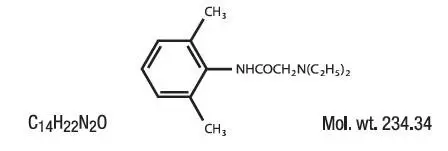
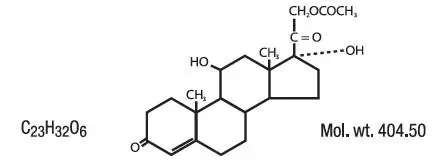
PHARMACOKINETICS:
Lidocaine may be absorbed following topical
administration to mucous membranes, its rate and extent of absorption
depending upon the specific site of application, duration of exposure,
concentration, and total dosage. In general, the rate of absorption of
local anesthetic agents following topical application occurs most
rapidly after intratracheal administration. Lidocaine is also
well-absorbed from the gastrointestinal tract, but little intact drug
appears in the circulation because of biotransformation of the liver.
Lidocaine
is metabolized rapidly by the liver, and metabolites and unchanged drug
are excreted by the kidneys. Biotransformation includes oxidative
N-dealkylation, ring hydroxylation, cleavage of the amide linkage, and
conjungation. N-dealkylation, a major pathway of biotransformation,
yields the metabolites monoethylglycinexylidide and glycinexylidide. The
pharmacological/toxicological actions of these metabolites are similar
to, but less potent than, those of lidocaine. Approximately 90% of
lidocaine administered is excreted in the form of various metabolites,
and less than 10% is excreted unchanged. The primary metabolite in
urine is a conjugate of 4-hydroxy-2, 6-dimethylaniline.
The
plasma binding of lidocaine is dependent of drug concentration, and the
fraction bound decreases with increasing concentration. At
concentrations of 1 to 4 g of free base per mL, 60 to 80 percent of
lidocaine is protein bound. Binding is also dependent on the plasma
concentration of the alpha-1-acid-glycoprotein.
Lidocaine crosses the blood-brain and placental barriers, presumably by passive diffusion.
Studies
of lidocaine metabolism following intravenous bolus injections have
shown that the elimination half-life of this agent is typically 1.5 to
2 hours. Because of the rapid rate at which lidocaine is metabolized,
any condition that affects liver function may alter lidocaine kinetics.
The half-life may be prolonged two-fold or more in patients with liver
dysfunction. Renal dysfunction does not affect lidocaine kinetics but
may increase the accumulation of metabolites.
Factors such as
acidosis and the use of CNS stimulants and depressants affect the CNS
levels of lidocaine required to produce overt systemic effects.
Objective adverse manifestations become increasingly apparent with
increasing venous plasma levels above 6 g free base per mL. In the
rhesus monkey arterial blood levels of 18-21 g/mL have been shown to be
the threshold for convulsive activity.
The extent of percutaneous
absorption of topical corticosteroids is determined by many factors
including the vehicle, the integrity of the epidermal barrier, and the
use of occlusive dressings.
Topical corticosteroids can be
absorbed from normal intact skin. Inflammation and/or other disease
processes in the skin increase percutaneous absorption. Occlusive
dressings substantially increase the percutaneous absorption of topical
corticosteroids. Thus, occlusive dressings may be a valuable
therapeutic adjunct for treatment of resistant dermatoses.
Once
absorbed through the skin, topical corticosteroids are handled through
pharmacokinetic pathways similar to systemically administered
corticosteroids. Corticosteroids are bound to plasma protein in varying
degrees. Corticosteroids are metabolized primarily in the liver and are
then excreted by the kidneys. Some of the topical corticosteroids and
their metabolites are also excreted into the bile.