Drug Detail:Kisqali (Ribociclib [ rye-boe-sye-klib ])
Drug Class: CDK 4/6 inhibitors
Highlights of Prescribing Information
KISQALI® (ribociclib) tablets, for oral use
Initial U.S. Approval: 2017
Recent Major Changes
| Indications and Usage (1) | 12/2021 |
| Dosage and Administration, Dosing and Administration (2.1) | 12/2021 |
| Warnings and Precautions (5) | 10/2022 |
Indications and Usage for Kisqali
KISQALI is a kinase inhibitor indicated for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer in combination with:
- an aromatase inhibitor as initial endocrine-based therapy; or
- fulvestrant as initial endocrine-based therapy or following disease progression on endocrine therapy in postmenopausal women or in men. (1)
Kisqali Dosage and Administration
KISQALI tablets are taken orally with or without food in combination with an aromatase inhibitor or fulvestrant. (2)
- Recommended starting dose: 600 mg orally (three 200 mg tablets) taken once daily with or without food for 21 consecutive days followed by 7 days off treatment. (2.1)
- Dose interruption, reduction, and/or discontinuation may be required based on individual safety and tolerability. (2.2)
Dosage Forms and Strengths
- Tablets: 200 mg (3)
Contraindications
None. (4)
Warnings and Precautions
- Interstitial Lung Disease (ILD)/Pneumonitis: Patients treated with CDK 4/6 inhibitors should be monitored for pulmonary symptoms indicative of ILD/pneumonitis. Interrupt and evaluate patients with new or worsening respiratory symptoms suspected to be due to ILD/pneumonitis. Permanently discontinue KISQALI in patients with recurrent symptomatic or severe ILD/pneumonitis. (2.2, 5.1)
- Severe Cutaneous Adverse Reactions (SCARs): Stevens-Johnson syndrome (SJS), Toxic epidermal necrolysis (TEN), and drug-reaction with eosinophilia and systemic symptoms (DRESS) can occur with KISQALI treatment. Permanently discontinue KISQALI in patients with SCARs or other life-threatening cutaneous reactions. (2.2, 5.2)
- QT Interval Prolongation: Monitor electrocardiograms (ECGs) and electrolytes prior to initiation of treatment with KISQALI. Repeat ECGs at approximately Day 14 of the first cycle and at the beginning of the second cycle, and as clinically indicated. Monitor electrolytes at the beginning of each cycle for 6 cycles, and as clinically indicated. Avoid using KISQALI with drugs known to prolong QT interval and/or strong CYP3A inhibitors. (2.2, 5.3, 7.1, 7.4)
- Increased QT Prolongation with Concomitant Use of Tamoxifen: KISQALI is not indicated for concomitant use with tamoxifen. (5.4)
- Hepatobiliary Toxicity: Increases in serum transaminase levels have been observed. Perform liver function tests (LFTs) before initiating treatment with KISQALI. Monitor LFTs every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated. (2.2, 5.5)
- Neutropenia: Perform complete blood count (CBC) before initiating therapy with KISQALI. Monitor CBC every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated. (2.2, 5.6)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of potential risk to a fetus and to use effective contraception during therapy. (5.7, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common (incidence ≥ 20%) adverse reactions, including laboratory abnormalities, are leukocytes decreased, neutrophils decreased, hemoglobin decreased, lymphocytes decreased, aspartate aminotransferase increased, gamma glutamyl transferase increased, alanine aminotransferase increased, infections, nausea, creatinine increased, fatigue, platelets decreased, diarrhea, vomiting, headache, constipation, alopecia, cough, rash, back pain, and glucose serum decreased. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- CYP3A4 Inhibitors: Avoid concomitant use of KISQALI with strong CYP3A inhibitors. If strong inhibitors cannot be avoided, reduce KISQALI dose. (2.2, 7.1)
- CYP3A4 Inducers: Avoid concomitant use of KISQALI with strong CYP3A inducers. (7.2)
- CYP3A Substrates: The dose of sensitive CYP3A substrates with narrow therapeutic indices may need to be reduced when given concurrently with KISQALI. (7.3)
- Drugs Known to Prolong QT Interval: Avoid concomitant use of drugs known to prolong QT interval, such as anti-arrhythmic medicines. (7.4)
Use In Specific Populations
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2022
Full Prescribing Information
1. Indications and Usage for Kisqali
KISQALI is indicated for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer in combination with:
- an aromatase inhibitor as initial endocrine-based therapy; or
- fulvestrant as initial endocrine-based therapy or following disease progression on endocrine therapy in postmenopausal women or in men.
2. Kisqali Dosage and Administration
2.1 Dosing and Administration
The recommended dose of KISQALI is 600 mg (three 200 mg film-coated tablets) taken orally, once daily for 21 consecutive days followed by 7 days off treatment resulting in a complete cycle of 28 days. KISQALI can be taken with or without food [see Clinical Pharmacology (12.3)].
When given with KISQALI, refer to the Full Prescribing Information for the recommended dose of the aromatase inhibitor being used.
When given with KISQALI, the recommended dose of fulvestrant is 500 mg administered on Days 1, 15, 29, and once monthly thereafter. Please refer to the Full Prescribing Information of fulvestrant.
Pre/perimenopausal women, or men, treated with the combination KISQALI plus an aromatase inhibitor should be treated with a luteinizing hormone-releasing hormone (LHRH) agonist according to current clinical practice standards.
Men treated with the combination of KISQALI plus fulvestrant should be treated with a luteinizing hormone-releasing hormone (LHRH) agonist according to current clinical practice standards.
Patients should take their dose of KISQALI at approximately the same time each day, preferably in the morning.
If the patient vomits after taking the dose, or misses a dose, no additional dose should be taken that day. The next prescribed dose should be taken at the usual time. KISQALI tablets should be swallowed whole (tablets should not be chewed, crushed or split prior to swallowing). No tablet should be ingested if it is broken, cracked, or otherwise not intact.
2.2 Dose Modifications
Dose Modifications for Adverse Reactions
The recommended dose modifications for adverse reactions are listed in Table 1.
| Level | KISQALI | |
| Dose | Number of tablets | |
| Starting dose | 600 mg/day | three 200 mg tablets |
| First dose reduction | 400 mg/day | two 200 mg tablets |
| Second dose reduction | 200 mg/day* | one 200 mg tablet |
| *If further dose reduction below 200 mg/day is required, discontinue the treatment. | ||
Tables 2, 3, 4, 5, 6, and 7 summarize recommendations for dose interruption, reduction, or discontinuation of KISQALI in the management of specific adverse reactions. Dose modification of KISQALI is recommended based on individual safety and tolerability.
| Grade 1 (asymptomatic) | Grade 2 (symptomatic) | Grade 3 (severe symptomatic) or 4 (life-threatening) |
|
| ILD/Pneumonitis
[see Warnings and Precautions (5.1)] | No dose interruption or adjustment is required. Initiate appropriate medical therapy and monitor as clinically indicated. | Dose interruption until recovery to Grade ≤ 1 then consider resuming KISQALI at the next lower dose level*. If Grade 2 recurs, discontinue KISQALI. | Discontinue KISQALI. |
| Abbreviation: ILD, interstitial lung disease. Grading according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. *An individualized benefit-risk assessment should be performed when considering resuming KISQALI. |
|||
| Grade 1
(< 10% body surface area (BSA) with active skin toxicity, no signs of systemic involvement) | Grade 2
(10%-30% BSA with active skin toxicity, no signs of systemic involvement) | Grade 3
(severe rash not responsive to medical management; > 30% BSA with active skin toxicity, signs of systemic involvement present; SJS*) | Grade 4
(any % BSA associated with extensive superinfection, with IV antibiotics indicated; life threatening consequences; TEN**) |
|
| Cutaneous adverse reactions, including SCARs
[see Warnings and Precautions (5.2)] | No dose adjustment is required.
Initiate appropriate medical therapy and monitor as clinically indicated. | Interrupt KISQALI until the etiology of the reaction has been determined. If the etiology is a SCAR, permanently discontinue KISQALI. If the etiology is not a SCAR, interrupt dose until recovery to Grade ≤ 1, then resume KISQALI at same dose level. If the cutaneous adverse reaction still recurs at Grade 3, resume KISQALI at the next lower dose level. | Permanently discontinue KISQALI. | |
| Abbreviations: BSA, body surface area; SCARs, severe cutaneous adverse reactions; SJS, stevens-johnson syndrome; TEN, toxic epidermal necrolysis. *SJS (Grade 3 and 4) is defined as skin sloughing covering < 10% BSA and 10%-30% BSA, respectively, with associated signs (e.g., erythema, purpura, epidermal detachment, and mucous membrane detachment). **TEN (Grade 4) is defined as skin sloughing covering ≥ 30% BSA with associated symptoms (e.g., erythema, purpura, epidermal detachment, and mucous membrane detachment). Grading according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. |
||||
| ECGs with QTcF* > 480 ms
[see Warnings and Precautions (5.3)] |
|
| ECGs with QTcF > 500 ms
[see Warnings and Precautions (5.3)] |
|
| Electrocardiograms (ECGs) should be assessed prior to initiation of treatment. Repeat ECGs at approximately Day 14 of the first cycle and at the beginning of the second cycle, and as clinically indicated. In case of (QTcF) prolongation at any given time during treatment, more frequent ECG monitoring is recommended. *QTcF = QT interval corrected by Fridericia’s formula. |
|
| Grade 1
(> ULN – 3 x ULN) | Grade 2
(> 3 to 5 x ULN) | Grade 3
(> 5 to 20 x ULN) | Grade 4
(> 20 x ULN) |
|
| AST and/or ALT elevations from baseline*, WITHOUT increase in total bilirubin above 2 x ULN
[see Warnings and Precautions (5.5)] | No dose adjustment is required. | Baseline* at < Grade 2:
Dose interruption until recovery to ≤ baseline grade, then resume KISQALI at same dose level. If Grade 2 recurs, resume KISQALI at next lower dose level. ----------------------------- Baseline* at Grade 2: No dose interruption. | Dose interruption until recovery to ≤ baseline* grade, then resume at next lower dose level. If Grade 3 recurs, discontinue KISQALI. | Discontinue KISQALI. |
| Combined elevations in AST and/or ALT WITH total bilirubin increase, in the absence of cholestasis
[see Warnings and Precautions (5.5)] | If patients develop ALT and/or AST > 3 x ULN along with total bilirubin > 2 x ULN irrespective of baseline grade, discontinue KISQALI. | |||
| Perform Liver Function Tests (LFTs) before initiating treatment with KISQALI. Monitor LFTs every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated. If Grade ≥ 2 abnormalities are noted, more frequent monitoring is recommended. |
||||
| Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; ULN, upper limit of normal. *Baseline = prior to treatment initiation. Grading according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. |
||||
| Grade 1 or 2
(ANC 1000/mm3 – < LLN) | Grade 3
(ANC 500 - < 1000/mm3) | Grade 3 febrile* neutropenia | Grade 4
(ANC < 500/mm3) |
|
| Neutropenia
[see Warnings and Precautions (5.6)] | No dose adjustment is required. | Dose interruption until recovery to Grade ≤ 2. Resume KISQALI at the same dose level. If toxicity recurs at Grade 3, dose interruption until recovery, then resume KISQALI at the next lower dose level. | Dose interruption until recovery of neutropenia to Grade ≤ 2. Resume KISQALI at the next lower dose level. | Dose interruption until recovery to Grade ≤ 2. Resume KISQALI at the next lower dose level. |
| Perform complete blood counts (CBCs) before initiating treatment with KISQALI. Monitor CBC every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated. |
||||
| Abbreviations: ANC, absolute neutrophil count; LLN, lower limit of normal. *Grade 3 neutropenia with single episode of fever > 38.3°C (or) above 38°C for more than one hour and/or concurrent infection. Grading according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. |
||||
| Grade 1 or 2 | Grade 3 | Grade 4 | |
| Other Toxicities | No dose adjustment is required. Initiate appropriate medical therapy and monitor as clinically indicated. | Dose interruption until recovery to Grade ≤ 1 then resume KISQALI at same dose level. If Grade 3 recurs, resume KISQALI at the next lower dose level. | Discontinue KISQALI. |
| *Excluding interstitial lung disease (ILD)/pneumonitis, cutaneous adverse reactions, including severe cutaneous adverse reactions (SCARs), QT interval prolongation, hepatobiliary toxicity, and neutropenia. Grading according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. |
|||
Refer to the Full Prescribing Information for the coadministered aromatase inhibitor or fulvestrant for dose modification guidelines in the event of toxicity and other relevant safety information.
Dose Modification for Use with Strong CYP3A Inhibitors
Avoid concomitant use of KISQALI with strong CYP3A inhibitors and consider an alternative concomitant medication with less potential for CYP3A inhibition [see Drug Interactions (7.1)]. If a strong CYP3A inhibitor must be coadministered, reduce the KISQALI dose to 400 mg once daily. If the strong inhibitor is discontinued, change the KISQALI dose (after at least 5 half-lives of the strong CYP3A inhibitor) to the dose used prior to the initiation of the strong CYP3A inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Dose Modification for Hepatic Impairment
No dose adjustment is necessary in patients with mild hepatic impairment (Child-Pugh class A). The recommended starting dose is 400 mg KISQALI once daily for patients with moderate (Child-Pugh class B) and severe hepatic impairment (Child-Pugh class C) [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Review the Full Prescribing Information for the coadministered aromatase inhibitor or fulvestrant for dose modifications related to hepatic impairment.
Dose Modification for Renal Impairment
No dose adjustment is necessary in patients with mild or moderate renal impairment. The recommended starting dose is 200 mg KISQALI once daily for patients with severe renal impairment [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
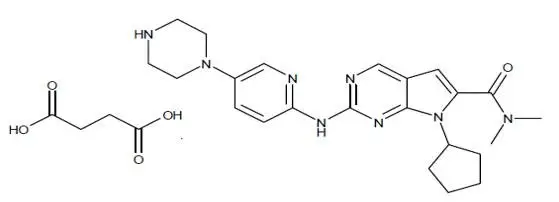
Tablet: 200 mg ribociclib (equivalent to 254.40 mg ribociclib succinate).
Film coated, light greyish violet, round, curved with beveled edges, debossed with “RIC” on one side and “NVR” on the other side.
5. Warnings and Precautions
5.1 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD) and/or pneumonitis can occur in patients treated with KISQALI and other CDK4/6 inhibitors.
Across clinical trials (MONALEESA-2, MONALEESA-3, MONALEESA-7), 1.6% of KISQALI-treated patients had ILD/pneumonitis of any grade, 0.4% had Grade 3 or 4, and 0.1% had a fatal outcome. Additional cases of ILD/pneumonitis have been observed in the postmarketing setting, with fatalities reported [see Adverse Reactions (6.2)].
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis which may include hypoxia, cough, and dyspnea. In patients who have new or worsening respiratory symptoms suspected to be due to ILD or pneumonitis, interrupt KISQALI immediately and evaluate the patient. Permanently discontinue KISQALI in patients with recurrent symptomatic or severe ILD/pneumonitis [see Dosage and Administration (2.2)].
5.2 Severe Cutaneous Adverse Reactions
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN), and drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS) can occur in patients treated with KISQALI [see Adverse Reactions (6.2)].
If signs or symptoms of severe cutaneous reactions occur, interrupt KISQALI until the etiology of the reaction has been determined [see Dosage and Administration (2.2)]. Early consultation with a dermatologist is recommended to ensure greater diagnostic accuracy and appropriate management.
If SJS, TEN, or DiHS/DRESS is confirmed, permanently discontinue KISQALI. Do not reintroduce KISQALI in patients who have experienced SCARs or other life-threatening cutaneous reactions during KISQALI treatment.
5.3 QT Interval Prolongation
KISQALI has been shown to prolong the QT interval in a concentration-dependent manner [see Clinical Pharmacology (12.2)]. Based on the observed QT prolongation during treatment, KISQALI may require dose interruption, reduction or discontinuation as described in Table 4 [see Dosage and Administration (2.2), Drug Interactions (7.4)].
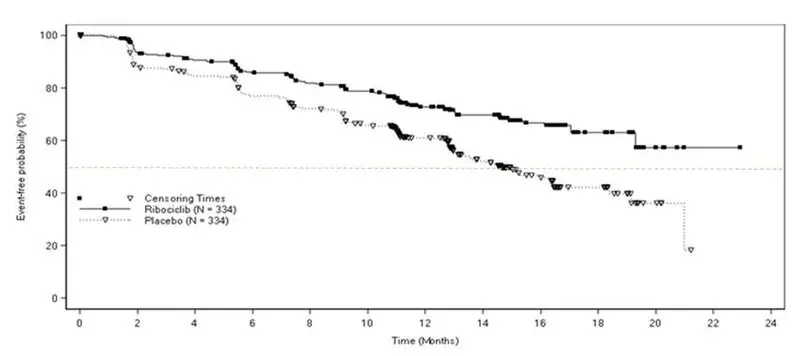
Across MONALEESA-2, MONALEESA-7, and MONALEESA-3 in patients with advanced or metastatic breast cancer who received the combination of KISQALI plus an aromatase inhibitor or fulvestrant, 15 out of 1054 patients (1.4%) had a > 500 ms post-baseline QTcF value, and 61 out of 1054 patients (6%) had a > 60 ms increase from baseline in QTcF intervals.
These ECG changes were reversible with dose interruption and the majority occurred within the first four weeks of treatment. There were no reported cases of Torsades de Pointes.
In MONALEESA-2, on the KISQALI plus letrozole treatment arm, there was one (0.3%) sudden death in a patient with Grade 3 hypokalemia and Grade 2 QT prolongation. No cases of sudden death were reported in MONALEESA-7 or MONALEESA-3 [see Adverse Reactions (6)].
Assess ECG prior to initiation of treatment. Initiate treatment with KISQALI only in patients with QTcF values less than 450 ms. Repeat ECG at approximately Day 14 of the first cycle and the beginning of the second cycle, and as clinically indicated.
Monitor serum electrolytes (including potassium, calcium, phosphorous and magnesium) prior to the initiation of treatment, at the beginning of the first 6 cycles, and as clinically indicated. Correct any abnormality before starting KISQALI therapy [see Dosage and Administration (2.2)].
Avoid the use of KISQALI in patients who already have or who are at significant risk of developing QT prolongation, including patients with:
- long QT syndrome
- uncontrolled or significant cardiac disease, including recent myocardial infarction, congestive heart failure, unstable angina, and bradyarrhythmias
- electrolyte abnormalities
Avoid using KISQALI with drugs known to prolong QT interval and/or strong CYP3A inhibitors as this may lead to prolongation of the QTcF interval.
5.4 Increased QT Prolongation With Concomitant Use of Tamoxifen
KISQALI is not indicated for concomitant use with tamoxifen. In MONALEESA-7, the observed mean QTcF increase from baseline was > 10 ms higher in the tamoxifen plus placebo subgroup compared with the non-steroidal aromatase inhibitors (NSAIs) plus placebo subgroup. In the placebo arm, an increase of > 60 ms from baseline occurred in 6/90 (7%) of patients receiving tamoxifen, and in no patients receiving an NSAI. An increase of > 60 ms from baseline in the QTcF interval was observed in 14/87 (16%) of patients in the KISQALI and tamoxifen combination and in 18/245 (7%) of patients receiving KISQALI plus an NSAI [see Clinical Pharmacology (12.2)].
5.5 Hepatobiliary Toxicity
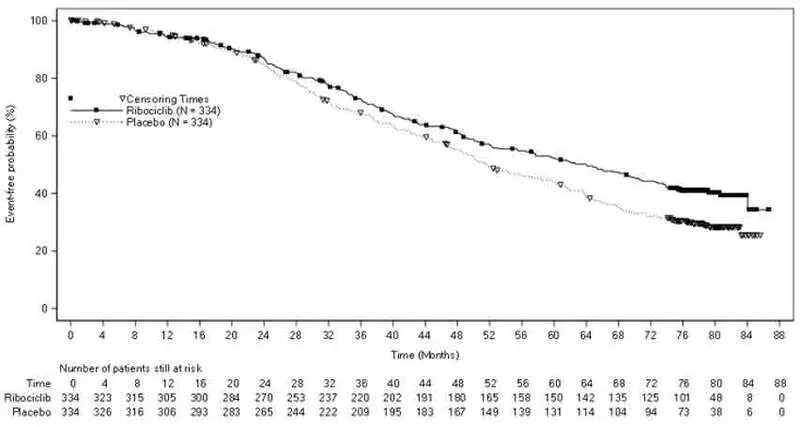
In MONALEESA-2, MONALEESA-7 and MONALEESA-3, increases in transaminases were observed. Across all studies, Grade 3 or 4 increases in alanine aminotransferase (ALT) (11% vs. 2.1%) and aspartate aminotransferase (AST) (8% vs. 2%) were reported in the KISQALI and placebo arms, respectively.
Among the patients who had Grade ≥ 3 ALT/AST elevation, the median time-to-onset was 92 days for the KISQALI plus aromatase inhibitor or fulvestrant treatment group. The median time to resolution to Grade ≤ 2 was 21 days in the KISQALI plus aromatase inhibitor or fulvestrant treatment group. In MONALEESA-2 and MONALEESA-3, concurrent elevations in ALT or AST greater than three times the ULN and total bilirubin greater than two times the ULN, with normal alkaline phosphatase, in the absence of cholestasis occurred in 6 (1%) patients and all patients recovered after discontinuation of KISQALI. No cases occurred in MONALEESA-7.
Perform liver function tests (LFTs) before initiating therapy with KISQALI. Monitor LFTs every 2 weeks for first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated [see Dosage and Administration (2.2)].
Based on the severity of the transaminase elevations, KISQALI may require dose interruption, reduction, or discontinuation as described in Table 5 (Dose Modification and Management for Hepatobiliary Toxicity) [see Dosage and Administration (2.2)]. Recommendations for patients who have elevated AST/ALT Grade ≥ 3 at baseline have not been established.
5.6 Neutropenia
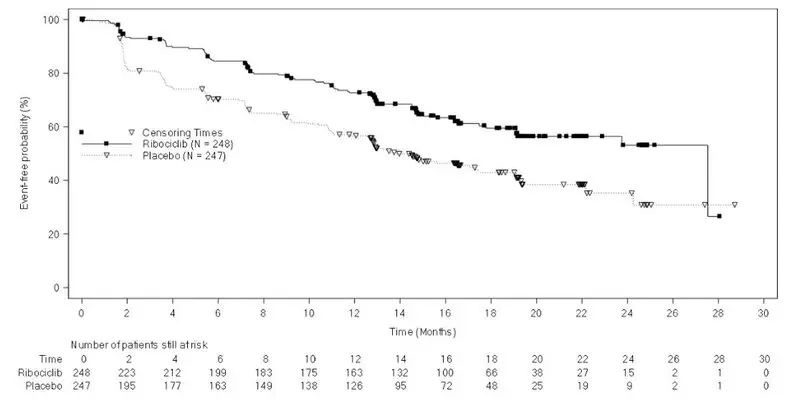
In MONALEESA-2, MONALEESA-7, and MONALEESA-3, neutropenia was the most frequently reported adverse reaction (75%), and a Grade 3/4 decrease in neutrophil count (based on laboratory findings) was reported in 62% of patients receiving KISQALI plus an aromatase inhibitor or fulvestrant. Among the patients who had Grade 2, 3, or 4 neutropenia, the median time to Grade ≥ 2 neutropenia was 17 days. The median time to resolution of Grade ≥ 3 (to normalization or Grade < 3) was 12 days in the KISQALI plus aromatase inhibitor or fulvestrant treatment group. Febrile neutropenia was reported in 1.7% of patients receiving KISQALI plus an aromatase inhibitor or fulvestrant. Treatment discontinuation due to neutropenia was 1%.
Perform complete blood count (CBC) before initiating therapy with KISQALI. Monitor CBC every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated.
Based on the severity of the neutropenia, KISQALI may require dose interruption, reduction or discontinuation as described in Table 6 [see Dosage and Administration (2.2)].
5.7 Embryo-Fetal Toxicity
Based on findings from animal studies and the mechanism of action, KISQALI can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of ribociclib to pregnant rats and rabbits during organogenesis caused embryo-fetal toxicities at maternal exposures that were 0.6 and 1.5 times the human clinical exposure, respectively, based on area under the curve (AUC). Advise pregnant women of the potential risk to a fetus. Advise women of reproductive potential to use effective contraception during therapy with KISQALI and for at least 3 weeks after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.1)]
- Severe Cutaneous Adverse Reactions [see Warnings and Precautions (5.2)]
- QT Interval Prolongation [see Warnings and Precautions (5.3, 5.4)]
- Hepatobiliary Toxicity [see Warnings and Precautions (5.5)]
- Neutropenia [see Warnings and Precautions (5.6)]
6.2 Postmarketing Experience
The following adverse events have been reported during post-approval use of KISQALI. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory disorders: Interstitial lung disease/pneumonitis
Skin and Subcutaneous Tissue Disorders: Stevens-Johnson Syndrome (SJS), Toxic epidermal necrolysis (TEN), Drug-induced hypersensitivity syndrome (DiHS)/Drug reaction with eosinophilia, and systemic symptoms (DRESS)
7. Drug Interactions
7.1 Drugs That May Increase Ribociclib Plasma Concentrations
CYP3A4 Inhibitors
Coadministration of a strong CYP3A4 inhibitor (ritonavir) increased ribociclib exposure in healthy subjects by 3.2-fold [see Clinical Pharmacology (12.3)]. Avoid concomitant use of strong CYP3A inhibitors (e.g., boceprevir, clarithromycin, conivaptan, grapefruit juice, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, and voriconazole) and consider alternative concomitant medications with less potential for CYP3A inhibition.
If coadministration of KISQALI with a strong CYP3A inhibitor cannot be avoided, reduce the dose of KISQALI to 400 mg once daily [see Dosage and Administration (2.2)].
Instruct patients to avoid grapefruit or grapefruit juice, which are known to inhibit cytochrome CYP3A enzymes and may increase the exposure to ribociclib [see Patient Counseling Information (17)].
7.2 Drugs That May Decrease Ribociclib Plasma Concentrations
CYP3A4 Inducers
Coadministration of a strong CYP3A4 inducer (rifampin) decreased the plasma exposure of ribociclib in healthy subjects by 89% [see Clinical Pharmacology (12.3)]. Avoid concomitant use of strong CYP3A inducers and consider an alternate concomitant medication with no or minimal potential to induce CYP3A (e.g., phenytoin, rifampin, carbamazepine, and St. John’s wort (Hypericum perforatum).
7.3 Effect of KISQALI on Other Drugs
CYP3A Substrates with Narrow Therapeutic Index
Coadministration of midazolam (a sensitive CYP3A4 substrate) with multiple doses of KISQALI (400 mg) increased the midazolam exposure by 3.8-fold in healthy subjects, compared with administration of midazolam alone [see Clinical Pharmacology (12.3)]. KISQALI given at the clinically relevant dose of 600 mg is predicted to increase the midazolam AUC by 5.2-fold. Therefore, caution is recommended when KISQALI is administered with CYP3A substrates with a narrow therapeutic index. The dose of a sensitive CYP3A substrate with a narrow therapeutic index, including but not limited to alfentanil, cyclosporine, dihydroergotamine, ergotamine, everolimus, fentanyl, pimozide, quinidine, sirolimus, and tacrolimus, may need to be reduced as ribociclib can increase their exposure.
7.4 Drugs That Prolong the QT Interval
Avoid coadministration of KISQALI with medicinal products with a known potential to prolong QT, such as antiarrhythmic medicines (including, but not limited to amiodarone, disopyramide, procainamide, quinidine, and sotalol), and other drugs that are known to prolong the QT interval (including, but not limited to, chloroquine, halofantrine, clarithromycin, haloperidol, methadone, moxifloxacin, bepridil, pimozide, and ondansetron) [see Warnings and Precautions (5.3), Clinical Pharmacology (12.2)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and the mechanism of action, KISQALI can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)].
There are no available human data informing the drug-associated risk. In animal reproduction studies, administration of ribociclib to pregnant animals during organogenesis resulted in increased incidences of post implantation loss and reduced fetal weights in rats and increased incidences of fetal abnormalities in rabbits at exposures 0.6 or 1.5 times the exposure in humans, respectively, at the highest recommended dose of 600 mg/day based on AUC (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk of major birth defects is 2%-4% and of miscarriage is 15%-20% of clinically recognized pregnancies in the U.S. general population.
Data
Animal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of ribociclib up to 1000 mg/kg/day and 60 mg/kg/day, respectively, during the period of organogenesis.
In rats, 300 mg/kg/day resulted in reduced maternal body weight gain and reduced fetal weights accompanied by skeletal changes related to the lower fetal weights. There were no significant effects on embryo-fetal viability or fetal morphology at 50 or 300 mg/kg/day.
In rabbits at doses ≥ 30 mg/kg/day, there were adverse effects on embryo-fetal development, including increased incidences of fetal abnormalities (malformations and external, visceral, and skeletal variants) and fetal growth (lower fetal weights). These findings included reduced/small lung lobes, additional vessel on the descending aorta, additional vessel on the aortic arch, small eyes, diaphragmatic hernia, absent accessory lobe or (partly) fused lung lobes, reduced/small accessory lung lobe, extra/rudimentary 13th ribs, misshapen hyoid bone, bent hyoid bone alae, and reduced number of phalanges in the pollex. There was no evidence of increased incidence of embryo-fetal mortality. There was no maternal toxicity observed at 30 mg/kg/day.
At 300 mg/kg/day in rats and 30 mg/kg/day in rabbits, the maternal systemic exposures (AUC) were approximately 0.6 and 1.5 times, respectively, the exposure in patients at the highest recommended dose of 600 mg/day.
8.2 Lactation
Risk Summary
It is not known if ribociclib is present in human milk. There are no data on the effects of ribociclib on the breastfed infant or on milk production. Ribociclib and its metabolites readily passed into the milk of lactating rats. Because of the potential for serious adverse reactions in breastfed infants from KISQALI, advise lactating women not to breastfeed while taking KISQALI and for at least 3 weeks after the last dose.
Data
In lactating rats administered a single dose of 50 mg/kg, exposure to ribociclib was 3.56-fold higher in milk compared to maternal plasma.
8.3 Females and Males of Reproductive Potential
Based on animal studies and mechanism of action, KISQALI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to starting treatment with KISQALI.
Contraception
Females
Advise females of reproductive potential to use effective contraception (methods that result in less than 1% pregnancy rates) during treatment with KISQALI and for at least 3 weeks after the last dose.
Infertility
Males
Based on animal studies, KISQALI may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
| KISQALI
ribociclib tablet, film coated |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| KISQALI
ribociclib tablet, film coated |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| KISQALI
ribociclib tablet, film coated |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
