Drug Detail:Kesimpta (Ofatumumab)
Drug Class: CD20 monoclonal antibodies Selective immunosuppressants
Highlights of Prescribing Information
KESIMPTA® (ofatumumab) injection, for subcutaneous use
Initial U.S. Approval: 2009
Indications and Usage for Kesimpta
KESIMPTA is a CD20-directed cytolytic antibody indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. (1)
Kesimpta Dosage and Administration
- Hepatitis B virus (HBV) and quantitative serum immunoglobulins screening are required before the first dose. (2.1)
- Administer KESIMPTA by subcutaneous injection only. (2.2, 2.3)
- Initial Dosing: 20 mg administered at Weeks 0, 1, and 2. (2.2)
- Subsequent Dosing: 20 mg administered monthly starting at Week 4. (2.2)
Dosage Forms and Strengths
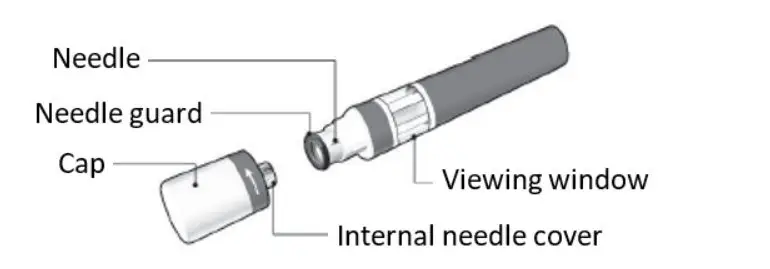
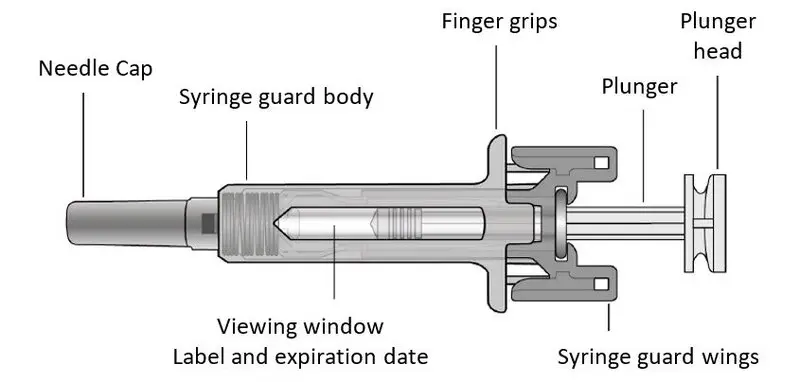
- Injection: 20 mg/0.4 mL solution in a single-dose prefilled Sensoready® Pen (3)
- Injection: 20 mg/0.4 mL solution in a single-dose prefilled syringe (3)
Contraindications
- Active HBV infection. (4)
Warnings and Precautions
- Infections: Delay KESIMPTA administration in patients with an active infection until the infection is resolved. Vaccination with live-attenuated or live vaccines is not recommended during treatment with KESIMPTA and after discontinuation, until B-cell repletion. (5.1)
- Injection-Related Reactions: Management for injection-related reactions depends on the type and severity of the reaction. (5.2)
- Reduction in Immunoglobulins: Monitor the level of immunoglobulins at the beginning, during, and after discontinuation of treatment with KESIMPTA until B-cell repletion. Consider discontinuing KESIMPTA if a patient develops a serious opportunistic infection or recurrent infections if immunoglobulin levels indicate immune compromise. (5.3)
- Fetal Risk: May cause fetal harm based on animal data. Advise females of reproductive potential of the potential risk to a fetus and to use an effective method of contraception during treatment and for 6 months after stopping KESIMPTA. (5.4, 8.1)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence greater than 10%) are upper respiratory tract infection, headache, injection-related reactions, and local injection site reactions. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2022
Full Prescribing Information
1. Indications and Usage for Kesimpta
KESIMPTA is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
2. Kesimpta Dosage and Administration
2.1 Assessments Prior to First Dose of KESIMPTA
Hepatitis B Virus Screening
Prior to initiating KESIMPTA, perform Hepatitis B virus (HBV) screening. KESIMPTA is contraindicated in patients with active HBV confirmed by positive results for Hepatitis B surface antigen [HBsAg] and anti-HBV tests. For patients who are negative for HBsAg and positive for Hepatitis B core antibody [HBcAb+] or are carriers of HBV [HBsAg+], consult liver disease experts before starting and during treatment with KESIMPTA [see Warnings and Precautions (5.1)].
Serum Immunoglobulins
Prior to initiating KESIMPTA, perform testing for quantitative serum immunoglobulins [see Warnings and Precautions (5.3)]. For patients with low serum immunoglobulins, consult immunology experts before initiating treatment with KESIMPTA.
Vaccinations
Because vaccination with live-attenuated or live vaccines is not recommended during treatment and after discontinuation until B-cell repletion, administer all immunizations according to immunization guidelines at least 4 weeks prior to initiation of KESIMPTA for live or live-attenuated vaccines, and whenever possible, at least 2 weeks prior to initiation of KESIMPTA for inactivated vaccines [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
The recommended dosage of KESIMPTA is:
- initial dosing of 20 mg by subcutaneous injection at Weeks 0, 1, and 2, followed by
- subsequent dosing of 20 mg by subcutaneous injection once monthly starting at Week 4.
Missed Doses
If an injection of KESIMPTA is missed, it should be administered as soon as possible without waiting until the next scheduled dose. Subsequent doses should be administered at the recommended intervals.
2.3 Administration Instructions
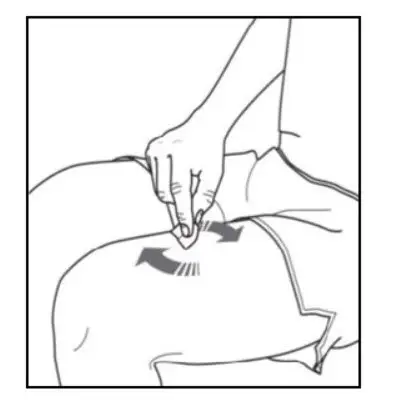
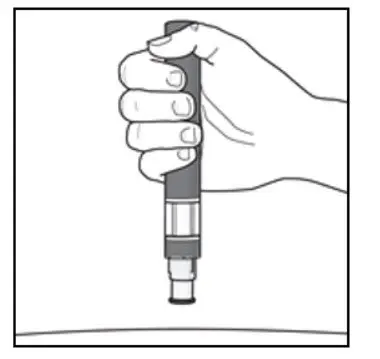
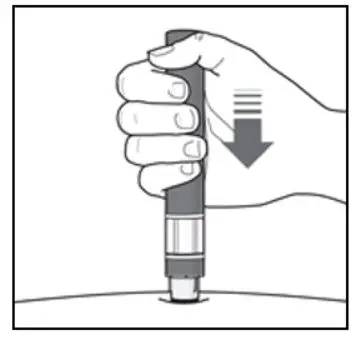
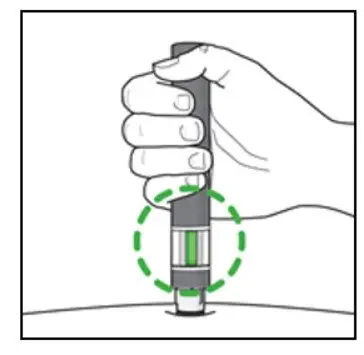
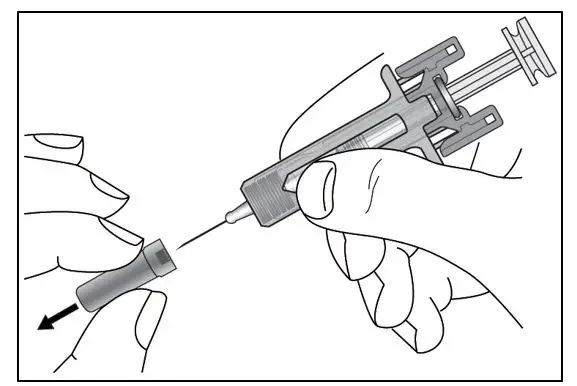
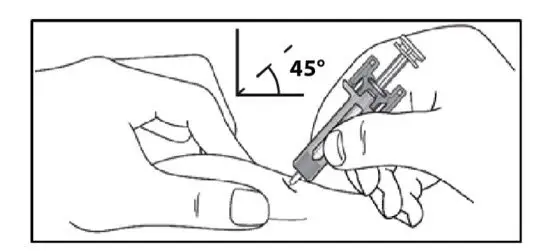
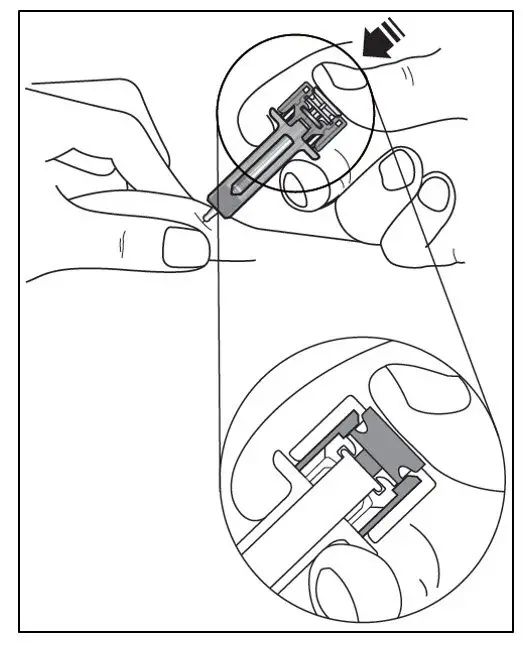
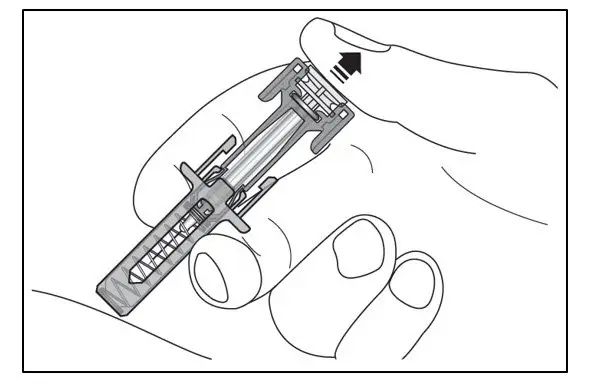
Administer by subcutaneous injection only.
KESIMPTA is intended for patient self-administration by subcutaneous injection.
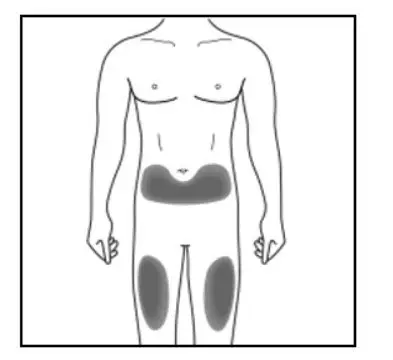
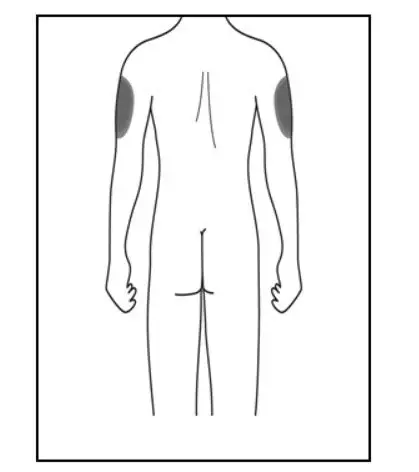
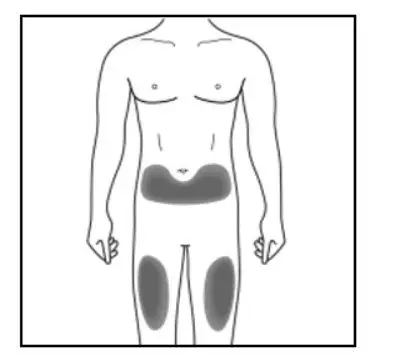
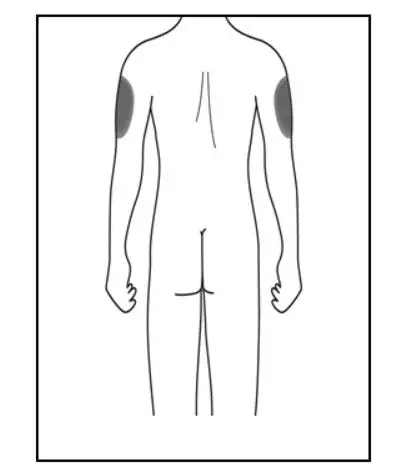
Administer KESIMPTA in the abdomen, thigh, or outer upper arm subcutaneously. Do not give injection into moles, scars, stretch marks, or areas where the skin is tender, bruised, red, scaly, or hard.
The first injection of KESIMPTA should be performed under the guidance of a healthcare professional [see Warnings and Precautions (5.2)].
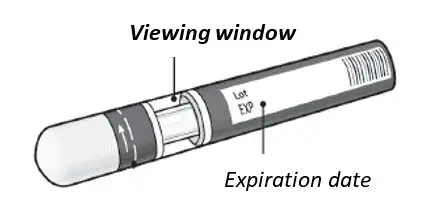
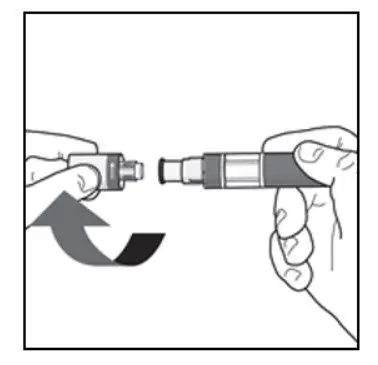
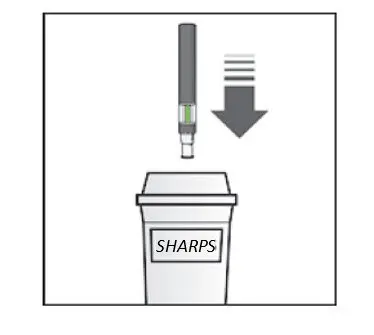
KESIMPTA Sensoready® pens and syringes are for one-time use only and should be discarded after use. See Instructions for Use for complete administration instructions.
2.4 Preparation of KESIMPTA
The KESIMPTA “Instructions for Use” for each presentation contains more detailed instructions on the preparation of KESIMPTA.
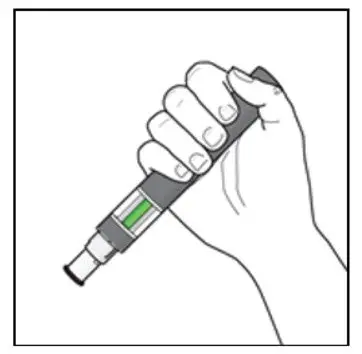
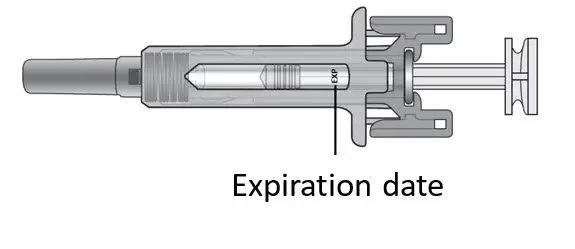
Before administration, remove KESIMPTA Sensoready Pen or KESIMPTA prefilled syringe from the refrigerator and allow KESIMPTA to reach room temperature for about 15 to 30 minutes. DO NOT remove the needle cover while allowing the prefilled syringe to reach room temperature.
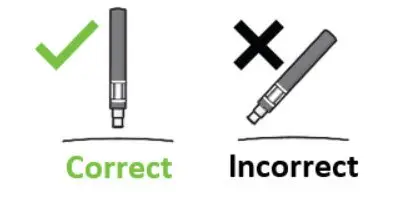
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if the liquid contains visible particles or is cloudy.
3. Dosage Forms and Strengths
KESIMPTA is a clear to slightly opalescent, and colorless to slightly brownish-yellow solution available as follows:
- Injection: 20 mg/0.4 mL in a single-dose prefilled Sensoready Pen
- Injection: 20 mg/0.4 mL in a single-dose prefilled syringe
4. Contraindications
KESIMPTA is contraindicated in patients with:
- Active HBV infection [see Warnings and Precautions (5.1)].
5. Warnings and Precautions
5.1 Infections
An increased risk of infections has been observed with other anti-CD20 B-cell depleting therapies.
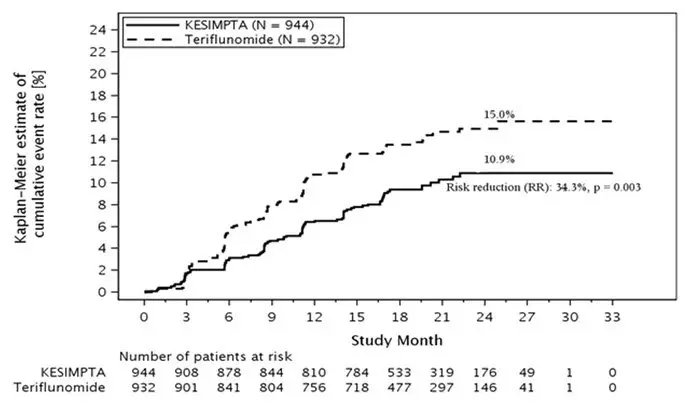
KESIMPTA has the potential for an increased risk of infections, including serious bacterial, fungal, and new or reactivated viral infections; some of these infections have been fatal in patients treated with other anti-CD20 antibodies. In Study 1 and Study 2 [see Clinical Studies (14)], the overall rate of infections and serious infections in patients treated with KESIMPTA was similar to patients who were treated with teriflunomide (51.6% vs 52.7%, and 2.5% vs 1.8%, respectively). The most common infections reported by KESIMPTA-treated patients in the randomized clinical relapsing MS (RMS) trials included upper respiratory tract infection (39%) and urinary tract infection (10%). Delay KESIMPTA administration in patients with an active infection until the infection is resolved.
Possible Increased Risk of Immunosuppressant Effects with Other Immunosuppressants
When initiating KESIMPTA after an immunosuppressive therapy or initiating an immunosuppressive therapy after KESIMPTA, consider the potential for increased immunosuppressive effects [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)]. KESIMPTA has not been studied in combination with other MS therapies.
Hepatitis B Virus
Reactivation
There were no reports of HBV reactivation in patients with MS treated with KESIMPTA. However, HBV reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, has occurred in patients being treated with ofatumumab for chronic lymphocytic leukemia (CLL) (at higher intravenous doses than the recommended dose in MS but for a shorter duration of treatment) and in patients treated with other anti-CD20 antibodies.
Infection
KESIMPTA is contraindicated in patients with active hepatitis B disease. Fatal infections caused by HBV in patients who have not been previously infected have occurred in patients being treated with ofatumumab for CLL (at higher intravenous doses than the recommended dose in MS but for a shorter duration of treatment). HBV screening should be performed in all patients before initiation of treatment with KESIMPTA. At a minimum, screening should include Hepatitis B surface antigen (HBsAg) and Hepatitis B Core Antibody (HBcAb) testing. These can be complemented with other appropriate markers as per local guidelines. For patients who are negative for HBsAg and positive for HB core antibody [HBcAb+] or are carriers of HBV [HBsAg+], consult liver disease experts before starting and during treatment with KESIMPTA. These patients should be monitored and managed following local medical standards to prevent HBV infection or reactivation.
Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically occurs in patients who are immunocompromised, and that usually leads to death or severe disability.
Although no cases of PML have been reported for KESIMPTA in the RMS clinical studies, PML resulting in death has occurred in patients being treated with ofatumumab for CLL (at substantially higher intravenous doses than the recommended dose in MS but for a shorter duration of treatment). In addition, JCV infection resulting in PML has also been observed in patients treated with other anti-CD20 antibodies and other MS therapies. At the first sign or symptom suggestive of PML, withhold KESIMPTA and perform an appropriate diagnostic evaluation. Magnetic resonance imaging (MRI) findings may be apparent before clinical signs or symptoms. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
If PML is confirmed, treatment with KESIMPTA should be discontinued.
Vaccinations
Administer all immunizations according to immunization guidelines at least 4 weeks prior to initiation of KESIMPTA for live or live-attenuated vaccines, and whenever possible, at least 2 weeks prior to initiation of KESIMPTA for inactivated vaccines.
KESIMPTA may interfere with the effectiveness of inactivated vaccines.
The safety of immunization with live or live-attenuated vaccines following KESIMPTA therapy has not been studied. Vaccination with live or live-attenuated vaccines is not recommended during treatment and after discontinuation until B-cell repletion [see Clinical Pharmacology (12.2)].
Vaccination of Infants Born to Mothers Treated with KESIMPTA During Pregnancy
In infants of mothers treated with KESIMPTA during pregnancy, do not administer live or live-attenuated vaccines before confirming the recovery of B-cell counts. Depletion of B-cells in these infants may increase the risks from live or live-attenuated vaccines.
Inactivated vaccines may be administered, as indicated, prior to recovery from B-cell depletion, but an assessment of vaccine immune responses, including consultation with a qualified specialist, should be considered to determine whether a protective immune response was mounted.
5.2 Injection-Related Reactions
In Study 1 and Study 2, systemic and local injection reactions were reported in 21% and 11% of patients treated with KESIMPTA compared to 15% and 6% of patients treated with teriflunomide who received matching placebo injections, respectively [see Adverse Reactions (6.1) and Clinical Studies (14)].
Injection-related reactions with systemic symptoms observed in clinical studies occurred most commonly within 24 hours of the first injection, but were also observed with later injections. Symptoms observed included fever, headache, myalgia, chills, and fatigue, and were predominantly (99.8%) mild to moderate in severity. There were no life-threatening injection reactions in the RMS clinical studies.
Local injection-site reaction symptoms observed in clinical studies included erythema, swelling, itching, and pain.
Only limited benefit of premedication with corticosteroids, antihistamines, or acetaminophen was observed in RMS clinical studies. The first injection of KESIMPTA should be performed under the guidance of an appropriately trained healthcare professional. If injection-related reactions occur, symptomatic treatment is recommended.
5.3 Reduction in Immunoglobulins
As expected with any B-cell depleting therapy, decreased immunoglobulin levels were observed. Decrease in immunoglobulin M (IgM) was reported in 7.7% of patients treated with KESIMPTA compared to 3.1% of patients treated with teriflunomide in RMS clinical trials [see Adverse Reactions (6.1)]. Treatment was discontinued because of decreased immunoglobulins in 3.4% of patients treated with KESIMPTA and in 0.8% of patients treated with teriflunomide. No decline in immunoglobulin G (IgG) was observed at the end of the study. Monitor the levels of quantitative serum immunoglobulins during treatment, especially in patients with opportunistic or recurrent infections, and after discontinuation of therapy until B-cell repletion. Consider discontinuing KESIMPTA therapy if a patient with low immunoglobulins develops a serious opportunistic infection or recurrent infections, or if prolonged hypogammaglobulinemia requires treatment with intravenous immunoglobulins.
5.4 Fetal Risk
Based on animal data, KESIMPTA can cause fetal harm due to B-cell lymphopenia and reduce antibody response in offspring exposed to KESIMPTA in utero. Transient peripheral B-cell depletion and lymphocytopenia have been reported in infants born to mothers exposed to other anti-CD20 B-cell depleting antibodies during pregnancy. Advise females of reproductive potential to use effective contraception while receiving KESIMPTA and for at least 6 months after the last dose [see Use in Specific Populations (8.1)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are discussed in greater detail elsewhere in the labeling:
- Infections [see Warnings and Precautions (5.1)]
- Injection-Related Reactions [see Warnings and Precautions (5.2)]
- Reduction in Immunoglobulins [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Approximately 1500 patients with RMS received KESIMPTA in clinical studies. In Study 1 and Study 2, 1882 patients with RMS were randomized, 946 of whom were treated with KESIMPTA for a median duration of 85 weeks; 33% of patients receiving KESIMPTA were treated for up to 120 weeks [see Clinical Studies (14.1)]. The most common adverse reactions occurring in greater than 10% of patients treated with KESIMPTA and more frequently than in patients treated with teriflunomide were upper respiratory tract infections, injection-related reactions (systemic), headache, and injection-site reactions (local). The most common cause of discontinuation in patients treated with KESIMPTA was low immunoglobulin M (3.3%), defined in trial protocols as IgM at 10% below the lower limit of normal (LLN).
Table 1 summarizes the adverse drug reactions that occurred in Study 1 and Study 2.
| aIncludes the following: nasopharyngitis, upper respiratory tract infection, influenza, sinusitis, pharyngitis, rhinitis, viral upper respiratory infection, tonsillitis, acute sinusitis, pharyngotonsillitis, laryngitis, pharyngitis streptococcal, viral rhinitis, sinusitis bacterial, tonsillitis bacterial, viral pharyngitis, viral tonsillitis, chronic sinusitis, nasal herpes, tracheitis. |
||
| Adverse Reactions | KESIMPTA 20 mg N = 946 % | Teriflunomide 14 mg N = 936 % |
| Upper respiratory tract infectionsa | 39 | 38 |
| Injection-related reactions (systemic) | 21 | 15 |
| Headache | 13 | 12 |
| Injection-site reactions (local) | 11 | 6 |
| Urinary tract infection | 10 | 8 |
| Back pain | 8 | 6 |
| Blood immunoglobulin M decreased | 6 | 2 |
Injection-Related Reactions and Injection-Site Reactions
The incidence of injection-related reactions (systemic) was highest with the first injection (14.4%), decreasing with subsequent injections (4.4% with second, less than 3% with third injection). Injection-related reactions were mostly (99.8%) mild to moderate in severity. Two (0.2%) patients treated with KESIMPTA reported serious injection-related reactions. There were no life-threatening injection-related reactions. Most frequently reported symptoms (2% or greater) included fever, headache, myalgia, chills, and fatigue.
In addition to systemic injection-related reactions, local reactions at the administration site were very common. Local injection-site reactions were all mild to moderate in severity. The most frequently reported symptoms (2% or greater) included erythema, pain, itching, and swelling [see Warnings and Precautions (5.2)].
Laboratory Abnormalities
Immunoglobulins
In Study 1 and Study 2, a decrease in the mean level of IgM was observed in KESIMPTA-treated patients but was not associated with an increased risk of infections [see Warnings and Precautions (5.3)]. In 14.3% of patients in Study 1 and Study 2, treatment with KESIMPTA resulted in a decrease in a serum IgM that reached a value below 0.34 g/L. KESIMPTA was associated with a decrease of 4.3% in mean IgG levels after 48 weeks of treatment and an increase of 2.2% after 96 weeks.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medication, and the underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other ofatumumab products may be misleading.
Treatment-induced anti-drug antibodies (ADAs) were detected in 2 of 914 (0.2%) KESIMPTA-treated patients; no patients with treatment-enhancing or neutralizing ADAs were identified. There was no impact of positive ADA titers on PK, safety profile or B-cell kinetics in any patient; however, these data are not adequate to assess the impact of ADAs on the safety and efficacy of KESIMPTA.
7. Drug Interactions
7.1 Immunosuppressive or Immune-Modulating Therapies
Concomitant usage of KESIMPTA with immunosuppressant drugs, including systemic corticosteroids, may increase the risk of infection. Consider the risk of additive immune system effects when coadministering immunosuppressive therapies with KESIMPTA.
When switching from therapies with immune effects, the duration and mechanism of action of these therapies should be taken into account because of potential additive immunosuppressive effects when initiating KESIMPTA.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of KESIMPTA in pregnant women. Ofatumumab may cross the placenta and cause fetal B-cell depletion based on findings from animal studies (see Data).
Transient peripheral B-cell depletion and lymphocytopenia have been reported in infants born to mothers exposed to other anti-CD20 antibodies during pregnancy. B-cell levels in infants following maternal exposure to KESIMPTA have not been studied in clinical trials. The potential duration of B-cell depletion in infants exposed to ofatumumab in utero, and the impact of B-cell depletion on the safety and effectiveness of vaccines, are unknown. Avoid administering live vaccines to neonates and infants exposed to KESIMPTA in utero until B-cell recovery occurs [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.2)].
Following administration of ofatumumab to pregnant monkeys, increased mortality, depletion of B-cell populations, and impaired immune function were observed in the offspring, in the absence of maternal toxicity, at plasma levels substantially higher than that in humans (see Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Intravenous administration of ofatumumab (weekly doses of 0, 20, or 100 mg/kg) to pregnant monkeys during the period of organogenesis (gestations days 20 to 50) resulted in no adverse effects on embryofetal development; however, B-cell depletion was observed in fetuses at both doses when assessed on gestation day 100. Plasma exposure (Cave) at the no-effect dose (100 mg/kg) for adverse effects on embryofetal development was greater than 5000 times that in humans at the recommended human maintenance dose of 20 mg. A no-effect dose for effects on B-cells was not identified; plasma exposure (Cave) at the low-effect dose (20 mg/kg) was approximately 780 times that in humans at the recommended human maintenance dose (RHMD) of 20 mg/month.
Intravenous administration of ofatumumab (5 weekly doses of 0, 10, and 100 mg/kg, followed by biweekly doses of 0, 3, and 20 mg/kg) to pregnant monkeys throughout pregnancy resulted in no adverse effects on the development of the offspring. However, postnatal death, B-cell depletion, and impaired immune function were observed in the offspring at the high dose. The deaths at the high dose were considered secondary to B-cell depletion. Plasma exposure (Cave) in dams at the no-effect dose (100/20 mg/kg) for adverse developmental effects was approximately 500 times that in humans at RHMD. A no-effect level for mortality and immune effects in offspring was not established because of the limited number of evaluable offspring at the low dose.
8.2 Lactation
Risk Summary
There are no data on the presence of ofatumumab in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. Human IgG is excreted in human milk, and the potential for absorption of ofatumumab to lead to B-cell depletion in the infant is unknown. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for KESIMPTA and any potential adverse effects on the breastfed infant from KESIMPTA or from the underlying maternal condition.
12. Kesimpta - Clinical Pharmacology
12.3 Pharmacokinetics
Absorption
A subcutaneous dose of 20 mg every 4 weeks leads to a mean AUCtau of 483 mcg h/mL and a mean Cmax of 1.43 mcg/mL at steady state.
After subcutaneous administration, ofatumumab is believed to be predominantly absorbed via the lymphatic system similarly to other therapeutic monoclonal antibodies.
Distribution
The volume of distribution at steady-state was estimated to be 5.42 L following subcutaneous administration of repeated KESIMPTA 20 mg dose.
Elimination
Ofatumumab is eliminated by both linear catabolic pathways and a non-linear B-cell mediated pathway, resulting in non-constant elimination half-life. Initially, a higher baseline B-cell count results in greater B-cell-mediated elimination and shorter ofatumumab half-life. As the B-cells are depleted, the catabolic pathway predominates, resulting in a relatively longer ofatumumab half-life compared to earlier in therapy when both elimination pathways were available. Following B-cell depletion, clearance was estimated to be 0.34 L/day following repeated subcutaneous administration of KESIMPTA 20 mg injections. The half-life at steady state was estimated to be approximately 16 days following subcutaneous administration of repeated KESIMPTA 20 mg dosage.
Metabolism
Ofatumumab is a protein for which the expected metabolic pathway is degradation to small peptides and amino acids by ubiquitous proteolytic enzymes.
Excretion
Ofatumumab, a monoclonal antibody, is not likely to undergo renal excretion.
Specific Populations
The following population characteristics do not have a clinically meaningful effect on the pharmacokinetics of ofatumumab: body weight, sex, age, race, or baseline B-cell count.
Patients with Renal/Hepatic Impairment
Pharmacokinetics of ofatumumab in patients with renal or hepatic impairment have not been studied.
Drug Interaction Studies
Ofatumumab does not share a common clearance pathway with chemical drugs that are metabolized by the cytochrome P450 system or other drug metabolizing enzymes. Additionally, there is no evidence that CD20 monoclonal antibodies are involved in the regulation of the expression of drug metabolizing enzymes. Interactions between KESIMPTA and other medicinal products have not been investigated in formal studies.
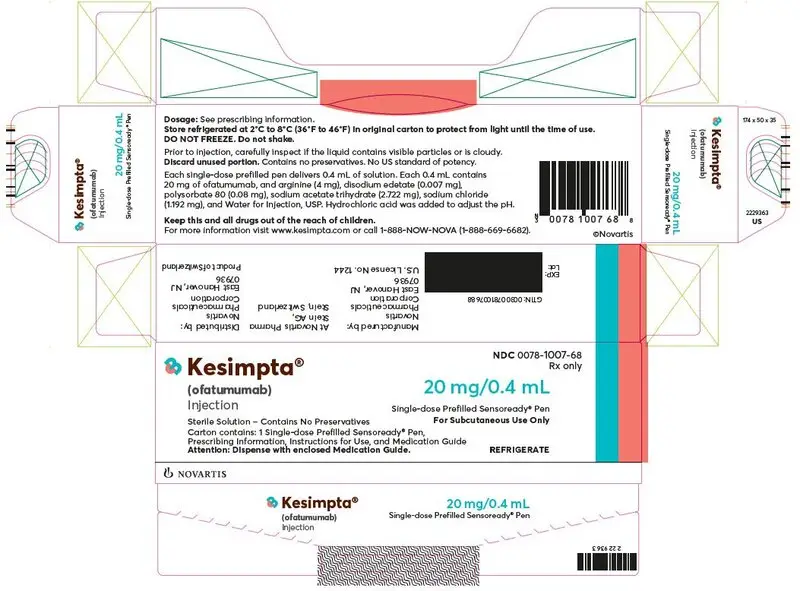
16. How is Kesimpta supplied
16.1 How Supplied
KESIMPTA (ofatumumab) injection is a preservative-free, clear to slightly opalescent and colorless to slightly brownish-yellow solution for subcutaneous administration, which is supplied as follows:
KESIMPTA Sensoready Pen:
Carton of one 20 mg/0.4 mL single-dose prefilled Sensoready Pen NDC 0078-1007-68
KESIMPTA Prefilled Syringe:
Carton of one 20 mg/0.4 mL single-dose prefilled syringe NDC 0078-1007-69
| KESIMPTA
ofatumumab injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
