Drug Detail:Ilaris (Canakinumab [ kan-a-kin-ue-mab ])
Drug Class: Interleukin inhibitors
Highlights of Prescribing Information
ILARIS® (canakinumab) injection, for subcutaneous use
Initial U.S. Approval: 2009
Recent Major Changes
| Indications and Usage, Still’s disease (AOSD and SJIA) (1.2) |
6/2020 |
| Dosage and Administration, Still’s disease (AOSD and SJIA) (2.4) |
6/2020 |
| Warnings and Precautions, Hypersensitivity (5.3) |
6/2020 |
| Warnings and Precautions, Macrophage Activation Syndrome (5.5) |
6/2020 |
Indications and Usage for Ilaris
ILARIS is an interleukin-1β blocker indicated for the treatment of:
Periodic Fever Syndromes:
-
Cryopyrin-Associated Periodic Syndromes (CAPS), in adults and children 4 years of age and older, including:
- Familial Cold Auto-inflammatory Syndrome (FCAS). (1.1)
- Muckle-Wells Syndrome (MWS). (1.1)
-
Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS) in adult and pediatric patients. (1.1)
-
Hyperimmunoglobulin D Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD) in adult and pediatric patients. (1.1)
- Familial Mediterranean Fever (FMF) in adult and pediatric patients. (1.1)
- Active Still’s disease, including Adult-Onset Still’s Disease (AOSD) and Systemic Juvenile Idiopathic Arthritis (SJIA) in patients aged 2 years and older. (1.2)
Ilaris Dosage and Administration
- Administer by subcutaneous injection. (2.1)
CAPS:
150 mg for CAPS patients with body weight greater than 40 kg and 2 mg/kg for CAPS patients with body weight greater than or equal to 15 kg and less than or equal to 40 kg. For children 15 to 40 kg with an inadequate response, the dose can be increased to 3 mg/kg. Administer subcutaneously every 8 weeks. (2.2)
TRAPS, HIDS/MKD, and FMF:
-
Body weight less than or equal to 40 kg
- The recommended starting dose is 2 mg/kg every 4 weeks. The dose can be increased to 4 mg/kg every 4 weeks if the clinical response is not adequate. (2.3)
-
Body weight greater than 40 kg
- The recommended starting dose is 150 mg every 4 weeks. The dose can be increased to 300 mg every 4 weeks if the clinical response is not adequate. (2.3)
Still’s disease (AOSD and SJIA):
4 mg/kg (with a maximum of 300 mg) for patients with a body weight greater than or equal to 7.5 kg. Administer subcutaneously every 4 weeks. (2.4)
Dosage Forms and Strengths
- Injection: 150 mg/mL solution in single-dose vials. (3)
Contraindications
Confirmed hypersensitivity to the active substance or to any of the excipients. (4)
Warnings and Precautions
- Interleukin-1 blockade may interfere with immune response to infections. Treatment with medications that work through inhibition of IL-1 has been associated with an increased risk of serious infections. ILARIS has been associated with an increased incidence of serious infections. Physicians should exercise caution when administering ILARIS to patients with infections, a history of recurring infections or underlying conditions which may predispose them to infections. Discontinue treatment with ILARIS if a patient develops a serious infection. Do not administer ILARIS to patients during an active infection requiring medical intervention. (5.1)
- Live vaccines should not be given concurrently with ILARIS. Prior to initiation of therapy with ILARIS, patients should receive all recommended vaccinations. (5.4)
Adverse Reactions/Side Effects
CAPS: The most common adverse reactions greater than 10% reported by patients treated with ILARIS are nasopharyngitis, diarrhea, influenza, rhinitis, nausea, headache, bronchitis, gastroenteritis, pharyngitis, weight increased, musculoskeletal pain, and vertigo. (6)
TRAPS, HIDS/MKD, and FMF: The most common adverse reactions greater than 10% reported by patients treated with ILARIS are injection-site reactions and nasopharyngitis. (6)
Still’s disease: The most common adverse drug reactions greater than or equal to 10% reported by patients treated with ILARIS are infections (nasopharyngitis and upper respiratory tract infections), abdominal pain, and injection-site reactions. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
No formal drug interaction studies have been conducted with ILARIS. (7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2020
Full Prescribing Information
1. Indications and Usage for Ilaris
1.1 Periodic Fever Syndromes
ILARIS® (canakinumab) is an interleukin-1β (IL-1β) blocker indicated for the treatment of the following autoinflammatory Periodic Fever Syndromes:
Cryopyrin-Associated Periodic Syndromes (CAPS)
ILARIS is indicated for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), in adults and children 4 years of age and older, including:
- Familial Cold Autoinflammatory Syndrome (FCAS)
- Muckle-Wells Syndrome (MWS)
Tumor Necrosis Factor Receptor (TNF) Associated Periodic Syndrome (TRAPS)
ILARIS is indicated for the treatment of Tumor Necrosis Factor (TNF) Receptor Associated Periodic Syndrome (TRAPS) in adult and pediatric patients.
Hyperimmunoglobulin D Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD)
ILARIS is indicated for the treatment of Hyperimmunoglobulin D (Hyper-IgD) Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD) in adult and pediatric patients.
Familial Mediterranean Fever (FMF)
ILARIS is indicated for the treatment of Familial Mediterranean Fever (FMF) in adult and pediatric patients.
1.2 Still’s Disease (Adult-Onset Still’s Disease [AOSD] and Systemic Juvenile Idiopathic Arthritis [SJIA])
ILARIS is indicated for the treatment of active Still’s disease, including Adult-Onset Still’s Disease (AOSD) and Systemic Juvenile Idiopathic Arthritis (SJIA) in patients aged 2 years and older.
2. Ilaris Dosage and Administration
2.2 Cryopyrin-Associated Periodic Syndromes (CAPS)
The recommended dose of ILARIS is 150 mg for CAPS patients with body weight greater than 40 kg. For CAPS patients with body weight greater than or equal to 15 kg and less than or equal to 40 kg, the recommended dose is 2 mg/kg. For children 15 to 40 kg with an inadequate response, the dose can be increased to 3 mg/kg.
ILARIS is administered every 8 weeks.
2.3 Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS), Hyperimmunoglobulin D Syndrome/Mevalonate Kinase Deficiency (HIDS/MKD), and Familial Mediterranean Fever (FMF)
The recommended dose of ILARIS for TRAPS, HIDS/MKD, and FMF patients is based on body weight.
For patients with body weight less than or equal to 40 kg, the recommended dose is 2 mg/kg administered every 4 weeks. The dose can be increased to 4 mg/kg every 4 weeks if the clinical response is not adequate.
For patients with body weight greater than 40 kg, the recommended dose is 150 mg administered every 4 weeks. The dose can be increased to 300 mg every 4 weeks if the clinical response is not adequate.
2.4 Still’s Disease, Including Adult-Onset Still’s Disease (AOSD) and Systemic Juvenile Idiopathic Arthritis (SJIA)
The recommended dose of ILARIS for patients with Still’s disease (AOSD and SJIA) with a body weight greater than or equal to 7.5 kg is 4 mg/kg (with a maximum of 300 mg) administered every 4 weeks.
2.5 Administration of ILARIS Solution
STEP 1: ILARIS solution has a concentration of 150 mg/mL. Do not shake. The solution should be essentially free from particulates, clear to opalescent, colorless to slightly brownish-yellow tint. If the solution has a distinctly brown discoloration, is highly opalescent or contains visible particles, do not use.
STEP 2: Using a sterile 1-mL syringe and 18-gauge x 2” needle, carefully withdraw the required volume depending on the dose to be administered and subcutaneously inject using a 27-gauge x 0.5” needle.
Injection into scar tissue should be avoided as this may result in insufficient exposure to ILARIS.
Discard unused product or waste material in accordance with the local requirements.
3. Dosage Forms and Strengths
Injection: 150 mg/mL solution in single-dose vials. The solution is a clear to slightly opalescent, colorless to a slightly brownish yellow tint.
4. Contraindications
Confirmed hypersensitivity to the active substance or to any of the excipients [see Warnings and Precautions (5.3) and Adverse Reactions (6.2)].
5. Warnings and Precautions
5.1 Serious Infections
ILARIS has been associated with an increased risk of serious infections. Physicians should exercise caution when administering ILARIS to patients with infections, a history of recurring infections or underlying conditions which may predispose them to infections. ILARIS should not be administered to patients during an active infection requiring medical intervention. Administration of ILARIS should be discontinued if a patient develops a serious infection.
Infections, predominantly of the upper respiratory tract, in some instances serious, have been reported with ILARIS. Generally, the observed infections responded to standard therapy. Isolated cases of unusual or opportunistic infections (e.g., aspergillosis, atypical mycobacterial infections, cytomegalovirus, herpes zoster) were reported during ILARIS treatment. A causal relationship of ILARIS to these events cannot be excluded. In clinical trials, ILARIS has not been administered concomitantly with tumor necrosis factor (TNF) inhibitors. An increased incidence of serious infections has been associated with administration of another IL-1 blocker in combination with TNF inhibitors. Coadministration of ILARIS with TNF inhibitors is not recommended because this may increase the risk of serious infections [see Drug Interactions (7.1)].
Drugs that affect the immune system by blocking TNF have been associated with an increased risk of new tuberculosis and reactivation of latent tuberculosis (TB). It is possible that use of IL-1 inhibitors, such as ILARIS, increases the risk of reactivation of tuberculosis or of opportunistic infections.
Prior to initiating immunomodulatory therapies, including ILARIS, patients should be evaluated for active and latent tuberculosis infection. Appropriate screening tests should be performed in all patients. ILARIS has not been studied in patients with a positive tuberculosis screen, and the safety of ILARIS in individuals with latent tuberculosis infection is unknown. Patients testing positive in tuberculosis screening should be treated according to standard medical practice prior to therapy with ILARIS. All patients should be instructed to seek medical advice if signs, symptoms, or high risk exposure suggestive of tuberculosis (e.g., persistent cough, weight loss, subfebrile temperature) appear during or after ILARIS therapy.
Healthcare providers should follow current CDC guidelines both to evaluate for and to treat possible latent tuberculosis infections before initiating therapy with ILARIS.
5.3 Hypersensitivity
Hypersensitivity reactions have been reported with ILARIS therapy. During clinical trials, no anaphylactic reactions attributable to treatment with canakinumab have been reported. It should be recognized that symptoms of the underlying disease being treated may be similar to symptoms of hypersensitivity. ILARIS should not be administered to any patients with known clinical hypersensitivity to ILARIS. If a severe hypersensitivity reaction occurs, administration of ILARIS should be discontinued and appropriate therapy initiated [see Contraindications (4) and Adverse Reactions (6.2)].
5.4 Immunizations
Live vaccines should not be given concurrently with ILARIS [see Drug Interactions (7.2)]. Since no data are available on either the efficacy or on the risks of secondary transmission of infection by live vaccines in patients receiving ILARIS, live vaccines should not be given concurrently with ILARIS. In addition, because ILARIS may interfere with normal immune response to new antigens, vaccinations may not be effective in patients receiving ILARIS. Limited data are available on the response to vaccinations with inactivated (killed) antigens in patients receiving ILARIS [see Drug Interactions (7.2)].
Because IL-1 blockade may interfere with immune response to infections, it is recommended that prior to initiation of therapy with ILARIS, adult and pediatric patients receive all recommended vaccinations, as appropriate and if feasible, including pneumococcal vaccine and inactivated influenza vaccine. See current recommended immunization schedules at the website of the Centers for Disease Control, http://www.cdc.gov/vaccines/schedules/index.html.
5.5 Macrophage Activation Syndrome
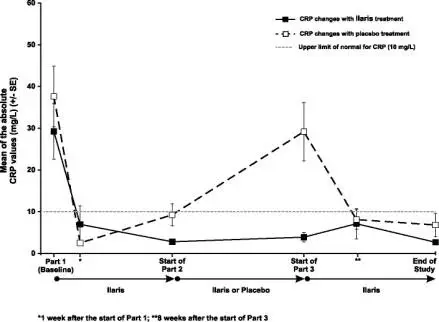
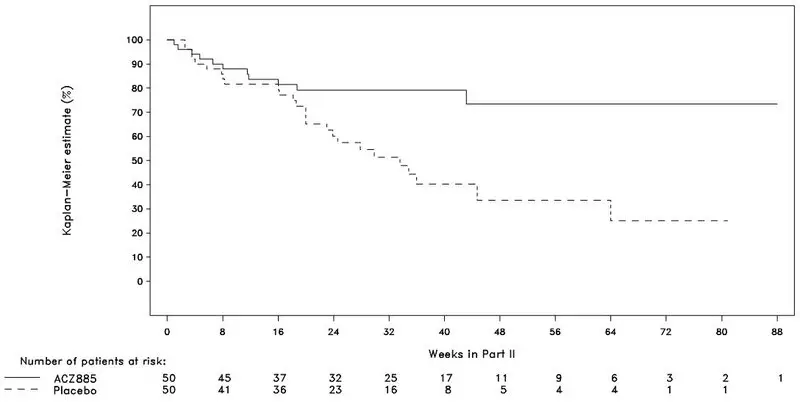
Macrophage activation syndrome (MAS) is a known, life-threatening disorder that may develop in patients with rheumatic conditions, in particular Still’s disease, and should be aggressively treated. Physicians should be attentive to symptoms of infection or worsening of Still’s disease, as these are known triggers for MAS. Eleven cases of MAS were observed in 201 SJIA patients treated with canakinumab in clinical trials. Based on the clinical trial experience, ILARIS does not appear to increase the incidence of MAS in Still’s disease patients, but no definitive conclusion can be made.
6. Adverse Reactions/Side Effects
In patients who have been treated with ILARIS in interventional trials in CAPS, TRAPS, HIDS/MKD, FMF or Still’s disease, the most frequently reported adverse drug reactions were infections predominantly of the upper respiratory tract. The majority of the events were mild to moderate although serious infections were observed.
Opportunistic infections have also been reported in patients treated with ILARIS [see Warnings and Precautions (5.1)].
7. Drug Interactions
Interactions between ILARIS and other medicinal products have not been investigated in formal studies.

| ILARIS
canakinumab injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
