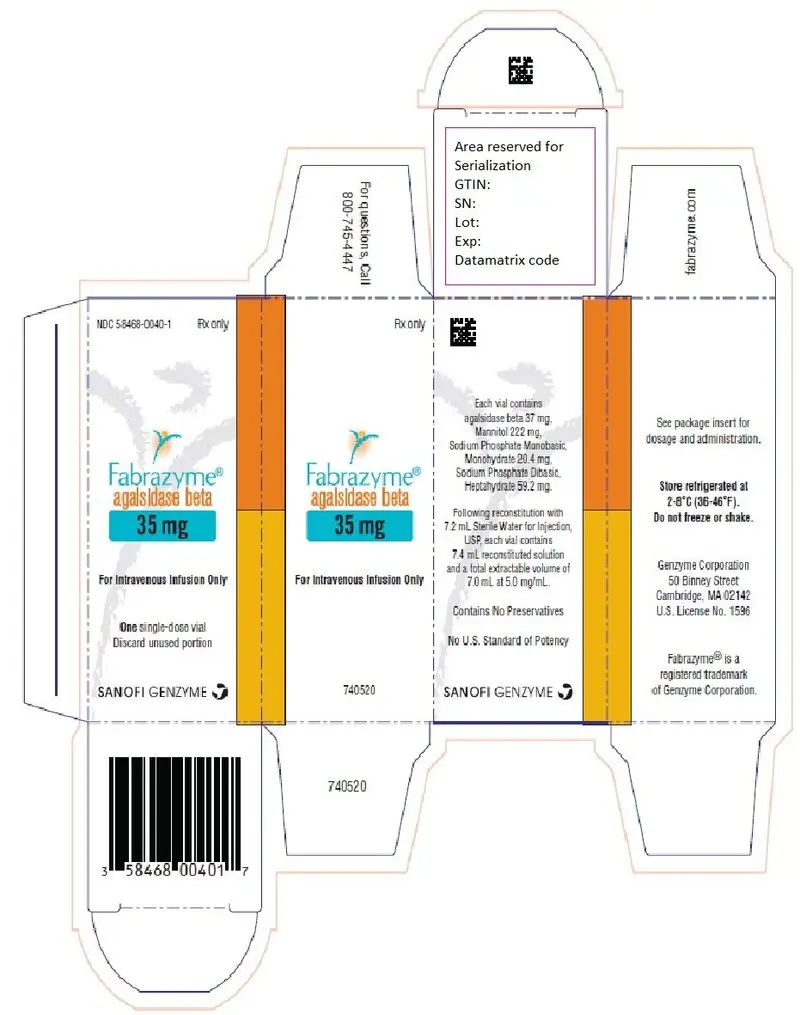
Fabrazyme (agalsidase beta) for injection is intended for intravenous infusion. It is supplied as a sterile, nonpyrogenic, preservative-free, white to off-white, lyophilized cake or powder for reconstitution with Sterile Water for Injection, USP. Each 35 mg vial contains 37 mg of agalsidase beta, as well as 222 mg mannitol, 20.4 mg sodium phosphate monobasic monohydrate, and 59.2 mg sodium phosphate dibasic heptahydrate. Following reconstitution as directed, 35 mg of agalsidase beta (7 mL) may be extracted from each 35 mg vial.
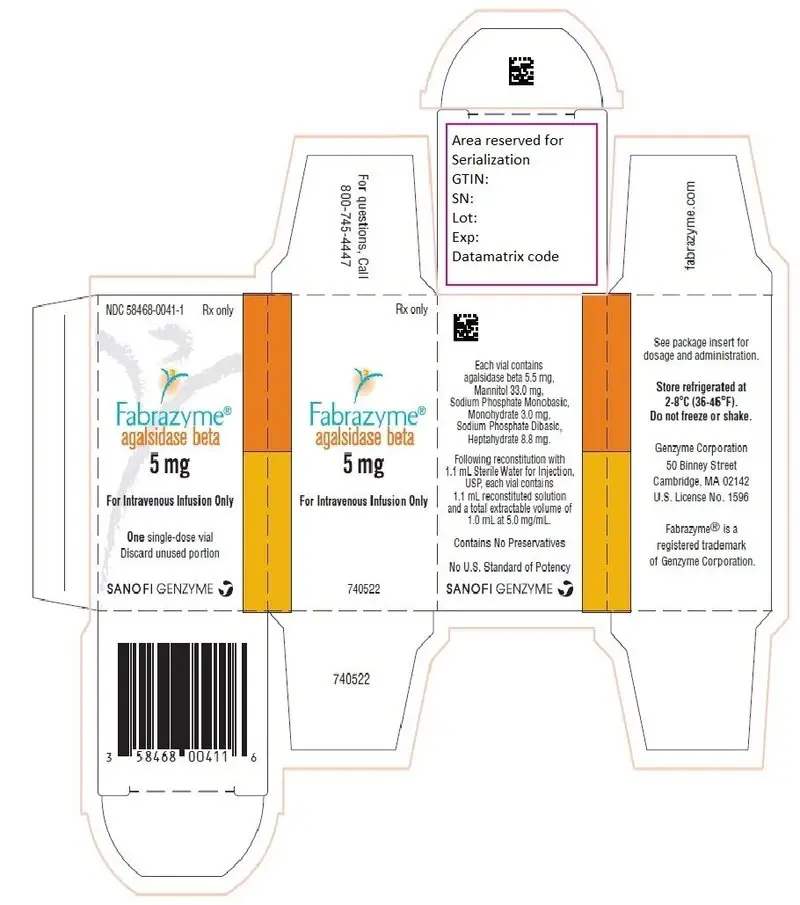
Each 5 mg vial contains 5.5 mg of agalsidase beta, as well as 33.0 mg mannitol, 3.0 mg sodium phosphate monobasic monohydrate, and 8.8 mg sodium phosphate dibasic heptahydrate. Following reconstitution as directed, 5 mg of agalsidase beta (1 mL) may be extracted from each 5 mg vial.
12. Fabrazyme - Clinical Pharmacology
12.1 Mechanism of Action
Fabrazyme (agalsidase beta) provides an exogenous source of α-galactosidase A in Fabry disease patients. Agalsidase beta is internalized and transported into lysosomes where it exerts enzymatic activity and reduces accumulated GL-3.
12.2 Pharmacodynamics
In Study 1, baseline mean values for plasma GL-3 were similar in the Fabrazyme (14.4 µg/mL) and the placebo (14.7 µg/mL) treatment groups. In the Fabrazyme treatment group, all 29 patients experienced normalization of plasma GL-3 levels (≤7.03 µg/mL) and they maintained normal plasma GL-3 levels for up to 60 months of treatment. Follow-up heart and kidney biopsies were assessed at month 54 in only 8 of the 44 patients, which showed sustained GL-3 clearance in the capillary endothelium of the kidney in 8 patients, and sustained GL-3 clearance in the capillary endothelium of the heart in 6 patients. The reduction in tissue GL-3 is summarized in the clinical studies section (Table 4) [see Clinical Studies (14)].
In Study 2, patients in the Fabrazyme treatment group had mean plasma GL-3 levels that decreased from 9.0 µg/mL at baseline (N=49) to 4.8 µg/mL at one year (N=37) and 4.8 µg/mL at two years (N=18). In the placebo group, the mean plasma GL-3 was 9.1 µg/mL at baseline (N=31), 8.8 µg/mL at one year (N=21), and 9.4 µg/mL at two years (N=7).
In Study 3, at baseline, all 14 males had elevated plasma GL-3 levels (i.e., >7.03 µg/mL), whereas the two female patients had normal plasma GL-3 levels. At weeks 24 and 48 of treatment, all 14 males had plasma GL-3 within the normal range. The two female patients' plasma GL-3 levels remained normal through study week 48. Histological evaluation of the capillary endothelium (vasculature), deep vessel endothelium, deep vessel smooth muscle cells, and perineurium of biopsied skin was conducted using histochemistry with light microscopy. Scoring was on a scale of 0 to 3 (0 defined as none; 1 as mild, 2 as moderate, and 3 as severe). At baseline, 12 of the 14 males had GL-3 inclusions present on skin biopsy (scores 1, 2, or 3) and all 12 achieved GL-3 inclusion scores of 0 at weeks 24 and 48 of treatment. The two females had no GL-3 inclusions in skin at baseline.
In Study 5, in an analysis of 24 Fabrazyme-treated pediatric patients with Fabry disease aged 2 to <8 years at Fabrazyme initiation and with elevated plasma GL-3 levels (i.e., >7.03 μg/mL) at baseline, plasma GL-3 levels fell within the normal range (i.e., ≤7.03 μg/mL) in 91% (20/22), 95% (18/19), and 92% (12/13) of patients at 6, 12, and 24 months, respectively.
12.3 Pharmacokinetics
The pharmacokinetics of Fabrazyme in clinical studies with adult and pediatric patients with Fabry disease is summarized in Table 3.
Fabrazyme exhibited nonlinear pharmacokinetics following intravenous infusions at 0.3 (30% of the approved recommended dosage), 1 mg/kg, and 3 mg/kg (3 times the approved recommended dosage) in adult patients. The area under the plasma concentration-time curve (AUCinf) and the maximum plasma concentration (Cmax) increased greater than dose proportional with increasing doses. The AUCinf and Cmax following multiple dose administrations were comparable to their values at the first dose.
In pediatric patients 8 to 16 years of age with body weight ranging from 27 to 65 kg, the AUCinf and Cmax following multiple dose administrations were higher compared to their values at the first dose. The increased plasma concentrations following multiple dose administrations in pediatric patients could be due to formation of antidrug antibodies; however, such impact was not observed in adult patients [see Adverse Reactions (6.2) and Use in Specific Populations (8.4)].
| Dose | Regimen | Mean Infusion Length (min) | Infusion number (n=patients) | AUCinf
µg min/mL | Cmax
µg/mL | Half-life min | CL mL/min/kg | Vss*
mL/kg |
|---|---|---|---|---|---|---|---|---|
| All data reported as the mean ± standard deviation. | ||||||||
|
||||||||
| Study FB9702-01: Phase 1/2 Study in Adult Patients with Fabry Disease | ||||||||
| 0.3 mg/kg | q14 days × 5 | 132 | 1 (n=3) | 79 ± 24 | 0.6 ± 0.2 | 92 ± 27 | 4.1 ± 1.2 | 225 ± 62 |
| 128 | 5 (n=3) | 74 ± 30 | 0.6 ± 0.2 | 78 ± 67 | 4.6 ± 2.2 | 330 ± 231 | ||
| 1 mg/kg | q14 days × 5 | 115 | 1 (n=3) | 496 ± 137 | 5.0 ± 1.1 | 67 ± 12 | 2.1 ± 0.7 | 112 ± 13 |
| 120 | 5 (n=2) | 466 ± 382 | 4.74 ± 4.3 | 45 ± 3 | 3.2 ± 2.6 | 243 ± 236 | ||
| 3 mg/kg | q14 days × 5 | 129 | 1 (n=2) | 4168 ± 1401 | 29.7 ± 14.6 | 102 ± 4 | 0.8 ± 0.3 | 81 ± 45 |
| 300 | 5 (n=2) | 4327 ± 2074 | 19.8 ± 5.8 | 87 ± 21 | 0.8 ± 0.4 | 165 ± 80 | ||
| Study 1: Phase 3 Study in Adult Patients with Fabry Disease | ||||||||
| 1 mg/kg | q14 days × 11 | 280 | 1–3 (n=11) | 649 ± 226 | 3.5 ± 1.6 | 89 ± 20 | 1.8 ± 0.8 | 120 ± 80 |
| 280 | 7 (n=11) | 372 ± 223 | 2.1 ± 1.14 | 82 ± 25 | 4.9 ± 5.6 | 570 ± 710 | ||
| 300 | 11 (n=11) | 784 ± 521 | 3.5 ± 2.2 | 119 ± 49 | 2.3 ± 2.2 | 280 ± 230 | ||
| Study 3: Phase 2 Study in Pediatric Patients with Fabry Disease | ||||||||
| 1 mg/kg | q14 days × 24 | 208 | 1 (n=8–9) | 344 ± 307 | 2.2 ± 1.9 | 86 ± 27 | 5.8 ± 4.6 | 1097 ± 912 |
| 111 | 12 (n=15) | 1007 ± 688 | 4.9 ± 2.4 | 130 ± 41 | 1.6 ± 1.2 | 292 ± 185 | ||
| 108 | 24 (n=9–10) | 1238 ± 547 | 7.1 ± 4.4 | 151 ± 59 | 1.1 ± 0.8 | 247 ± 146 | ||
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
There are no animal or human studies to assess the carcinogenic or mutagenic potential of Fabrazyme. A study to evaluate the effects of agalsidase beta on fertility and general reproduction was performed in male and female rats at doses up to 10 mg/kg/day (23 times the human dose, on a body surface area basis). There were no adverse effects of agalsidase beta on fertility and early embryonic development in rats.
14. Clinical Studies
The safety and efficacy of Fabrazyme were assessed in four clinical studies in patients with Fabry disease and one matched analysis based on data from observational studies.
Study 1 was a randomized, double-blind, placebo-controlled, multinational, multicenter study of 58 patients with Fabry disease (56 males and 2 females), ages 16 to 61 years, all naive to enzyme replacement therapy [see Clinical Pharmacology (12.2)]. Patients were randomized 1:1 to receive either Fabrazyme 1 mg/kg every 2 weeks or placebo for 20 weeks. Patients had a median age of 24 years in the placebo group and 33 years in the Fabrazyme group at baseline. At baseline, all patients had plasma αGAL activity below the detection limit and 79% had leukocyte αGAL activity below the detection limit. The median plasma GL-3 at baseline was 14.4 ng/uL in the placebo group and 14.7 ng/uL in the Fabrazyme group with the overall range of <1.2 to 36 ng/uL. The median eGFR at baseline was 98.5 mL/hr in the placebo group and 83.0 mL/hr in the Fabrazyme group (overall range 24 to 153 mL/hr). All patients were pretreated with acetaminophen and an antihistamine. Oral steroids were an additional option to the pretreatment regimen for patients who exhibited severe or recurrent infusion-associated reactions. Tissue biopsy specimens (kidney, heart, skin) were evaluated at baseline and at week 20 by light microscopy for the presence and number of GL-3 inclusions using a semi-quantitative methodology. Renal interstitial capillaries were scored based on the number of GL-3 inclusions on a scale of 0 to 3 (0 defined as "nearly none" or "trace," 1 defined as "mild," 2 defined as "moderate," and 3 defined as "severe"). The primary endpoint was the proportion of patients in either group with a renal capillary GL-3 inclusion score of zero at week 20. In the Fabrazyme group, 20 of 29 (69%) patients achieved a score of zero while 0 of 29 placebo-treated patients achieved a score of zero (p<0.001). Similar reductions in GL-3 inclusions were observed in the capillary endothelium of the heart and skin (Table 4). All 58 patients who completed Study 1 were subsequently treated with Fabrazyme 1 mg/kg every two weeks in an open-label extension study. After six months of open-label treatment, most patients with available biopsy data achieved a GL-3 inclusion score of 0 in capillary endothelium (Table 4).
| 20 weeks of randomized treatment in study 1 | 6 months of Fabrazyme open-label treatment | |||
|---|---|---|---|---|
| Placebo (n=29) | Fabrazyme (n=29) | Placebo/Fabrazyme (n=29)* | Fabrazyme/Fabrazyme (n=29)* |
|
|
||||
| Kidney | 0/29 | 20/29 | 24/24 | 23/25 |
| Heart | 1/29 | 21/29 | 13/18 | 19/22 |
| Skin | 1/29 | 29/29 | 25/26 | 26/27 |
Study 2 was a randomized (2:1 Fabrazyme to placebo), double-blind, placebo-controlled, multinational, multicenter study of 82 patients (72 males and 10 females) with Fabry disease, all naive to enzyme replacement therapy [see Clinical Pharmacology (12.2)]. Of the 82 enrolled patients, 51 and 31 patients were randomized to the Fabrazyme and placebo groups, respectively. Patients were 20 to 72 years of age with a median age of 45 years at baseline, a median age of 36 years at Fabry disease diagnosis, and at a median of 10 years at symptom onset. The median plasma GL-3 at baseline was 9.3 ug/mL in the placebo group and 8.9 ug/mL in the Fabrazyme group with the overall range of 2.8 to 18.9 ug/mL. At baseline, patients had median plasma αGAL activity 1.5 nmol/hour/mL (range: 0 to 1.5), leukocyte αGAL activity 1.8 nmol/hour/mL (range: 0 to 4.0), eGFR 52 mL/min/1.73 m2 (range: 25 to 113), and protein to creatinine ratio 0.9 mg/mg (range: 0 to 7.3). Patients received either 1 mg/kg Fabrazyme IV or placebo every two weeks for up to 35 months (median follow-up 18.5 months). The primary efficacy endpoint was the time to first occurrence of a clinically significant event (renal, cardiac, or cerebrovascular event, or death). A total of 14 of 51 (28%) Fabrazyme-treated patients and 13 of 31 (42%) placebo-treated patients experienced a clinically significant event (HR 0.57, 95% CI: 0.27, 1.22).
Study 3 (Pediatric Study) was an open-label, single-arm, multinational, multicenter study in 16 pediatric patients with Fabry disease (14 males, 2 females), aged 8 to 16 years (median 12 years) [see Clinical Pharmacology (12.2)]. At baseline, patients had median plasma αGAL activity 0.2 nmol/hour/mL (range: 0.0, 2.0) and median leukocyte αGAL activity 0.5 nmol/hour/mg (range: 0.0, 12.5). All 14 males had elevated plasma GL-3 levels (i.e., >7.03 µg/mL) at baseline, whereas the two females had normal plasma GL-3 levels. Median eGFR was normal (112.1 mL/min/1.73 m2) at baseline and did not change during treatment, and median urinary protein was 151.0 mg/24 hr (range: 70.0, 431.0). All patients received Fabrazyme 1 mg/kg every two weeks for up to 48 weeks.
Study 5 was a long-term, observational study assessing the rate of decline in renal function (eGFR slope) in 122 patients with Fabry disease aged 16 years and older treated with Fabrazyme. Treated patients were matched 1:1 based on age (at Fabrazyme initiation), sex, Fabry disease subtype (classic or non-classic), and baseline eGFR to a historical cohort of untreated patients with Fabry disease. The median follow-up time was 3 years in the untreated group and 4.5 years in the treated group (maximum follow-up time 5 years in both groups). In the matched cohort, the median age (at Fabrazyme initiation) was 35 years, 72% of patients were male, 84% of patients had the classic Fabry disease subtype, and the median baseline eGFR was 93 mL/min/1.73 m2. The estimated mean eGFR slope was -1.5 mL/min/1.73 m2/year in the Fabrazyme-treated group and -3.2 mL/min/1.73 m2/year in the untreated group (eGFR slope difference: 1.7 mL/min/1.73 m2/year; 95% CI: 0.5, 3.0).
16. How is Fabrazyme supplied
Fabrazyme (agalsidase beta) for injection is supplied as a sterile, nonpyrogenic, white to off-white lyophilized cake or powder in single-dose vials.
35 mg vial: NDC 58468-0040-1
5 mg vial: NDC 58468-0041-1
| FABRAZYME
agalsidase beta injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FABRAZYME
agalsidase beta injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Genzyme Corporation (025322157) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 968278916 | ANALYSIS(58468-0040, 58468-0041) , API MANUFACTURE(58468-0040, 58468-0041) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 050424395 | PACK(58468-0040, 58468-0041) , LABEL(58468-0040, 58468-0041) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 968278932 | API MANUFACTURE(58468-0040, 58468-0041) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 943130096 | ANALYSIS(58468-0040, 58468-0041) , API MANUFACTURE(58468-0040, 58468-0041) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 968302658 | ANALYSIS(58468-0040, 58468-0041) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Ireland Limited | 985127419 | ANALYSIS(58468-0040, 58468-0041) , MANUFACTURE(58468-0040, 58468-0041) , PACK(58468-0040, 58468-0041) , LABEL(58468-0040, 58468-0041) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 117450412 | ANALYSIS(58468-0040, 58468-0041) , API MANUFACTURE(58468-0040, 58468-0041) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Resilience US, Inc | 118999964 | ANALYSIS(58468-0040, 58468-0041) | |
