Drug Detail:Fintepla (Fenfluramine)
Drug Class: CNS stimulants Miscellaneous anticonvulsants
Highlights of Prescribing Information
FINTEPLA® (fenfluramine) oral solution
Initial U.S. Approval: 1973
WARNING: VALVULAR HEART DISEASE and PULMONARY ARTERIAL HYPERTENSION
See full prescribing information for complete boxed warning.
- There is an association between serotonergic drugs with 5-HT2B receptor agonist activity, including fenfluramine (the active ingredient in FINTEPLA), and valvular heart disease and pulmonary arterial hypertension. (5.1)
- Echocardiogram assessments are required before, during, and after treatment with FINTEPLA. (2.1, 2.6, 5.1)
- FINTEPLA is available only through a restricted program called the FINTEPLA REMS. (5.2)
Recent Major Changes
| Dosage and Administration (2.2, 2.3, 2.4, 2.5) | 1/2023 |
Indications and Usage for Fintepla
FINTEPLA is indicated for the treatment of seizures associated with Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age and older. (1)
Fintepla Dosage and Administration
- FINTEPLA is to be administered orally and may be taken with or without food. (2.2)
- Dravet Syndrome
- The initial starting and maintenance dosage is 0.1 mg/kg twice daily, which can be increased weekly based on efficacy and tolerability. (2.2)
- The maximum daily maintenance dosage of FINTEPLA is 0.35 mg/kg twice daily (maximum daily dosage of 26 mg). (2.2)
- Lennox-Gastaut Syndrome
- The initial starting dosage is 0.1 mg/kg twice daily, which should be increased weekly based on tolerability. (2.2)
- The recommended maintenance dosage of FINTEPLA is 0.35 mg/kg twice daily (maximum daily dosage of 26 mg). (2.2)
- Dravet Syndrome and Lennox-Gastaut Syndrome
- Dose adjustment is required in patients taking concomitant stiripentol plus clobazam: the maximum daily maintenance dosage of FINTEPLA is 0.2 mg/kg twice daily (maximum daily dosage of 17 mg). (2.2, 2.3, 2.4, 7.1)
- Dosage adjustment is recommended in patients:
- Taking strong CYP1A2 or CYP2D6 inhibitors (2.3, 7.1)
- With severe renal impairment (2.4, 8.6)
- With mild, moderate, and severe hepatic impairment (2.5, 8.7)
Dosage Forms and Strengths
Oral solution: 2.2 mg/mL fenfluramine (3)
Contraindications
- Hypersensitivity to fenfluramine or any of the excipients in FINTEPLA (4)
- Within 14 days of the administration of monoamine oxidase inhibitors due to an increased risk of serotonin syndrome (4)
Warnings and Precautions
- Decreased Appetite and Decreased Weight: Advise patients that FINTEPLA can cause decreased appetite and decreased weight. (5.3)
- Somnolence, Sedation, and Lethargy: Monitor for somnolence and sedation. Advise patients not to drive or operate machinery until they have gained sufficient experience on FINTEPLA. (5.4)
- Suicidal Behavior and Ideation: Monitor patients for suicidal behavior and thoughts. (5.5)
- Withdrawal of Antiepileptic Drugs: FINTEPLA should be gradually withdrawn to minimize the risk of increased seizure frequency and status epilepticus. (5.6)
- Serotonin Syndrome: Advise patients that serotonin syndrome is a potentially life-threatening condition and may occur with FINTEPLA, particularly with concomitant administration of FINTEPLA with other serotonergic drugs. (5.7)
- Increase in Blood Pressure: Monitor blood pressure during treatment. (5.8)
- Glaucoma: Discontinue therapy in patients with acute decrease in visual acuity or ocular pain. (5.9)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence at least 10% and greater than placebo) in patients with Dravet Syndrome were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; blood pressure increased; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; status epilepticus. (6.1)
The most common adverse reactions (incidence at least 10% and greater than placebo) in patients with Lennox-Gastaut syndrome were diarrhea; decreased appetite; fatigue; somnolence; vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact UCB, Inc. at 1 844-599-2273 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Dose adjustment is required for patients taking stiripentol plus clobazam. (2.2, 2.3, 7.1)
- Strong CYP1A2 or CYP2D6 inhibitors: a dose adjustment is recommended (2.3, 7.1)
- Strong CYP1A2, CYP2B6, or CYP3A4 inducers: it is recommended to avoid coadministration with FINTEPLA. If coadministration is necessary, consider a FINTEPLA dosage increase. (7.1)
Use In Specific Populations
- Pregnancy: Based on animal data, may cause fetal harm (8.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2023
Full Prescribing Information
WARNING: VALVULAR HEART DISEASE and PULMONARY ARTERIAL HYPERTENSION
There is an association between serotonergic drugs with 5-HT2B receptor agonist activity, including fenfluramine (the active ingredient in FINTEPLA), and valvular heart disease and pulmonary arterial hypertension [see Warnings and Precautions (5.1)].
Echocardiogram assessments are required before, during, and after treatment with FINTEPLA. The benefits versus the risks of initiating or continuing FINTEPLA must be considered, based on echocardiogram findings [see Dosage and Administration (2.1, 2.6) and Warnings and Precautions (5.1)].
Because of the risks of valvular heart disease and pulmonary arterial hypertension, FINTEPLA is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the FINTEPLA REMS [see Warnings and Precautions (5.2)].
1. Indications and Usage for Fintepla
FINTEPLA is indicated for the treatment of seizures associated with Dravet syndrome (DS) and Lennox-Gastaut syndrome (LGS) in patients 2 years of age and older.
2. Fintepla Dosage and Administration
2.1 Assessments Prior to Initiating FINTEPLA
Prior to starting treatment with FINTEPLA, obtain an echocardiogram assessment to evaluate for valvular heart disease and pulmonary arterial hypertension [see Dosage and Administration (2.6) and Warnings and Precautions (5.1)].
2.2 Dosing Information
FINTEPLA is to be administered orally and may be taken with or without food.
Lennox-Gastaut Syndrome
- The initial starting dosage for patients with Lennox-Gastaut syndrome is 0.1 mg/kg twice daily, which should be increased weekly based on tolerability. Table 1 provides the recommended titration schedule.
- Patients with Lennox-Gastaut syndrome not on concomitant stiripentol who are tolerating FINTEPLA should be titrated to the recommended maintenance dosage of 0.35 mg/kg twice daily (maximum daily dosage of 26 mg).
- Patients with Lennox-Gastaut syndrome taking concomitant stiripentol plus clobazam who are tolerating FINTEPLA should be titrated to the recommended maintenance dosage of 0.2 mg/kg twice daily (maximum daily dosage of 17 mg) [see Drug Interactions (7.1)].
| Without concomitant stiripentol* | With concomitant stiripentol plus clobazam | |||
|---|---|---|---|---|
| Weight-based Dosage | Maximum Total Daily Dosage† | Weight-based Dosage | Maximum Total Daily Dosage† | |
|
||||
| Initial Dosage‡ | 0.1 mg/kg twice daily | 26 mg | 0.1 mg/kg twice daily | 17 mg |
| Day 7 | 0.2 mg/kg twice daily | 26 mg | 0.15 mg/kg twice daily | 17 mg |
| Day 14§ | 0.35 mg/kg twice daily | 26 mg | 0.2 mg/kg twice daily | 17 mg |
2.3 Dosage Modifications for Patients with Concomitant Use of Strong CYP1A2 or CYP2D6 Inhibitors (DS and LGS)
For patients with concomitant use of FINTEPLA with a strong CYP1A2 or CYP2D6 inhibitor, a maximum total daily dosage of 20 mg without concomitant stiripentol and 17 mg with concomitant stiripentol plus clobazam is recommended [see Drug Interactions (7.1)].
2.4 Dosage Modifications for Patients with Severe Renal Impairment (DS and LGS)
For patients with severe renal impairment (estimated glomerular filtration rate (eGFR) 15 to 29 mL/min/1.73m2), a maximum total daily dosage of 20 mg without concomitant stiripentol and 17 mg with concomitant stiripentol plus clobazam is recommended [see Use in Specific Populations (8.6)].
2.5 Dosage Modifications for Patients with Mild, Moderate, and Severe Hepatic Impairment (DS and LGS)
See Table 2 for dosage adjustments and recommendations for patients with hepatic impairment [see Use in Specific Populations (8.7)].
| Hepatic Impairment Classification | Without concomitant stiripentol* | With concomitant stiripentol plus clobazam |
|---|---|---|
| Maximum total daily dosage | Maximum total daily dosage | |
|
||
| Mild (Child-Pugh A) | 20 mg | 13 mg* |
| Moderate (Child-Pugh B) | 20 mg | Use not recommended |
| Severe (Child-Pugh C) | 17 mg | Use not recommended |
2.6 Assessments During and After Administration of FINTEPLA
To evaluate for valvular heart disease and pulmonary arterial hypertension, obtain an echocardiogram assessment every 6 months during treatment with FINTEPLA, and 3 to 6 months after the final dose of FINTEPLA [see Warnings and Precautions (5.1)].
2.7 Administration Instructions
A calibrated measuring device (either a 3 mL or 6 mL oral syringe) will be provided by the pharmacy and is recommended to measure and administer the prescribed dose accurately [see How Supplied/Storage and Handling (16.1)]. A household teaspoon or tablespoon is not an adequate measuring device and should not be used.
Discard any unused FINTEPLA oral solution remaining after 3 months of first opening the bottle or the "Discard After" date on the bottle, whichever is sooner.
FINTEPLA is compatible with commercially available gastric and nasogastric feeding tubes.
2.8 Discontinuation of FINTEPLA
When discontinuing FINTEPLA, the dose should be decreased gradually. As with all antiepileptic drugs, abrupt discontinuation should be avoided when possible, to minimize the risk of increased seizure frequency and status epilepticus [see Warnings and Precautions (5.6)].
3. Dosage Forms and Strengths
Oral solution: 2.2 mg/mL fenfluramine as a clear, colorless, cherry flavored liquid.
4. Contraindications
FINTEPLA is contraindicated in patients with:
- Hypersensitivity to fenfluramine or any of the excipients in FINTEPLA [see Description (11)]
- Concomitant use, or within 14 days of the administration, of monoamine oxidase inhibitors because of an increased risk of serotonin syndrome [see Warnings and Precautions (5.7)]
5. Warnings and Precautions
5.1 Valvular Heart Disease and Pulmonary Arterial Hypertension
Because of the association between serotonergic drugs with 5-HT2B receptor agonist activity, including fenfluramine (the active ingredient in FINTEPLA), and valvular heart disease (VHD) and pulmonary arterial hypertension (PAH), cardiac monitoring is required prior to starting treatment, during treatment, and after treatment with FINTEPLA concludes. Cardiac monitoring via echocardiogram can identify evidence of valvular heart disease and pulmonary arterial hypertension prior to a patient becoming symptomatic, aiding in early detection of these conditions. In clinical trials for DS and LGS of up to 3 years in duration, no patient receiving FINTEPLA developed valvular heart disease or pulmonary arterial hypertension [see Boxed Warning and Adverse Reactions (6.1)].
5.2 FINTEPLA REMS Program
FINTEPLA is available only through a restricted distribution program called the FINTEPLA REMS program because of the risk of valvular heart disease and pulmonary arterial hypertension [see Warnings and Precautions (5.1)].
Notable requirements of the FINTEPLA REMS Program include:
- Prescribers must be certified by enrolling in the FINTEPLA REMS program.
- Prescribers must counsel patients receiving FINTEPLA about the risk of valvular heart disease and pulmonary arterial hypertension, how to recognize signs and symptoms of valvular heart disease and pulmonary arterial hypertension, the need for baseline (pretreatment) and periodic cardiac monitoring via echocardiogram during FINTEPLA treatment, and cardiac monitoring after FINTEPLA treatment.
- Patients must enroll in the REMS program and comply with ongoing monitoring requirements [see Warnings and Precautions (5.1)].
- The pharmacy must be certified by enrolling in the REMS program and must only dispense to patients who are authorized to receive FINTEPLA.
- Wholesalers and distributors must only distribute to certified pharmacies.
Further information is available at www.FinteplaREMS.com or by telephone at 1-877-964-3649.
5.3 Decreased Appetite and Decreased Weight
FINTEPLA can cause decreases in appetite and weight. In placebo-controlled studies for DS (Study 1 and Study 2 combined), approximately 37% of patients treated with FINTEPLA reported, as an adverse reaction, decreased appetite and approximately 9% reported decreased weight, as compared to 8% and 1%, respectively, of patients on placebo. In the placebo- controlled study for LGS (Study 3), approximately 28% of patients treated with FINTEPLA reported, as an adverse reaction, decreased appetite and approximately 5% reported decreased weight, as compared to 15% and 2%, respectively, of patients on placebo [see Adverse Reactions (6.1)]. By the end of the controlled studies, 19% (Studies 1 and 2 combined) of DS patients and 7% (Study 3) of LGS patients treated with FINTEPLA had a measured decrease in weight of 7% or greater from their baseline weight, compared to 2% (Study 1 and 2) and 0% (Study 3) of patients on placebo. This measured decrease in weight appeared to be dose-related. In the controlled studies for DS, 26% of patients on FINTEPLA 0.7 mg/kg/day (Study 1), 19% of patients on FINTEPLA 0.4 mg/kg/day in combination with stiripentol (Study 2), and 13% of patients taking FINTEPLA 0.2 mg/kg/day (Study 1) experienced at least a 7% decrease in weight from baseline. In the controlled study for LGS, 9% of patients on FINTEPLA 0.7 mg/kg/day (Study 3) and 6% of patients on FINTEPLA 0.2 mg/kg/day (Study 3) experienced at least a 7% decrease in weight from baseline. Approximately half of the patients with LGS and most patients with DS resumed the expected measured increases in weight during the open-label extension studies. Given the frequency of these adverse reactions, the growth of pediatric patients treated with FINTEPLA should be carefully monitored. Weight should be monitored regularly during treatment with FINTEPLA, and dose modifications should be considered if a decrease in weight is observed.
5.4 Somnolence, Sedation, and Lethargy
FINTEPLA can cause somnolence, sedation, and lethargy. In controlled studies for DS (Study 1 and Study 2 combined), the incidence of somnolence, sedation, and lethargy was 25% in patients treated with FINTEPLA, compared with 11% of patients on placebo. In the controlled study for LGS (Study 3), the incidence of somnolence, sedation, and lethargy was 19% in patients treated with FINTEPLA, compared with 16% of patients on placebo. In general, these effects may diminish with continued treatment [see Adverse Reactions (6.1)].
Other central nervous system (CNS) depressants, including alcohol, could potentiate these effects of FINTEPLA. Prescribers should monitor patients for somnolence and sedation and should advise patients not to drive or operate machinery until they have gained sufficient experience on FINTEPLA to gauge whether it adversely affects their ability to drive or operate machinery.
5.5 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including FINTEPLA, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with an AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs that did not include FINTEPLA showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed. Table 3 shows absolute and relative risk by indication for all evaluated AEDs.
| Indication | Placebo Patients with Events per 1000 Patients | Drug Patients with Events per 1000 Patients | Relative Risk: Incidence of Events in Drug Patients/ Incidence in Placebo Patients | Risk Difference: Additional Drug Patients with Events per 1000 Patients |
|---|---|---|---|---|
| Epilepsy | 1.0 | 3.4 | 3.5 | 2.4 |
| Psychiatric | 5.7 | 8.5 | 1.5 | 2.9 |
| Other | 1.0 | 1.8 | 1.9 | 0.9 |
| Total | 2.4 | 4.3 | 1.8 | 1.9 |
The relative risk for suicidal thoughts or behavior was higher in clinical trials in patients with epilepsy than in clinical trials in patients with psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing FINTEPLA or any other AED must balance the risk of suicidal thoughts or behaviors with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
5.6 Withdrawal of Antiepileptic Drugs
As with most AEDs, FINTEPLA should generally be withdrawn gradually because of the risk of increased seizure frequency and status epilepticus. If withdrawal is needed because of a serious adverse reaction, rapid discontinuation can be considered.
5.7 Serotonin Syndrome
Serotonin syndrome, a potentially life-threatening condition, may occur with FINTEPLA, particularly with concomitant administration of FINTEPLA with other serotonergic drugs, including, but not limited to, selective serotonin-norepinephrine reuptake inhibitors (SNRIs), selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), bupropion, triptans, dietary supplements (e.g., St. John's Wort, tryptophan), drugs that impair metabolism of serotonin (including monoamine oxidase inhibitors [MAOIs], which are contraindicated with FINTEPLA [see Contraindications (4)], dextromethorphan, lithium, tramadol, and antipsychotics with serotonergic agonist activity. Patients should be monitored for the emergence of signs and symptoms of serotonin syndrome, which include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular signs (e.g., hyperreflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). If serotonin syndrome is suspected, treatment with FINTEPLA should be stopped immediately and symptomatic treatment should be started.
5.8 Increase in Blood Pressure
FINTEPLA can cause an increase in blood pressure [see Adverse Reactions (6.1)]. Rare cases of significant elevation in blood pressure, including hypertensive crisis, has been reported in adult patients treated with fenfluramine, including patients without a history of hypertension. In clinical trials of up to 3 years in duration, no pediatric or adult patient receiving FINTEPLA developed a hypertensive crisis. Monitor blood pressure in patients treated with FINTEPLA.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in labeling:
- Valvular Heart Disease and Pulmonary Arterial Hypertension [see Warnings and Precautions (5.1)]
- Decreased Appetite and Decreased Weight [see Warnings and Precautions (5.3)]
- Somnolence, Sedation, and Lethargy [see Warnings and Precautions (5.4)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.5)]
- Withdrawal of Antiepileptic Drugs [see Warnings and Precautions (5.6)]
- Serotonin Syndrome [see Warnings and Precautions (5.7)]
- Increase in Blood Pressure [see Warnings and Precautions (5.8)]
- Glaucoma [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In controlled and uncontrolled trials in patients with Dravet syndrome (DS), 341 patients were treated with FINTEPLA, including 312 patients treated for more than 6 months, 284 patients treated for more than 1 year, and 138 patients treated for more than 2 years.
In controlled and uncontrolled trials in patients with Lennox-Gastaut syndrome (LGS), 262 patients were treated with FINTEPLA, including 219 patients treated for more than 6 months, 172 patients treated for more than 1 year, and 127 patients treated for more than 2 years.
Dravet Syndrome
In placebo-controlled trials of patients with DS taking concomitant standard of care AEDs, 122 patients were treated with FINTEPLA and 84 patients received placebo [see Clinical Studies (14.1)]. The duration of treatment in these trials was 16 weeks (Study 1) or 17 weeks (Study 2).
In Study 1 and Study 2, the mean age was 9 years (range 2 to 19 years) and approximately 46% of patients were female and 74% were White. All patients were receiving at least one other AED.
In Study 1 and Study 2, the rates of discontinuation as a result of any adverse reaction were 13%, 0%, and 7% for patients treated with FINTEPLA 0.7 mg/kg/day, 0.2 mg/kg/day, and 0.4 mg/kg/day in combination with stiripentol, respectively, compared to 6% for patients on placebo. The most frequent adverse reaction leading to discontinuation in the patients treated with any dose of FINTEPLA was somnolence (3%).
The most common adverse reactions that occurred in patients treated with FINTEPLA (incidence at least 10% and greater than placebo) were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; blood pressure increased; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; status epilepticus.
Table 4 lists the adverse reactions that were reported in 5% or more of patients treated with FINTEPLA and at a rate greater than those on placebo during the titration and maintenance phases of Study 1 and Study 2.
| Adverse Reaction | FINTEPLA Dose Group | Combined Placebo Group* | ||
|---|---|---|---|---|
| Study 1 | Study 2 | |||
| 0.2 mg/kg/day | 0.7 mg/kg/day | 0.4 mg/kg/day† | ||
| N=39 % | N=40 % | N=43 % | N=84 % |
|
|
||||
| Decreased appetite | 23 | 38 | 49 | 8 |
| Somnolence, sedation, lethargy | 26 | 25 | 23 | 11 |
| Abnormal echocardiogram‡ | 18 | 23 | 9 | 6 |
| Diarrhea | 31 | 15 | 23 | 6 |
| Constipation | 3 | 10 | 7 | 0 |
| Fatigue, malaise, asthenia | 15 | 10 | 30 | 5 |
| Ataxia, balance disorder, gait disturbance | 10 | 10 | 7 | 1 |
| Abnormal behavior | 0 | 8 | 9 | 0 |
| Blood pressure increased | 13 | 8 | 0 | 5 |
| Drooling, salivary hypersecretion | 13 | 8 | 2 | 0 |
| Hypotonia | 0 | 8 | 0 | 0 |
| Rash | 8 | 8 | 5 | 4 |
| Blood prolactin increased | 0 | 5 | 0 | 0 |
| Chills | 0 | 5 | 2 | 0 |
| Decreased activity | 0 | 5 | 0 | 1 |
| Dehydration | 0 | 5 | 0 | 0 |
| Insomnia | 0 | 5 | 5 | 2 |
| Pyrexia | 15 | 5 | 21 | 14 |
| Stereotypy | 0 | 5 | 0 | 0 |
| Upper respiratory tract infection | 21 | 5 | 7 | 10 |
| Vomiting | 10 | 5 | 5 | 8 |
| Weight decreased | 13 | 5 | 7 | 1 |
| Croup | 5 | 3 | 0 | 1 |
| Ear infection | 8 | 3 | 9 | 5 |
| Gastroenteritis | 8 | 3 | 2 | 0 |
| Increased heart rate | 5 | 3 | 0 | 2 |
| Irritability | 0 | 3 | 9 | 2 |
| Rhinitis | 8 | 3 | 7 | 2 |
| Tremor | 3 | 3 | 9 | 0 |
| Urinary incontinence | 5 | 3 | 0 | 0 |
| Decreased blood glucose | 0 | 0 | 9 | 1 |
| Bronchitis | 3 | 0 | 9 | 1 |
| Contusion | 5 | 0 | 0 | 0 |
| Eczema | 0 | 0 | 5 | 0 |
| Enuresis | 5 | 0 | 0 | 0 |
| Fall | 10 | 0 | 0 | 4 |
| Headache | 8 | 0 | 0 | 2 |
| Laryngitis | 0 | 0 | 5 | 0 |
| Negativism | 5 | 0 | 0 | 0 |
| Status epilepticus | 3 | 0 | 12 | 2 |
| Urinary tract infection | 5 | 0 | 5 | 0 |
| Viral infection | 0 | 0 | 5 | 1 |
Lennox-Gastaut Syndrome
In the placebo-controlled trial of patients with LGS taking concomitant standard of care AEDs (Study 3), 176 patients were treated with FINTEPLA and 87 patients received placebo [see Clinical Studies (14.2)]. The duration of treatment in this trial was 16 weeks. The mean age was 13.7 years (range 2 to 35 years) and 29% of patients were at least 18 years of age, 45% of patients were female, and 79% were White. All patients were receiving at least one other AED.
The rates of discontinuation as a result of any adverse reaction were 6% and 5% for patients treated with FINTEPLA 0.7 mg/kg/day and 0.2 mg/kg/day, respectively, compared to 1% for patients on placebo. The most frequent adverse reactions leading to discontinuation in the patients treated with any dose of FINTEPLA were seizure (2%) and somnolence (2%).
The common adverse reactions that occurred in patients treated with FINTEPLA (incidence at least 10% and greater than placebo) were diarrhea; decreased appetite; fatigue; somnolence; vomiting.
Table 5 lists the adverse reactions that were reported in 5% or more of patients treated with FINTEPLA and at a rate greater than those on placebo during the titration and maintenance phases of Study 3.
| Adverse Reaction | FINTEPLA Dose Group | ||
|---|---|---|---|
| Study 3 | Placebo Group | ||
| 0.2 mg/kg/day | 0.7mg/kg/day | ||
| N=89 % | N=87 % | N=87 % |
|
| Decreased appetite | 20 | 36 | 12 |
| Fatigue, malaise, asthenia | 14 | 24 | 16 |
| Somnolence, sedation, lethargy | 12 | 22 | 16 |
| Diarrhea | 11 | 13 | 5 |
| Constipation | 6 | 9 | 6 |
| Vomiting | 14 | 8 | 6 |
| Weight decreased | 2 | 8 | 2 |
| Upper respiratory tract infection | 8 | 7 | 3 |
| Seizure | 9 | 5 | 7 |
| Irritability | 8 | 3 | 6 |
7. Drug Interactions
7.2 Effects of Serotonin Receptor Antagonists
Cyproheptadine and potent 5-HT1A, 5-HT1D, 5-HT2A, and 5-HT2C serotonin receptor antagonists may decrease the efficacy of FINTEPLA. If cyproheptadine or potent 5--HT1A, 5--HT1D, 5-HT2A, or 5-HT2C serotonin receptor antagonists are coadministered with FINTEPLA, patients should be monitored appropriately.
7.3 Serotonergic Drugs
Concomitant administration of FINTEPLA and drugs (e.g., SSRIs, SNRIs, TCAs, MAO inhibitors, trazodone, etc.), over-the-counter medications (e.g., dextromethorphan), or herbal supplements (e.g., St. John's Wort) that increase serotonin may increase the risk of serotonin syndrome [see Warnings and Precautions (5.7)]. Concomitant use of FINTEPLA is contraindicated within 14 days of taking MAOIs. Use FINTEPLA with caution in patients taking other medications that increase serotonin.
8. Use In Specific Populations
8.4 Pediatric Use
The safety and effectiveness of FINTEPLA for the treatment of seizures associated with DS and LGS have been established in patients 2 years of age and older.
Use of FINTEPLA for the treatment of seizures associated with DS in patients 2 years of age and older is supported by two randomized, double-blind, placebo-controlled trials in 202 patients 2 to 18 years of age. Use of FINTEPLA for the treatment of seizures associated with LGS is supported by a randomized, double-blind, placebo-controlled study in 263 patients aged 2 to 35 years, including 187 patients less than 18 years [see Boxed Warning, Warnings and Precautions (5), Adverse Reaction (6.1), and Clinical Studies (14)].
FINTEPLA can cause decreases in appetite and weight. The growth of pediatric patients treated with FINTEPLA should be carefully monitored.
Safety and effectiveness in patients less than 2 years of age have not been established.
8.5 Geriatric Use
Clinical studies of FINTEPLA for the treatment of DS or LGS did not include patients 65 years of age and over to determine whether they respond differently from younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
In patients with estimated glomerular filtration rate (eGFR) 15 to 29 mL/min/1.73m2, do not exceed the maximum daily dosage of FINTEPLA of 20 mg. In patients with eGFR 15 to 29 ml/min/1.73m2 and concomitant stiripentol use, do not exceed the maximum daily dosage of FINTEPLA of 17 mg [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)]. FINTEPLA has not been studied in patients with eGFR < 15 mL/min/1.73m2.
8.7 Hepatic Impairment
Combined molar exposures of fenfluramine and norfenfluramine were increased in subjects with various degrees of hepatic impairment (Child-Pugh Class A, B, and C), necessitating a dosage adjustment in these patients [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
10. Overdosage
Overdose has not been observed in the FINTEPLA clinical trial program. However, overdose of fenfluramine, the active ingredient in FINTEPLA, has been reported at higher doses than those included in the clinical trial program. Some of the cases were fatal. Events reported after overdose include mydriasis, tachycardia, flushing, tremors/twitching/muscle spasms, agitation/restlessness/anxiety, increased muscle tone/rigor/opisthotonos, respiratory distress or failure, and seizure. Seizure, coma, and cardiorespiratory arrest were reported in most of the fatal overdoses.
There is no available specific antidote to the overdose reactions of FINTEPLA. In the event of overdose, standard medical practice for the management of drug overdosage should be used. An adequate airway, oxygenation, and ventilation should be ensured; monitoring of cardiac rhythm and vital sign measurement is recommended. A certified poison control center should be contacted for updated information on the management of overdose with FINTEPLA.
11. Fintepla Description
FINTEPLA oral solution contains 2.2 mg/mL fenfluramine, equivalent to 2.5 mg/mL of the hydrochloride salt.
The active ingredient, fenfluramine hydrochloride, is designated chemically as N-ethyl-α- methyl-3-(trifluoromethyl)phenethylamine hydrochloride.
The structural formula is:
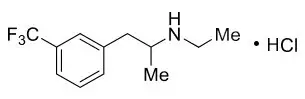
Fenfluramine hydrochloride is a white to off-white crystalline solid. The pKa of fenfluramine is 10.2.
FINTEPLA is a clear, colorless solution, pH 5.
FINTEPLA contains the following inactive ingredients: cherry flavor, citric acid, ethylparaben hydroxyethylcellulose, methylparaben, potassium citrate, sucralose, and water.
FINTEPLA contains no ingredient made from gluten-containing grain (wheat, barley, or rye), and contains not more than 0.1% of carbohydrates, which is solely derived from the cherry flavor.
12. Fintepla - Clinical Pharmacology
12.1 Mechanism of Action
The precise mechanism by which fenfluramine exerts its therapeutic effects in the treatment of seizures associated with Dravet syndrome and Lennox-Gastaut syndrome is unknown.
Fenfluramine and the metabolite, norfenfluramine, exhibit agonist activity at serotonin 5-HT2 receptors. There is an association between serotonergic drugs with 5-HT2B receptor agonist activity, including fenfluramine and norfenfluramine, and valvular heart disease and pulmonary arterial hypertension.
12.3 Pharmacokinetics
The pharmacokinetics of fenfluramine and norfenfluramine were studied in healthy subjects, in pediatric patients with DS, and in pediatric and adult patients with LGS. The steady-state systemic exposure (Cmax and AUC) of fenfluramine was slightly greater than dose proportional over the dose range of 13 to 51.8 mg twice-daily fenfluramine (i.e., 1 to 4 times the maximum recommended dose). In pediatric patients with DS who received FINTEPLA 0.7 mg/kg/day, up to a total daily dose of 26 mg fenfluramine, the geometric mean steady-state fenfluramine (coefficient of variation) Cmax was 68.0 (41%) ng/mL and AUC0-24h was 1390 (44%) ng*h/mL.
14. Clinical Studies
14.1 Dravet Syndrome
The effectiveness of FINTEPLA for the treatment of seizures associated with DS in patients 2 years of age and older was established in two randomized, double-blind, placebo-controlled trials in patients 2 to 18 years of age.
Study 1 (N=117) compared a 0.7 mg/kg/day and a 0.2 mg/kg/day dose of FINTEPLA with placebo in patients who were not receiving stiripentol (NCT02682927 and NCT02826863). Study 2 (N=85) compared a 0.4 mg/kg/day dose of FINTEPLA with placebo in patients who were receiving stiripentol and either clobazam, valproate, or both (NCT02926898). In both studies, patients had a clinical diagnosis of DS and were inadequately controlled on at least one AED or other antiseizure treatment including vagal nerve stimulation or a ketogenic diet. Both trials had a 6-week baseline period, during which patients were required to have a minimum of 6 convulsive seizures while on stable AED therapy. Convulsive seizures included tonic, clonic, generalized tonic-clonic, tonic-atonic, secondarily generalized tonic-clonic, hemiclonic, and focal with observable motor signs. The baseline period was followed by randomization into a 2-week (Study 1) or 3-week (Study 2) titration period and a subsequent 12-week maintenance period, where the dose of FINTEPLA remained stable.
In Study 1, 98% of patients were taking between 1 and 4 concomitant AEDs. The most frequently used concomitant AEDs (in at least 25% of patients), were valproate (61%), clobazam (59%), and topiramate (25%). In Study 2, 100% of patients were taking between 2 and 4 concomitant AEDs. The most frequently used concomitant AEDs (in at least 25% of patients), were stiripentol (100%), clobazam (94%), and valproate (89%).
The primary efficacy endpoint in both studies was the change from baseline in the frequency of convulsive seizures per 28 days during the combined 14-week (Study 1) or 15-week (Study 2) titration and maintenance periods (i.e., treatment period). The median longest interval between convulsive seizures was also assessed.
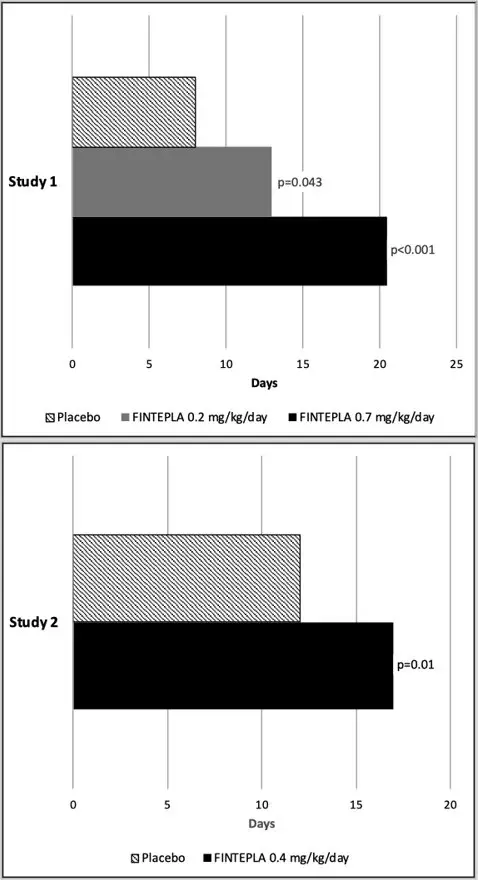
In Study 1 and Study 2, the reduction in convulsive seizure frequency per 28 days was statistically significantly greater for all dose groups of FINTEPLA compared to placebo (Table 6). A reduction in convulsive seizures was observed within 3 to 4 weeks of starting FINTEPLA, and the effect remained generally consistent over the 14- or 15-week treatment period.
| Convulsive Seizure Frequency (per 28 days) | Placebo | FINTEPLA 0.2 mg/kg/day | FINTEPLA 0.7 mg/kg/day | FINTEPLA 0.4 mg/kg/day |
|---|---|---|---|---|
| ±All 0.4 mg/kg/day patients were also taking concomitant stiripentol, which increases the exposure of FINTEPLA. | ||||
|
||||
| Study 1 | N=39 | N=38 | N=40 | NA |
| Baseline Period Median | 29.4 | 18.1 | 18.7 | NA |
| % Difference Relative to Placebo* | -31.7% | -70.0% | NA | |
| p-value compared to placebo | 0.043 | <0.001 | ||
| Study 2 | N=42 | NA | NA | N=43 |
| Baseline Period Median | 11.5 | NA | NA | 15.0 |
| % Difference Relative to Placebo* | NA | NA | -59.5% | |
| p-value compared to placebo | <0.001 | |||
Figure 1 and Figure 2 display the percentage of patients by category of seizure response from baseline in convulsive seizure frequency (per 28 days) during the treatment period in Study 1 and Study 2, respectively.
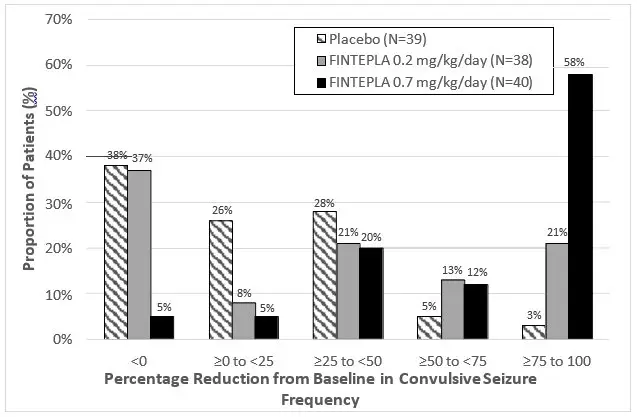
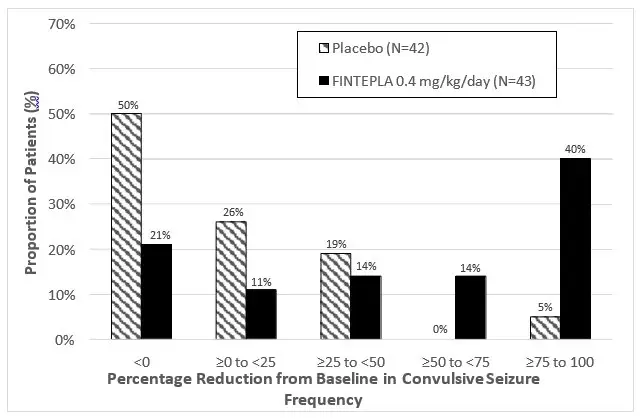
In Study 1, 3 of 40 (8%) patients in the FINTEPLA 0.7 mg/kg/day group and 3 of 38 (8%) patients in the FINTEPLA 0.2 mg/kg/day group reported no convulsive seizures during the 14-week treatment period, compared to 0 patients in the placebo group. In Study 2, 1 of 43 (2%) patients in the FINTEPLA 0.4 mg/kg/day group reported no convulsive seizures during the 15-week treatment period, compared to 0 patients in the placebo group.
In Study 1 and Study 2, FINTEPLA was associated with a statistically significant longer interval between convulsive seizures compared to placebo (Figure 3).
14.2 Lennox-Gastaut Syndrome
The effectiveness of FINTEPLA for the treatment of seizures associated with LGS in patients 2 years of age and older was established in a randomized, double-blind, placebo-controlled study in 263 patients 2 to 35 years of age (Study 3; NCT03355209).
Study 3 compared a 0.7 mg/kg/day and a 0.2 mg/kg/day dose of FINTEPLA with placebo. Patients had a diagnosis of LGS and were inadequately controlled on at least one AED, with or without vagal nerve stimulation and/or ketogenic diet. The study had a 4-week baseline period, during which patients were required to have a minimum of 8 drop seizures while on stable AED therapy. Drop seizures were generalized tonic-clonic, secondarily generalized tonic-clonic, tonic, atonic, or tonic-atonic seizures that were confirmed to result in drops. The baseline period was followed by randomization into a 2-week titration period and a subsequent 12-week maintenance period, where the dose of FINTEPLA remained stable.
In Study 3, 99% of patients were taking between 1 and 4 concomitant AEDs. The most frequently used concomitant AEDs (in at least 25% of patients) were clobazam (45%), lamotrigine (34%), and valproate (56%).
The primary efficacy endpoint in Study 3 was the median percent change from baseline in the frequency of drop seizures per 28 days during the combined 14-week titration and maintenance periods (i.e., treatment period). The proportion of patients who achieve improvement (minimally, much, or very much improved) in the Clinical Global Impression of Change (CGI-I) as assessed by Principal Investigator was a secondary endpoint.
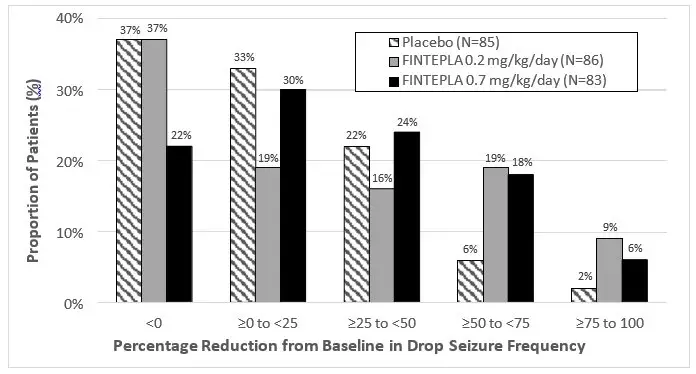
In Study 3, the median percent change from baseline (reduction) in the frequency of drop seizures per 28 days was significantly greater for the 0.7 mg/kg/day dose group of FINTEPLA compared with placebo (Table 7). A reduction in drop seizures was observed within 2 weeks of initiating treatment with FINTEPLA, and the effect remained generally consistent over the 14-week treatment period.
The median percent reduction from baseline in drop seizure frequency per 28 days for the lower dose of FINTEPLA (0.2 mg/kg/day) did not reach statistical significance compared to placebo (Table 7).
| Drop Seizure Frequency (per 28 days) | Placebo | FINTEPLA 0.2 mg/kg/day | FINTEPLA 0.7 mg/kg/day |
|---|---|---|---|
|
|||
| Study 3 | N=85* | N=86* | N=83* |
| Baseline Period Median Seizure Frequency | 55.0 | 77.8 | 80.0 |
| Median Percentage Change from Baseline During Treatment | -8.7% | -13.2% | -23.7% |
| p-value compared to placebo | 0.1917† | 0.0037 | |
Figure 4 displays the percentage of patients by category of reduction from baseline in drop seizure frequency per 28 days during the treatment period in Study 3.
Numerically greater improvements on the CGI-I by Investigator were observed in patients treated with FINTEPLA compared with placebo.
16. How is Fintepla supplied
16.1 How Supplied
FINTEPLA oral solution is a clear, colorless, cherry flavored liquid containing 2.2 mg/mL fenfluramine and is supplied in a white plastic bottle with a child resistant closure as follows:
- Carton containing one 360 mL bottle (NDC 43376-322-36)
- Carton containing one 30 mL bottle (NDC 43376-322-30)
Before dispensing, the pharmacist will insert a press-in bottle adapter into the dispensing bottle. The pharmacy will provide 3 mL or 6 mL calibrated oral dosing syringes.
16.2 Storage and Handling
Store FINTEPLA at room temperature between 20°C to 25°C (68°F to 77°F); excursions are permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Do not refrigerate or freeze. Store the bottle and syringe together.
Discard any unused portion 3 months after first opening the bottle or the "Discard After" date on the bottle, whichever is sooner.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
| MEDICATION GUIDE FINTEPLA® (fin-TEP-la) (fenfluramine) oral solution |
|||
|---|---|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 3/2023 | ||
| Read this Medication Guide before you start taking FINTEPLA and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. | |||
| What is the most important information I should know about FINTEPLA? FINTEPLA can cause serious side effects, including:
|
|||
|
| ||
| Because of the risk of heart valve problems (valvular heart disease) and high blood pressure in arteries of lungs (pulmonary arterial hypertension) FINTEPLA is only available through a restricted program called the FINTEPLA Risk Evaluation and Mitigation Strategy (REMS) Program. Before you or your child receives FINTEPLA, your healthcare provider or pharmacist will make sure you understand how to take FINTEPLA safely. If you have any questions about FINTEPLA, ask your healthcare provider, visit www.FinteplaREMS.com, or call 1-877-964-3649. | |||
|
|||
|
| ||
How can I watch for early symptoms of suicidal thoughts and actions?
|
|||
|
|||
What is FINTEPLA?
|
|||
Do not take FINTEPLA if you:
|
|||
Before taking FINTEPLA, tell your healthcare provider about all of your medical conditions, including if you:
Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine. |
|||
How should I take FINTEPLA?
|
|||
What should I avoid while taking FINTEPLA?
|
|||
| What are the possible side effects of FINTEPLA? FINTEPLA may cause serious side effects, including:
|
|||
|
| ||
| Call your healthcare provider right away if you have any of the following symptoms of serotonin syndrome. | |||
|
| ||
|
|||
|
| ||
| If you have any of these symptoms, call your healthcare provider right away. The most common side effects of FINTEPLA when used to treat Dravet syndrome (DS) include: |
|||
|
| ||
| The most common side effects of FINTEPLA when used to treat Lennox-Gastaut syndrome (LGS) include: | |||
|
| ||
| These are not all the possible side effects of FINTEPLA. For more information, ask your healthcare provider or pharmacist. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
How should I store FINTEPLA?
|
|||
| General information about the safe and effective use of FINTEPLA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FINTEPLA for a condition for which it was not prescribed. Do not give FINTEPLA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about FINTEPLA that is written for health professionals. |
|||
| What are the ingredients in FINTEPLA?
Active ingredient: fenfluramine hydrochloride Inactive ingredients: cherry flavor, citric acid, ethylparaben, hydroxyethylcellulose, methylparaben, potassium citrate, sucralose, and water. FINTEPLA contains no ingredient made from gluten-containing grain (wheat, barley, or rye) and contains not more than 0.1% of carbohydrates, which is from the cherry flavoring. Manufactured for: UCB, Inc., Smyrna, GA 30080 FINTEPLA® is a registered trademark of the UCB Group of Companies. ©2023. All rights reserved. For more information about FINTEPLA, go to www.fintepla.com or call 1-866-964-3649. |
|||
INSTRUCTIONS FOR USEFINTEPLA ® (fin-TEP-la)(fenfluramine) oral solution 2.2 mg/mL
Be sure that you read, understand, and follow these instructions before you start using FINTEPLA oral solution and each time you get a refill. There may be new information.
This Instructions for Use contains information on how to take FINTEPLA. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
What is included with FINTEPLA?
The following items are included to prepare and give an oral dose of FINTEPLA:
- 1 bottle of FINTEPLA oral solution (2.2 mg/mL)
- 2 reusable oral syringes
1 Bottle of FINTEPLA Oral Solution (2.2 mg/mL) 2 Oral Syringes 
Call the pharmacist at 1-844-288-5007 if you did not receive the items listed above, or if you need help using them.
Important information about FINTEPLA
- FINTEPLA is an oral medicine (taken by mouth) and is given 2 times each day. Follow your healthcare provider's instructions for taking or giving doses of FINTEPLA.
- If you have questions about how to prepare or give FINTEPLA, contact your healthcare provider or call your pharmacist.
- Always use the oral syringes provided with FINTEPLA to make sure the right dose is given. If you need a new syringe contact your pharmacist. Do not use a household teaspoon or tablespoon.
Oral syringes provided with FINTEPLA by the pharmacy.
With FINTEPLA you will receive 2 reusable oral syringes.
| 2 oral syringes that can measure up to 3 mL | ||
| OR | ||
| 2 oral syringes that can measure up to 6 mL | ||
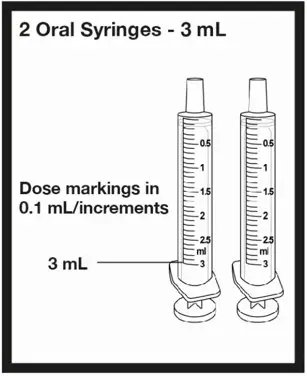 | or | 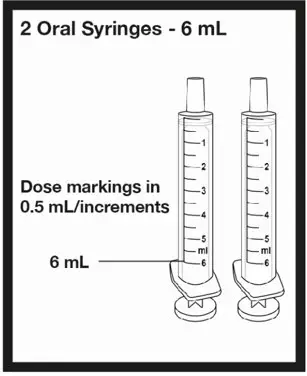 |
Call the pharmacist at 1-844-288-5007 if you have any questions about the syringes provided with FINTEPLA.
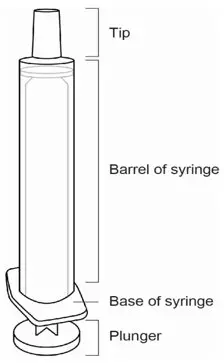
| Parts of the Oral Syringe |
|
| Preparing a Dose | |
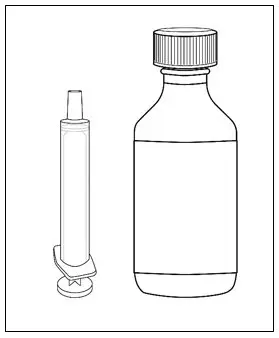 | Step 1. Make sure you have:
|
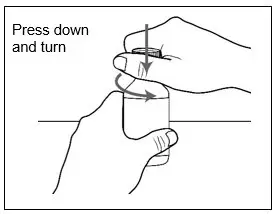 | Step 3. Press down and turn the childproof cap to the left (counterclockwise) and remove it from the bottle.
|
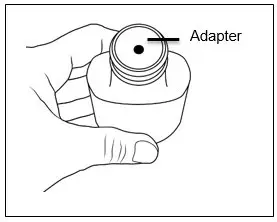 | Step 4. Make sure the adapter is on the bottle.
|
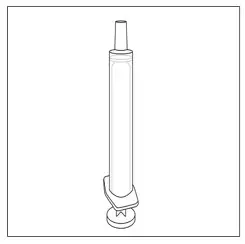 | Step 5. Remove an oral syringe from its packaging, if needed. Only use the oral syringes provided with FINTEPLA. If an oral syringe is damaged, or you cannot read the dose markings:
|
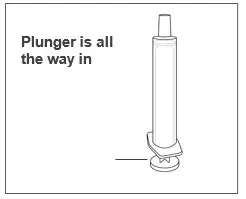 | Step 6. Make sure the plunger is pushed all the way into the oral syringe. |
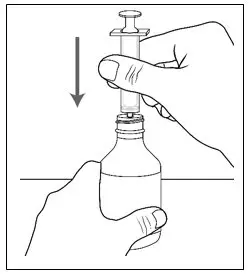 | Step 7. Hold the bottle of medicine firmly on a hard, flat surface. Step 8. Push the tip of the oral syringe into the opening of the adapter until it cannot be pushed further. |
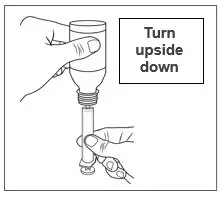 | Step 9. Hold the oral syringe and bottle together and turn upside down. |
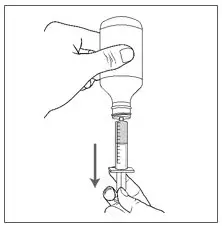 | Step 10. Slowly pull the plunger of the oral syringe to withdraw the prescribed dose. |
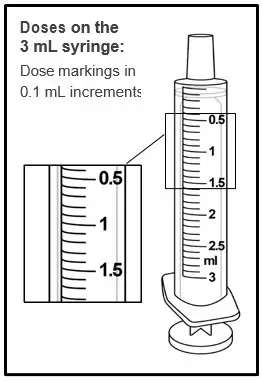 | 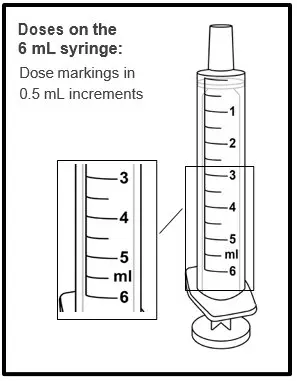 |
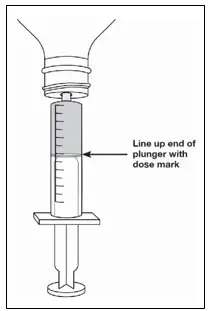 | Step 11. Line up the end of the plunger with the mark for the prescribed dose on the oral syringe. Tips to Getting the Correct Dose
|
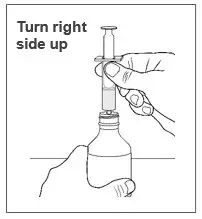 | Step 12. Hold the oral syringe and bottle together and then turn the bottle right side up. |
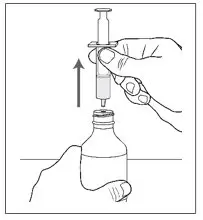 | Step 13. Holding the bottle firmly, gently pull the oral syringe out of the bottle adapter. |
| Step 14. Make sure the dose in the oral syringe still matches the prescribed dose. If the dose does not match:
|
|
| Giving FINTEPLA | |
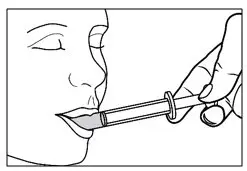 | Step 15. Place the tip of the oral syringe against the inside of the cheek. Step 16. Gently push the plunger in until all the medicine in the oral syringe is given.
|
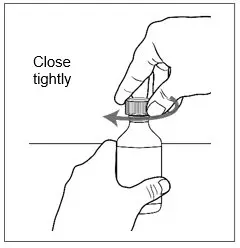 | Step 17. Place the cap back on the bottle tightly by turning the cap right (clockwise) until it stops.
|
| Cleaning the syringe | |
 |
|
How should I store FINTEPLA?
- Store FINTEPLA at room temperature between 68°F to 77°F (20°C to 25°C).
- Do not refrigerate or freeze.
- Keep the cap tightly closed and the bottle upright.
- Store the FINTEPLA bottle and syringe together in a clean area.
- Throw away (discard) any unused FINTEPLA 3 months after first opening the bottle or if the Discard After date on the package or bottle has passed. Whichever one comes first.
- Keep FINTEPLA and all medicines out of the reach of children.
Manufactured for: UCB, Inc., Smyrna, GA 30080
FINTEPLA® is a registered trademark of the UCB Group of Companies. ©2023. All rights reserved.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 3/2023



| FINTEPLA
fenfluramine solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - UCB, Inc. (028526403) |