Drug Detail:Fotivda (Tivozanib)
Drug Class: VEGF/VEGFR inhibitors
Highlights of Prescribing Information
FOTIVDA® (tivozanib) capsules, for oral use
Initial U.S. Approval: 2021
Indications and Usage for Fotivda
FOTIVDA is a kinase inhibitor indicated for the treatment of adult patients with relapsed or refractory advanced renal cell carcinoma (RCC) following two or more prior systemic therapies. (1)
Fotivda Dosage and Administration
- Recommended Dose: 1.34 mg once daily with or without food for 21 days on treatment followed by 7 days off treatment (28-day cycle) until disease progression or unacceptable toxicity. (2.1)
- Dose interruptions and/or dose reduction may be needed to manage adverse reactions. (2.2)
- For patients with moderate hepatic impairment, reduce the dose to 0.89 mg for 21 days on treatment followed by 7 days off treatment (28-day cycle). (2.3)
Dosage Forms and Strengths
Capsules: 1.34 mg and 0.89 mg (3)
Contraindications
None. (4)
Warnings and Precautions
- Hypertension and Hypertensive Crisis: Control blood pressure prior to initiating FOTIVDA. Monitor for hypertension and treat as needed. For persistent hypertension despite use of anti-hypertensive medications, reduce the FOTIVDA dose. (5.1)
- Cardiac Failure: Monitor for signs or symptoms of cardiac failure throughout treatment with FOTIVDA. (5.2)
- Cardiac Ischemia and Arterial Thromboembolic Events: Closely monitor patients who are at increased risk for these events. Permanently discontinue FOTIVDA for severe arterial thromboembolic events, such as myocardial infarction and stroke. (5.3)
- Venous Thromboembolic Events: Closely monitor patients who are at increased risk for these events. Permanently discontinue FOTIVDA for severe venous thromboembolic events. (5.4)
- Hemorrhagic Events: Closely monitor patients who are at risk for or who have a history of bleeding. (5.5)
- Proteinuria: Monitor throughout treatment with FOTIVDA. For moderate to severe proteinuria, reduce the dose or temporarily interrupt treatment with FOTIVDA. (5.6)
- Thyroid Dysfunction: Monitor before initiation and throughout treatment with FOTIVDA. (5.7)
- Risk of Impaired Wound Healing: Withhold FOTIVDA for at least 24 days before elective surgery. Do not administer for at least 2 weeks following major surgery and adequate wound healing. The safety of resumption of FOTIVDA after resolution of wound healing complications has not been established. (5.8)
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS): Discontinue FOTIVDA if signs or symptoms of RPLS occur. (5.9)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.10, 8.1, 8.3)
- Allergic Reactions to Tartrazine: The 0.89 mg capsule of FOTIVDA contains FD&C Yellow No.5 (tartrazine) which may cause allergic-type reactions (including bronchial asthma) in certain susceptible patients. (5.11)
Adverse Reactions/Side Effects
The most common (≥20%) adverse reactions were fatigue, hypertension, diarrhea, decreased appetite, nausea, dysphonia, hypothyroidism, cough, and stomatitis, and the most common Grade 3 or 4 laboratory abnormalities (≥5%) were sodium decreased, lipase increased, and phosphate decreased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AVEO Pharmaceuticals, Inc. at 1-833-FOTIVDA (1-833-368-4832) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
CYP3A Inducers: Avoid concomitant use of strong CYP3A inducers. (7.1)
Use In Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
- Females and Males of Reproductive Potential: Can impair fertility. (8.3)
- Hepatic Impairment: Adjust dosage in patients with moderate hepatic impairment. Avoid use in patients with severe hepatic impairment. (2.3, 8.7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2021
Full Prescribing Information
1. Indications and Usage for Fotivda
FOTIVDA is indicated for the treatment of adult patients with relapsed or refractory advanced renal cell carcinoma (RCC) following two or more prior systemic therapies.
2. Fotivda Dosage and Administration
2.1 Recommended Dosing
The recommended dosage of FOTIVDA is 1.34 mg taken orally once daily for 21 days on treatment followed by 7 days off treatment for a 28-day cycle.
Continue treatment until disease progression or until unacceptable toxicity occurs.
Take FOTIVDA with or without food. Swallow the FOTIVDA capsule whole with a glass of water. Do not open the capsule.
If a dose is missed, the next dose should be taken at the next scheduled time. Do not take two doses at the same time.
2.2 Dose Modifications for Adverse Reactions
Initiate medical management for diarrhea, nausea, or vomiting prior to dose interruption or reduction.
If dose modifications are required for adverse reactions, reduce the dosage of FOTIVDA to 0.89 mg for 21 days on treatment followed by 7 days off treatment for a 28-day cycle.
Recommendations for dosage modifications are provided in Table 1.
| Adverse Reaction | Severity* | Dosage Modifications for FOTIVDA |
|---|---|---|
|
||
| Hypertension [see Warnings and Precautions (5.1)] | Grade 3 |
|
| Grade 4 |
|
|
| Cardiac Failure [see Warnings and Precautions (5.2)] | Grade 3 |
|
| Grade 4 |
|
|
| Arterial Thromboembolic Events [see Warnings and Precautions (5.3)] | Any Grade |
|
| Hemorrhagic Events [see Warnings and Precautions (5.5)] | Grade 3 or 4 |
|
| Proteinuria [see Warnings and Precautions (5.6)] | 2 grams or greater proteinuria in 24 hours |
|
| Reverse Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.9)] | Any Grade |
|
| Other Adverse Reactions | Persistent or intolerable Grade 2 or 3 adverse reaction Grade 4 laboratory abnormality |
|
| Grade 4 adverse reaction |
|
|
2.3 Dosage Modifications for Moderate Hepatic Impairment
Reduce the recommended dosage of FOTIVDA to 0.89 mg capsule taken orally once daily for 21 days on treatment followed by 7 days off treatment for a 28-day cycle for patients with moderate hepatic impairment [see Use in Specific Populations (8.7)].
3. Dosage Forms and Strengths
Capsules:
- 1.34 mg: bright yellow opaque cap imprinted with "TIVZ" in dark blue ink and a bright yellow opaque body imprinted with "SD" in dark blue ink.
- 0.89 mg: dark blue opaque cap imprinted with "TIVZ" in yellow ink and a bright yellow opaque body imprinted with "LD" in dark blue ink.
5. Warnings and Precautions
5.1 Hypertension and Hypertensive Crisis
FOTIVDA can cause severe hypertension and hypertensive crisis. Hypertension occurred in 45% of patients treated with FOTIVDA, with 22% of the events ≥ Grade 3. Median time to onset of hypertension was 2 weeks (range: 0 – 192 weeks).
Hypertensive crisis occurred in 0.8% of patients. One patient (0.1%) died due to hypertensive emergency after FOTIVDA overdose [see OVERDOSAGE (10)].
FOTIVDA has not been studied in patients with systolic blood pressure > 150 mmHg or diastolic blood pressure > 100 mmHg.
Control blood pressure prior to treatment with FOTIVDA. Monitor blood pressure after 2 weeks and at least monthly thereafter during treatment with FOTIVDA. Treat patients with anti-hypertensive therapy when hypertension occurs during treatment with FOTIVDA.
Withhold FOTIVDA for severe hypertension despite optimal anti-hypertensive therapy. For persistent hypertension despite use of anti-hypertensive medications, reduce the FOTIVDA dose [see DOSAGE AND ADMINISTRATION (2.2)].
Discontinue FOTIVDA if hypertension is severe and persistent despite anti-hypertensive therapy and dose reduction of FOTIVDA, or in patients who experience hypertensive crisis.
If FOTIVDA is interrupted, monitor patients receiving anti-hypertensive medications for hypotension.
5.2 Cardiac Failure
FOTIVDA can cause serious, sometimes fatal, cardiac failure. Cardiac failure in FOTIVDA-treated patients occurred in 1.6%, with 1% of events ≥ Grade 3, and 0.6% events were fatal.
FOTIVDA has not been studied in patients with symptomatic cardiac failure within the preceding 6 months before FOTIVDA treatment initiation.
Periodically monitor patients for symptoms of cardiac failure throughout treatment with FOTIVDA.
Management of cardiac failure events may require interruption, dose reduction, or permanent discontinuation of FOTIVDA therapy [see DOSAGE AND ADMINISTRATION (2.2)].
5.3 Cardiac Ischemia and Arterial Thromboembolic Events
FOTIVDA can cause serious, sometimes fatal, cardiac ischemia and arterial thromboembolic events. Cardiac ischemia in FOTIVDA-treated patients occurred in 3.2%, with 1.5% of events ≥ Grade 3, and 0.4% events were fatal. Arterial thromboembolic events were reported in 2% of FOTIVDA-treated patients, including death due to ischemic stroke (0.1%).
FOTIVDA has not been studied in patients who had an arterial thrombotic event, myocardial infarction, or unstable angina within the preceding 6 months before FOTIVDA treatment initiation.
Closely monitor patients who are at risk for, or who have a history of these events (such as myocardial infarction and stroke), during treatment with FOTIVDA.
Discontinue FOTIVDA in patients who develop any severe or life-threatening arterial thromboembolic event [see DOSAGE AND ADMINISTRATION (2.2)].
5.4 Venous Thromboembolic Events
FOTIVDA can cause serious, sometimes fatal, venous thromboembolic events. Venous thromboembolic events occurred in 2.4% of patients treated with FOTIVDA, including death (0.3%).
Closely monitor patients who are at risk for, or who have a history of these events during treatment with FOTIVDA.
Discontinue FOTIVDA in patients who develop any severe or life-threatening venous thromboembolic event [see DOSAGE AND ADMINISTRATION (2.2)].
5.5 Hemorrhagic Events
FOTIVDA can cause serious, sometimes fatal, hemorrhagic events. Hemorrhagic events occurred in 11% of patients treated with FOTIVDA, including death (0.2%).
FOTIVDA has not been studied in patients with significant bleeding within the preceding 6 months before FOTIVDA treatment initiation.
Closely monitor patients who are at risk for or who have a history of bleeding during treatment with FOTIVDA.
Discontinue FOTIVDA in patients who develop severe or life-threatening hemorrhagic events [see DOSAGE AND ADMINISTRATION (2.2)].
5.6 Proteinuria
FOTIVDA can cause proteinuria. Proteinuria occurred in 8% of FOTIVDA-treated patients with 2% of events Grade 3. Of the patients who developed proteinuria, 3/81 (3.7%) had acute kidney injury either concurrently or later during treatment.
Monitor patients for proteinuria before initiation of, and periodically throughout, treatment with FOTIVDA.
For patients who develop moderate to severe proteinuria, reduce the dose or interrupt FOTIVDA treatment.
Discontinue FOTIVDA in patients who develop nephrotic syndrome [see DOSAGE AND ADMINISTRATION (2.2)].
5.7 Thyroid Dysfunction
FOTIVDA can cause thyroid dysfunction. Thyroid dysfunction events in FOTIVDA-treated patients occurred in 11%, with 0.3% Grade 3 or 4 events. Hypothyroidism was reported in 8% of patients and hyperthyroidism was reported in 1% of patients.
Monitor thyroid function before initiation of, and periodically throughout, treatment with FOTIVDA.
Treat hypothyroidism and hyperthyroidism to maintain euthyroid state before and during treatment with FOTIVDA.
5.8 Risk of Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway, such as FOTIVDA. Therefore, FOTIVDA has the potential to adversely affect wound healing.
Withhold FOTIVDA for at least 24 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of FOTIVDA after resolution of wound healing complications has not been established.
5.9 Reversible Posterior Leukoencephalopathy Syndrome
Reversible posterior leukoencephalopathy syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by MRI, can occur with FOTIVDA. Perform an evaluation for RPLS in any patient presenting with seizures, headaches, visual disturbances, confusion, or altered mental function.
Discontinue FOTIVDA in patients who develop RPLS [see DOSAGE AND ADMINISTRATION (2.2)].
5.10 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, FOTIVDA can cause fetal harm when administered to a pregnant woman. In embryo-fetal developmental studies, oral administration of tivozanib to pregnant animals during the period of organogenesis caused maternal toxicity, fetal malformations and embryo-fetal death at doses below the maximum recommended clinical dose on a mg/m2 basis.
Advise pregnant woman of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with FOTIVDA and for one month after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with FOTIVDA and for one month after the last dose [see USE IN SPECIFIC POPULATIONS (8.1, 8.3) and CLINICAL PHARMACOLOGY (12.1)].
5.11 Allergic Reactions to Tartrazine (FD&C Yellow No.5)
FOTIVDA 0.89 mg capsule contains FD&C Yellow No.5 (tartrazine) as an imprint ink which may cause allergic-type reactions (including bronchial asthma) in certain susceptible patients. Although the overall incidence of FD&C Yellow No.5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin hypersensitivity.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are also described elsewhere in the labeling:
- Hypertension and Hypertensive Crisis [see WARNINGS AND PRECAUTIONS (5.1)]
- Cardiac Failure [see WARNINGS AND PRECAUTIONS (5.2)]
- Cardiac Ischemia and Arterial Thromboembolic Events [see WARNINGS AND PRECAUTIONS (5.3)]
- Venous Thromboembolic Events [see WARNINGS AND PRECAUTIONS (5.4)]
- Hemorrhagic Events [see WARNINGS AND PRECAUTIONS (5.5)]
- Proteinuria [see WARNINGS AND PRECAUTIONS (5.6)]
- Thyroid Dysfunction [see WARNINGS AND PRECAUTIONS (5.7)]
- Risk of Impaired Wound Healing [see WARNINGS AND PRECAUTIONS (5.8)]
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS) [see WARNINGS AND PRECAUTIONS (5.9)]
6.1 Clinical Trial Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The pooled safety population described in WARNINGS AND PRECAUTIONS reflect exposure to FOTIVDA administered at 1.34 mg orally once daily with or without food for 21 days on treatment followed by 7 days off treatment for a 28-day cycle in 1008 patients with advanced RCC in TIVO-3 and five other monotherapy studies. Among 1008 patients who received FOTIVDA, 52% were exposed for 6 months or longer and 34% were exposed for greater than one year.
8. Use In Specific Populations
8.3 Females and Males of Reproductive Potential
FOTIVDA can cause fetal harm when administered to a pregnant woman [see USE IN SPECIFIC POPULATIONS (8.1)].
8.4 Pediatric Use
The safety and effectiveness of FOTIVDA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 1008 patients with advanced RCC treated with FOTIVDA, 29% were ≥ 65 years of age and 4% were ≥ 75 of age. No overall differences in safety were observed between patients ≥ 65 versus < 65 years of age.
Of the 175 patients with advanced RCC following two or more prior systemic therapies randomized to FOTIVDA, 44% were ≥ 65 years of age and 9% were ≥ 75 of age. No overall differences in effectiveness were observed between patients ≥ 65 versus < 65 years of age.
8.6 Renal Impairment
No dosage modification is recommended for patients with mild to severe renal impairment (creatinine clearance [CLcr] 15-89 mL/min, estimated by Cockcroft-Gault). The recommended dosage for patients with end-stage renal disease has not been established [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Reduce the dosage when administering FOTIVDA in patients with moderate (total bilirubin greater than 1.5 to 3 times ULN with any AST) hepatic impairment [see Dosage and Administration (2.3)]. No dosage modification is recommended for patients with mild (total bilirubin less than or equal to ULN with AST greater than ULN or total bilirubin greater than 1 to 1.5 times ULN with any AST) hepatic impairment. The recommended dosage of FOTIVDA in patients with severe (total bilirubin greater than 3 to 10 times ULN with any AST) hepatic impairment has not been established [see Clinical Pharmacology (12.3)].
10. Overdosage
Overdosage with FOTIVDA can cause severe hypertension and hypertensive crisis that may result in death [see WARNINGS AND PRECAUTIONS (5.1)].
During clinical studies, three patients inadvertently received doses ≥ 2.68 mg (≥ 2 times the recommended dose) of FOTIVDA. One patient who received two daily doses of 8.9 mg of FOTIVDA experienced hypertensive crisis with severe hypertensive retinopathy; a second patient who received three doses of 1.34 mg in one day experienced fatal uncontrolled hypertension; and a third patient who received two doses of 1.34 mg FOTIVDA in one day experienced persistent hypertension lasting over 5 days.
There is no specific treatment or antidote for FOTIVDA overdose.
In cases of suspected overdose, withhold FOTIVDA, closely monitor patients for hypertension and hypertensive crisis and other potential adverse reactions. Immediately manage signs or symptoms of hypertension and provide other supportive care as clinically indicated.
11. Fotivda Description
Tivozanib is a kinase inhibitor. Tivozanib hydrochloride, the active ingredient, has the chemical name 1-{2-chloro-4-[(6,7-dimethoxyquinolin-4-yl)oxy]phenyl}-3-(5-methylisoxazol-3-yl)urea hydrochloride hydrate. The molecular formula is C22H19ClN4O5 ∙ HCl ∙ H2O and the molecular weight is 509.34 Daltons. The chemical structure is:
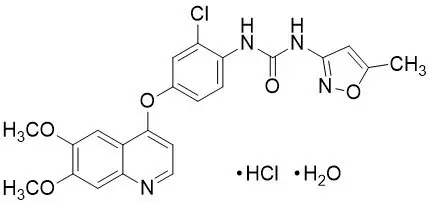
Tivozanib hydrochloride is a white to light brown crystalline powder that is practically insoluble in water (0.09 mg/mL).
FOTIVDA 1.34 mg capsule contains 1.5 mg of tivozanib hydrochloride (equivalent to 1.34 mg tivozanib) with inactive ingredients: mannitol and magnesium stearate. Capsule composition: gelatin, titanium dioxide, FDA yellow iron oxide, and Blue SB-6018 (ink).
FOTIVDA 0.89 mg capsule contains 1.0 mg of tivozanib hydrochloride (equivalent to 0.89 mg tivozanib) with inactive ingredients: mannitol and magnesium stearate. Capsule composition: gelatin, titanium dioxide, FDA yellow iron oxide, FD&C Blue #2, Blue SB-6018 (ink) and Yellow SB-3017 (ink). The Yellow SB-3017 ink contains FD&C Yellow No.5 (tartrazine).
12. Fotivda - Clinical Pharmacology
12.1 Mechanism of Action
Tivozanib is a tyrosine kinase inhibitor. In vitro cellular kinase assays demonstrated that tivozanib inhibits phosphorylation of vascular endothelial growth factor receptor (VEGFR)-1, VEGFR-2 and VEGFR-3 and inhibits other kinases including c-kit and PDGFR β at clinically relevant concentrations. In tumor xenograft models in mice and rats, tivozanib inhibited angiogenesis, vascular permeability, and tumor growth of various tumor cell types including human renal cell carcinoma.
12.3 Pharmacokinetics
The pharmacokinetics of tivozanib were evaluated in patients with solid tumors administered 1.34 mg once daily unless otherwise specified. Steady-state tivozanib AUC and Cmax increased in a dose-proportional manner over the dose range of 0.89 to 1.78 mg once daily (0.67 to 1.3 times the recommended dose).
Steady-state was reached by 14 days and the accumulation ratio after administration of 1.34 mg once daily was approximately 6- to 7- fold. Mean steady-state tivozanib [coefficient of variation (CV%)] Cmax was 86.9 (44.7%) ng/mL and AUC0-24h was 1510 (46.1%) ng*h/mL.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with tivozanib.
Tivozanib was not mutagenic in a bacterial reverse mutation (Ames) assay and was not clastogenic in an in vitro cytogenetic assay in Chinese hamster ovary cells or in an in vivo mouse bone marrow micronucleus assay.
In animal studies assessing mating and fertility parameters, oral doses ≥ 0.03 mg/kg/day (0.2 times the maximum recommended clinical dose on a mg/m2 basis) in rats were associated with increased epididymis and testis weights, and doses ≥ 0.3 mg/kg/day (2 times the maximum recommended clinical dose on a mg/m2 basis) reduced mating and produced infertility. An increase in embryo lethality was noted at doses ≥ 0.1 mg/kg/day (0.7 times the maximum recommended clinical dose on a mg/m2 basis).
14. Clinical Studies
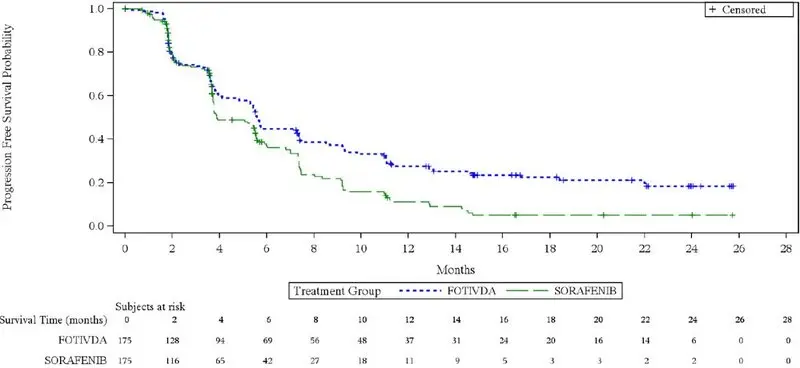
The efficacy of FOTIVDA was evaluated in TIVO-3 (NCT02627963), a randomized (1:1), open-label, multicenter trial of FOTIVDA versus sorafenib in patients with relapsed or refractory advanced RCC who received 2 or 3 prior systemic treatments including at least one VEGFR kinase inhibitor other than sorafenib or tivozanib. Patients were randomized to receive FOTIVDA 1.34 mg orally once daily for 21 days on treatment followed by 7 days off treatment for a 28-day cycle, or to receive sorafenib 400 mg orally twice a day continuously, until disease progression or unacceptable toxicity. Randomization was stratified by prior therapy [two kinase inhibitors (KIs), a KI plus an immune checkpoint inhibitor, or a KI plus other systemic agents] and by International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) prognostic score. Patients were excluded if they had more than 3 prior treatments or Central Nervous System metastases. The main efficacy outcome measure was progression-free survival (PFS) assessed by a blinded independent radiology review committee. Other efficacy endpoints were objective response rate (ORR) and overall survival (OS).
The median age was 63 years (range: 30 to 90 years), 73% were male, 95% were Caucasian, ECOG performance status was 0 in 48% and 1 in 49% of patients (respectively), and 98% of patients had clear cell or clear cell component histology. Prior therapy included two KIs (45%), a KI plus an immune checkpoint inhibitor (26%), and a KI plus another systemic agent (29%). At the time of study entry, 20% of patients had favorable, 61% intermediate, and 19% poor IMDC prognoses.
Efficacy results are summarized in Table 4 and Figure 1.
| Endpoint | FOTIVDA N= 175 | Sorafenib N= 175 |
|---|---|---|
| CI: Confidence interval; HR: Hazard ratio (FOTIVDA/sorafenib); NE: not estimable. | ||
|
||
| Progression Free Survival (PFS)* | ||
| Events, n (%) | 123 (70) | 123 (70) |
| Progressive Disease | 103 (59) | 109 (62) |
| Death | 20 (11) | 14 (8) |
| Median (95% CI), months | 5.6 (4.8, 7.3) | 3.9 (3.7, 5.6) |
| HR (95% CI) † | 0.73 (0.56, 0.95) | |
| P-value‡ | 0.016 | |
| Overall Survival | ||
| Deaths, n (%) | 125 (71) | 126 (72) |
| Median (95% CI), months | 16.4 (13.4, 21.9) | 19.2 (14.9, 24.2) |
| HR (95% CI) † | 0.97 (0.75, 1.24) | |
| Objective Response Rate % (95% CI)* | 18 (12, 24) | 8 (4, 13) |
| Median duration of response in months (95% CI) | NE (9.8, NE) | 5.7 (5.6, NE) |
Figure 1. Kaplan-Meier Plot of PFS in TIVO-3
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (PATIENT INFORMATION).
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Issued: 03/2021 | |||
| PATIENT INFORMATION
FOTIVDA® (fo-TIV-dah) (tivozanib) capsules |
||||
| What is FOTIVDA?
FOTIVDA is a prescription medicine used to treat adults with advanced kidney cancer (advanced renal cell carcinoma or RCC) that has been treated with 2 or more prior medicines and has come back or did not respond to treatment. It is not known if FOTIVDA is safe and effective in children. |
||||
Before taking FOTIVDA, tell your healthcare provider about all your medical conditions, including if you:
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
||||
How should I take FOTIVDA?
|
||||
| What are the possible side effects of FOTIVDA? FOTIVDA may cause serious side effects, including:
|
||||
|
|
| ||
|
||||
|
|
|||
|
||||
|
|
|||
|
||||
|
||||
|
|
| ||
|
||||
| The most common side effects of FOTIVDA include: | ||||
|
|
|||
| Other side effects include vomiting and weakness or lack of energy. FOTIVDA may cause fertility problems in males and females, which may affect your ability to have a child. Talk to your healthcare provider if this is a concern for you. Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with FOTIVDA if you have certain side effects. These are not all of the possible side effects of FOTIVDA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||
How should I store FOTIVDA?
|
||||
| General information about the safe and effective use of FOTIVDA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use FOTIVDA for a condition for which it was not prescribed. Do not give FOTIVDA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about FOTIVDA that is written for health professionals. |
||||
| What are the ingredients in FOTIVDA?
Active ingredient: tivozanib hydrochloride Inactive ingredients: mannitol and magnesium stearate. The capsule contains gelatin, titanium dioxide, FDA yellow iron oxide, and Blue SB-6018 (ink). The 0.89 mg capsule also contains FD&C Blue #2 and Yellow SB-3017 (ink). The Yellow SB-3017 ink contains FD&C Yellow No.5 (tartrazine). Manufactured for: AVEO Pharmaceuticals, Inc. Boston, MA 02108 Manufactured by: Catalent CTS, Inc. Kansas City, MO 64137 For more information go to www.fotivda.com or call 1-833-FOTIVDA (1-833-368-4832). |
||||


| FOTIVDA
tivozanib capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FOTIVDA
tivozanib capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - AVEO Pharmaceuticals, Inc. (111045360) |
