Drug Detail:Linzess (Linaclotide [ lin-a-kloe-tide ])
Drug Class: Guanylate cyclase-C agonists
Highlights of Prescribing Information
LINZESS® (linaclotide) capsules, for oral use
Initial U.S. Approval: 2012
WARNING: RISK OF SERIOUS DEHYDRATION IN PEDIATRIC PATIENTS LESS THAN 2 YEARS OF AGE
See full prescribing information for complete boxed warning.
- LINZESS is contraindicated in patients less than 2 years of age; in neonatal mice, linaclotide caused deaths due to dehydration. (4, 5.1, 8.4)
Recent Major Changes
| Indications and Usage, Functional Constipation (1) | 6/2023 |
| Dosage and Administration, Functional Constipation (2.1) | 6/2023 |
| Dosage and Administration (2.2) | 6/2023 |
| Warnings and Precautions (5.2) | 6/2023 |
Indications and Usage for Linzess
LINZESS is a guanylate cyclase-C agonist indicated for treatment of:
- Irritable bowel syndrome with constipation (IBS-C) in adults. (1)
- Chronic idiopathic constipation (CIC) in adults. (1)
- Functional constipation (FC) in pediatric patients 6 to 17 years of age. (1)
Linzess Dosage and Administration
The recommended dosage in adults is:
- IBS-C: 290 mcg orally once daily. (2.1)
- CIC: 145 mcg orally once daily or 72 mcg orally once daily based on individual presentation or tolerability. (2.1)
The recommended dosage in pediatric patients 6 to 17 years is:
- FC: 72 mcg orally once daily. (2.1)
Administration Instructions (2.2):
- Take on empty stomach at least 30 minutes prior to a meal at approximately the same time each day.
- Do not crush or chew LINZESS capsule or capsule contents.
- For patients who have difficulty swallowing capsules whole or those with a nasogastric or gastrostomy tube, see full prescribing information for instructions for opening the capsule and administering with applesauce or water.
Dosage Forms and Strengths
Capsules: 72 mcg, 145 mcg and 290 mcg (3)
Contraindications
- Patients less than 2 years of age. (4, 5.1, 8.4)
- Patients with known or suspected mechanical gastrointestinal obstruction. (4)
Warnings and Precautions
Diarrhea: Patients may experience severe diarrhea. If severe diarrhea occurs, suspend dosing and rehydrate the patient. (5.2)
Adverse Reactions/Side Effects
- Most common adverse reactions (≥2%) reported in adult patients with IBS-C or CIC are: diarrhea, abdominal pain, flatulence and abdominal distension. (6.1)
- Most common adverse reaction (≥2%) reported in pediatric patients 6 to 17 years of age with FC is diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie, Inc. at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Linzess
LINZESS is indicated for the treatment of:
• irritable bowel syndrome with constipation (IBS-C) in adults
• chronic idiopathic constipation (CIC) in adults
• functional constipation (FC) in pediatric patients 6 to 17 years of age
2. Linzess Dosage and Administration
2.1 Recommended Dosage
Irritable Bowel Syndrome with Constipation (IBS-C) in adults
The recommended dosage of LINZESS is 290 mcg orally once daily.
Chronic Idiopathic Constipation (CIC) in adults
The recommended dosage of LINZESS is 145 mcg orally once daily. A dosage of 72 mcg once daily may be used based on individual presentation or tolerability.
Functional Constipation (FC) in pediatric patients 6 to 17 years of age
The recommended dosage of LINZESS is 72 mcg orally once daily.
2.2 Preparation and Administration Instructions
• Take LINZESS on an empty stomach, at least 30 minutes prior to a meal, at approximately the same time each day.
• If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take 2 doses at the same time.
• Do not crush or chew LINZESS capsule or capsule contents.
• Swallow LINZESS capsule whole.
• For patients who are unable to swallow the capsule whole, LINZESS capsules can be opened and administered orally in either applesauce or with water or administered with water via a nasogastric or gastrostomy tube. Sprinkling of LINZESS beads on other soft foods or in other liquids has not been tested.
Oral Administration in Applesauce:
- Place one teaspoonful of room-temperature applesauce into a clean container.
- Open the capsule.
- Sprinkle the entire contents (beads) on applesauce.
- Consume the entire contents immediately. Do not chew the beads. Do not store the bead-applesauce mixture for later use.
Oral Administration in Water:
- Pour approximately 30 mL of room-temperature bottled water into a clean cup.
- Open the capsule.
- Sprinkle the entire contents (beads) into the water.
- Gently swirl beads and water for at least 20 seconds.
- Swallow the entire mixture of beads and water immediately.
- Add another 30 mL of water to any beads remaining in cup, swirl for 20 seconds, and swallow immediately.
- Do not store the bead-water mixture for later use.
Note: The drug is coated on the surface of the beads and will dissolve off the beads into the water. The beads will remain visible and will not dissolve. Therefore, it is not necessary to consume all the beads to deliver the complete dose.
Administration with Water via a Nasogastric or Gastrostomy Tube:
- Open the capsule and empty the beads into a clean container with 30 mL of room-temperature bottled water.
- Mix by gently swirling beads for at least 20 seconds.
- Draw-up the beads and water mixture into an appropriately sized catheter-tipped syringe and apply rapid and steady pressure (10 mL/10 seconds) to dispense the syringe contents into the tube.
- Add another 30 mL of water to any beads remaining in the container and repeat the process.
- After administering the bead-water mixture, flush nasogastric/ gastrostomy tube with a minimum of 10 mL of water.
Note: It is not necessary to flush all the beads through to deliver the complete dose.
3. Dosage Forms and Strengths
LINZESS capsules are white to off-white opaque:
- 72 mcg; gray imprint “FL 72”
- 145 mcg; gray imprint “FL 145”
- 290 mcg; gray imprint “FL 290”
4. Contraindications
LINZESS is contraindicated in:
- Patients less than 2 years of age due to the risk of serious dehydration [see Warnings and Precautions (5.1), Use in Specific Populations (8.4)].
- Patients with known or suspected mechanical gastrointestinal obstruction.
5. Warnings and Precautions
5.1 Risk of Serious Dehydration in Pediatric Patients Less Than 2 Years of Age
LINZESS is contraindicated in patients less than 2 years of age. In neonatal mice (human age equivalent of approximately 0 to 28 days), linaclotide increased fluid secretion as a consequence of age-dependent elevated GC-C agonism which was associated with increased mortality within the first 24 hours due to dehydration. There was no age-dependent trend in GC-C intestinal expression in a clinical study of children 2 to less than 18 years of age; however, there are insufficient data available on GC-C intestinal expression in children less than 2 years of age to assess the risk of developing diarrhea and its potentially serious consequences in these patients [see Warnings and Precautions (5.2) and Use in Specific Populations (8.4)].
5.2 Diarrhea
In adults, diarrhea was the most common adverse reaction of LINZESS-treated patients in the pooled IBS-C and CIC double-blind placebo-controlled trials. The incidence of diarrhea was similar between the IBS-C and CIC populations. Severe diarrhea was reported in 2% of 145 mcg and 290 mcg LINZESS-treated patients, and in <1% of 72 mcg LINZESS-treated CIC patients [see Adverse Reactions (6.1)].
Diarrhea has also been reported in pediatric patients 6 to 17 years of age with FC treated with LINZESS. In a double-blind placebo-controlled trial, diarrhea was the most common adverse reaction and was reported in 4% of pediatric patients 6 to 17 years of age treated with LINZESS 72 mcg once daily. Severe diarrhea was reported in one LINZESS-treated patient [see Adverse Reactions (6.1)].
In post-marketing experience, severe diarrhea associated with dizziness, syncope, hypotension and electrolyte abnormalities (hypokalemia and hyponatremia) requiring hospitalization or intravenous fluid administration have been reported in patients treated with LINZESS.
If severe diarrhea occurs, suspend dosing and rehydrate the patient.
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Exposure in clinical development included approximately 2570, 2040, and 1220 adult patients with either IBS-C or CIC, treated with LINZESS for 6 months or longer, 1 year or longer, and 18 months or longer, respectively (not mutually exclusive).
Demographic characteristics were comparable between treatment groups in all studies [see Clinical Studies (14.1, 14.2)].
Irritable Bowel Syndrome with Constipation (IBS-C) in Adults
Most Common Adverse Reactions
The data described below reflect exposure to LINZESS in the two placebo-controlled clinical trials involving 1605 adult patients with IBS-C (Trials 1 and 2) [see Clinical Studies (14.1)]. Patients were randomized to receive placebo or 290 mcg LINZESS once daily on an empty stomach for up to 26 weeks. Table 1 provides the incidence of adverse reactions reported in at least 2% of IBS-C patients in the LINZESS treatment group and at an incidence that was greater than in the placebo group.
| Adverse Reactions
| LINZESS
290 mcg [N=807] % | Placebo
[N=798] % |
| Gastrointestinal
Diarrhea Abdominal painb Flatulence Abdominal distension |
20 7 4 2 |
3 5 2 1 |
| Infections and Infestations
Viral Gastroenteritis |
3 |
1 |
| Nervous System Disorders
Headache |
4 |
3 |
a: Reported in at least 2% of LINZESS-treated patients and at an incidence greater than placebo
b: “Abdominal pain” term includes abdominal pain, upper abdominal pain, and lower abdominal pain.
Adverse reactions in an additional placebo-controlled trial in 614 IBS-C patients randomized to placebo or LINZESS 290 mcg once daily on an empty stomach for 12 weeks (Trial 6) were similar to those in Table 1.
Diarrhea
Diarrhea was the most commonly reported adverse reaction of the LINZESS-treated patients in the pooled IBS-C pivotal placebo-controlled trials. In these trials, 20% of LINZESS-treated patients reported diarrhea compared to 3% of placebo-treated patients. Severe diarrhea was reported in 2% of the LINZESS-treated patients versus less than 1% of the placebo-treated patients, and 5% of LINZESS-treated patients discontinued due to diarrhea vs less than 1% of placebo-treated patients. The majority of reported cases of diarrhea started within the first 2 weeks of LINZESS treatment [see Warnings and Precautions (5.2)].
Adverse Reactions Leading to Discontinuation
In placebo-controlled trials in patients with IBS-C, 9% of patients treated with LINZESS and 3% of patients treated with placebo discontinued prematurely due to adverse reactions. In the LINZESS-treatment group, the most common reasons for discontinuation due to adverse reactions were diarrhea (5%) and abdominal pain (1%). In comparison, less than 1% of patients in the placebo group withdrew due to diarrhea or abdominal pain.
Adverse Reactions Leading to Dose Reductions
In the open-label, long-term trials, 2147 patients with IBS-C received 290 mcg of LINZESS daily for up to 18 months. In these trials, 29% of patients had their dose reduced or suspended secondary to adverse reactions, the majority of which were diarrhea or other GI adverse reactions.
Less Common Adverse Reactions
Defecation urgency, fecal incontinence, vomiting, and gastroesophageal reflux disease were reported in <2% of patients in the LINZESS-treatment group and at an incidence greater than in the placebo treatment group.
Chronic Idiopathic Constipation (CIC) in Adults
Most Common Adverse Reactions
The data described below reflect exposure to LINZESS in the two double-blind placebo-controlled clinical trials of 1275 adult patients with CIC (Trials 3 and 4) [see Clinical Studies (14.2)]. Patients were randomized to receive placebo or 145 mcg LINZESS or 290 mcg LINZESS once daily on an empty stomach, for at least 12 weeks. Table 2 provides the incidence of adverse reactions reported in at least 2% of CIC patients in the 145 mcg LINZESS treatment group and at an incidence that was greater than in the placebo treatment group.
| Adverse Reactions | LINZESS
145 mcg [N=430] % | Placebo
[N=423] % |
| Gastrointestinal
Diarrhea Abdominal painb Flatulence Abdominal distension |
16 7 6 3 |
5 6 5 2 |
| Infections and Infestations
Upper respiratory tract infection Sinusitis |
5 3 |
4 2 |
a: Reported in at least 2% of LINZESS-treated patients and at an incidence greater than placebo
b: “Abdominal pain” term includes abdominal pain, upper abdominal pain, and lower abdominal pain.
The safety of a 72 mcg dose was evaluated in an additional placebo-controlled trial in which 1223 patients were randomized to LINZESS 72 mcg, 145 mcg, or placebo once daily for 12 weeks (Trial 5).
In Trial 5, adverse reactions that occurred at a frequency of ≥ 2% in LINZESS-treated patients (N=411 in each LINZESS 72 mcg and 145 mcg group) and at a higher rate than placebo (N=401) were:
- Diarrhea (LINZESS 72 mcg 19%; LINZESS 145 mcg 22%; placebo 7%)
- Abdominal distension (LINZESS 72 mcg 2%; LINZESS 145 mcg 1%; placebo < 1%)
Diarrhea
In Trials 3 and 4 (pooled) and Trial 5, diarrhea was the most commonly reported adverse reaction in LINZESS-treated patients in the CIC placebo-controlled studies.
In all trials, the majority of reported cases of diarrhea started within the first 2 weeks of LINZESS treatment.
Severe diarrhea was reported in less than 1% of the 72 mcg LINZESS-treated patients (Trial 5), in 2% of the 145 mcg LINZESS-treated patients (Trials 3, 4, and 5), and less than 1% of the placebo-treated patients (Trials 3, 4, and 5) [see Warnings and Precautions (5.2)].
Adverse Reactions Leading to Discontinuation
In placebo-controlled trials in patients with CIC, 3% of patients treated with 72 mcg (Trial 5) and between 5% and 8% (Trials 3, 4, and 5) of patients treated with 145 mcg of LINZESS discontinued prematurely due to adverse reactions compared to between less than 1% and 4% (Trials 3, 4, and 5) of patients treated with placebo.
In patients treated with 72 mcg LINZESS, the most common reason for discontinuation due to adverse reactions was diarrhea (2% in Trial 5) and, in patients treated with 145 mcg LINZESS, the most common reasons for discontinuation due to adverse reactions were diarrhea (between 3% and 5% in Trials 3, 4, and 5) and abdominal pain (1% in Trials 3 and 4). In comparison, less than 1% of patients in the placebo group withdrew due to diarrhea or abdominal pain (Trials 3, 4, and 5).
Adverse Reactions Leading to Dose Reductions
In the open-label, long-term trials, 1129 patients with CIC received 290 mcg of LINZESS daily for up to 18 months. In these trials, 27% of patients had their dose reduced or suspended secondary to adverse reactions, the majority of which were diarrhea or other GI adverse reactions.
Less Common Adverse Reactions
Defecation urgency, fecal incontinence, dyspepsia, and viral gastroenteritis were reported in less than 2% of patients in the LINZESS treatment group and at an incidence greater than in the placebo treatment group.
Functional Constipation (FC) in Pediatric Patients 6 to 17 Years of Age
The safety of LINZESS 72 mcg once daily was evaluated in pediatric patients 6 to 17 years of age with FC in a 12-week double-blind, placebo-controlled clinical trial (Trial 7) [see Clinical Studies (14.3)]. There were 164 patients per treatment group.
Diarrhea was the most common adverse reaction and was reported in 4% of LINZESS-treated patients compared to 2% of placebo-treated patients. One patient in the LINZESS-treated group reported severe diarrhea and discontinued treatment. No patient in the placebo-treated group discontinued treatment due to severe diarrhea. Most reported cases of diarrhea started within the first 2 weeks of LINZESS treatment [see Warnings and Precautions (5.2)].
Other adverse reactions reported at a higher incidence in the LINZESS group than the placebo group included nausea (2 patients) and abdominal discomfort and dehydration (1 patient each).
8. Use In Specific Populations
8.4 Pediatric Use
LINZESS is contraindicated in patients less than 2 years of age. In nonclinical studies, deaths occurred within 24 hours in neonatal mice (human age equivalent of approximately 0 to 28 days) following oral administration of linaclotide which increased fluid secretion as a consequence of age-dependent elevated GC-C agonism resulting in rapid and severe dehydration (see Juvenile Animal Toxicity Data).
A clinical GC-C ontogeny study in children 6 months to less than 18 years of age (N=99) was conducted to measure GC-C mRNA expression levels in duodenal and colonic samples to evaluate the risk of diarrhea and severe dehydration due to GC-C agonism. The results showed no age dependent trend in GC-C intestinal expression in children 2 to less than 18 years of age. There was insufficient data on GC-C intestinal expression to assess the risk of developing diarrhea and its potentially serious consequences in children less than 2 years of age [see Warnings and Precautions (5.1)].
The safety and effectiveness of LINZESS for the treatment of FC in pediatric patients 6 to 17 years of age have been established. Use of LINZESS for this indication is supported by evidence from adequate and well-controlled studies in adults and pediatric patients 6 years of age and older. The safety of LINZESS in adult and pediatric patients 6 to 17 years of age in clinical studies was similar [see Adverse Reactions (6.1) and Clinical Studies (14.3)].
The safety and effectiveness of LINZESS in patients with FC less than 6 years of age or in patients with IBS-C less than 18 years of age have not been established.
Juvenile Animal Toxicity Data
In toxicology studies in neonatal mice, oral administration of linaclotide at 10 mcg/kg/day caused deaths on post-natal day 7 (human age equivalent of approximately 0 to 28 days). These deaths were due to rapid and severe dehydration produced by significant fluid shifts into the intestinal lumen resulting from GC-C agonism in neonatal mice [see Contraindications (4) and Warnings and Precautions (5.1)].
Tolerability to linaclotide increases with age in juvenile mice. In 2-week-old mice, linaclotide was well tolerated at a dose of 50 mcg/kg/day, but deaths occurred after a single oral dose of 100 mcg/kg. In 3-week-old mice, linaclotide was well tolerated at 100 mcg/kg/day, but deaths occurred after a single oral dose of 600 mcg/kg.
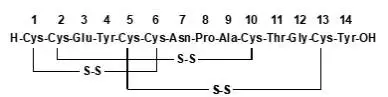
12. Linzess - Clinical Pharmacology
12.3 Pharmacokinetics
Absorption
LINZESS is minimally absorbed with negligible systemic availability following oral administration. Concentrations of linaclotide and its active metabolite in plasma are below the limit of quantitation after oral doses of 72 mcg, 145 mcg, or 290 mcg were administered. Therefore, standard pharmacokinetic parameters such as area under the curve (AUC), maximum concentration (Cmax), and half-life (t½) cannot be calculated.
Food Effect
Neither linaclotide nor its active metabolite were detected in the plasma following administration of LINZESS 290 mcg once daily for 7 days both in the non-fed and fed state in healthy subjects.
Distribution
Given that linaclotide plasma concentrations following recommended oral doses are not measurable, linaclotide is not expected to be distributed to tissues to any clinically relevant extent.
Elimination
Metabolism
Linaclotide is metabolized within the gastrointestinal tract to its principal, active metabolite by loss of the terminal tyrosine moiety. Both linaclotide and the metabolite are proteolytically degraded within the intestinal lumen to smaller peptides and naturally occurring amino acids.
Excretion
Active peptide recovery in the stool samples of fed and fasted healthy subjects following administration of LINZESS 290 mcg once daily for seven days averaged about 5% (fasted) and about 3% (fed) and all of it as the active metabolite.
Specific Populations
Renal and Hepatic Impairment
Renal or hepatic impairment is not expected to affect the clearance of linaclotide or the active metabolite because linaclotide metabolism occurs within the gastrointestinal tract and plasma concentrations are not measurable in plasma following administration of the recommended dosage.
Drug Interaction Studies
No drug-drug interaction studies have been conducted with LINZESS. Systemic exposures of drug and active metabolite are negligible following oral administration.
Linaclotide does not interact with the cytochrome P450 enzyme system based on the results of in vitro studies. In addition, linaclotide does not interact with common efflux and uptake transporters (including the efflux transporter P-glycoprotein (P-gp)). Based on these in vitro data no drug-drug interactions through modulation of CYP enzymes or common transporters are anticipated.
14. Clinical Studies
14.1 Irritable Bowel Syndrome with Constipation (IBS-C) in Adults
The efficacy of LINZESS for the treatment of IBS-C was established in two double-blind, placebo-controlled, randomized, multicenter trials in adult patients (Trials 1 (NCT00948818) and 2 (NCT00938717)). A total of 800 patients in Trial 1 and 804 patients in Trial 2 [overall mean age of 44 years (range 18 to 87 years), 90% female, 77% white, 19% black, and 12% Hispanic] received treatment with LINZESS 290 mcg or placebo once daily and were evaluated for efficacy. All patients met Rome II criteria for IBS and were required, during the 2-week baseline period, to meet the following criteria:
- a mean abdominal pain score of at least 3 on a 0-to-10-point numeric rating scale
- less than 3 complete spontaneous bowel movements (CSBMs) per week [a CSBM is a spontaneous bowel movement (SBM) that is associated with a sense of complete evacuation; a SBM is a bowel movement occurring in the absence of laxative use], and
- less than or equal to 5 SBMs per week.
The trial designs were identical through the first 12 weeks, and thereafter differed only in that Trial 1 included a 4-week randomized withdrawal (RW) period, and Trial 2 continued for 14 additional weeks (total of 26 weeks) of double-blind treatment. During the trials, patients were allowed to continue stable doses of bulk laxatives or stool softeners but were not allowed to take laxatives, bismuth, prokinetic agents, or other drugs to treat IBS-C or chronic constipation.
Efficacy of LINZESS was assessed using overall responder analyses and change-from-baseline endpoints. Results for endpoints were based on information provided daily by patients in diaries.
The 4 primary efficacy responder endpoints were based on a patient being a weekly responder for either at least 9 out of the first 12 weeks of treatment or at least 6 out of the first 12 weeks of treatment. For the 9 out of 12 weeks combined primary responder endpoint, a patient had to have at least a 30% reduction from baseline in mean abdominal pain, at least 3 CSBMs and an increase of at least 1 CSBM from baseline, all in the same week, for at least 9 out of the first 12 weeks of treatment. Each of the 2 components of the 9 out of 12 weeks combined responder endpoint, abdominal pain and CSBMs, was also a primary endpoint.
For the 6 out of 12 weeks combined primary responder endpoint, a patient had to have at least a 30% reduction from baseline in mean abdominal pain and an increase of at least 1 CSBM from baseline, all in the same week, for at least 6 out of the first 12 weeks of treatment. To be considered a responder for this analysis, patients did not have to have at least 3 CSBMs per week.
The efficacy results for the 9 out of 12 weeks and the 6 out of 12 weeks responder endpoints are shown in Tables 3 and 4, respectively. In both trials, the proportion of patients who were responders to LINZESS 290 mcg was statistically significantly higher than with placebo.
| Trial 1 | Trial 2 | |||||
| LINZESS
290 mcg Once Daily (N=405) | Placebo
(N=395) | Treatment Difference
[95% CI] | LINZESS
290 mcg Once Daily (N=401) | Placebo
(N=403) | Treatment Difference
[95% CI] |
|
| Combined Responder*
(Abdominal Pain and CSBM Responder) | 12% | 5% | 7% [3.2%, 10.9%] | 13% | 3% | 10% [6.1%, 13.4%] |
| Abdominal Pain Responder* (≥ 30% Abdominal Pain Reduction) | 34% | 27% | 7% [0.9%, 13.6%] | 39% | 20% | 19% [13.2%, 25.4%] |
| CSBM Responder* (≥ 3 CSBMs and Increase ≥1 CSBM from Baseline) | 20% | 6% | 13% [8.6%, 17.7%] | 18% | 5% | 13% [8.7%, 17.3%] |
| * Primary Endpoints Note: Analyses based on first 12 weeks of treatment for both Trials 1 and 2 CI =Confidence Interval |
||||||
| Trial 1 | Trial 2 | |||||
| LINZESS
290 mcg Once Daily (N=405) | Placebo
(N=395) | Treatment Difference
[95% CI] | LINZESS
290 mcg Once Daily (N=401) | Placebo
(N=403) | Treatment Difference
[95% CI] |
|
| Combined Responder*
(Abdominal Pain and CSBM Responder) | 34% | 21% | 13% [6.5%, 18.7%] | 34% | 14% | 20% [14.0%, 25.5%] |
| Abdominal Pain Responder** (≥ 30% Abdominal Pain Reduction) | 50% | 37% | 13% [5.8%, 19.5%] | 49% | 34% | 14% [7.6%, 21.1%] |
| CSBM Responder** (Increase ≥ 1 CSBM from Baseline) | 49% | 30% | 19% [12.4%, 25.7%] | 48% | 23% | 25% [18.7%, 31.4%] |
| * Primary Endpoint, ** Secondary Endpoints Note: Analyses based on first 12 weeks of treatment for both Trials 1 and 2 CI =Confidence Interval |
||||||
In each trial, improvement from baseline in abdominal pain and CSBM frequency was seen over the first 12-weeks of the treatment periods. For change from baseline in the 11-point abdominal pain scale, LINZESS 290 mcg began to separate from placebo in the first week. Maximum effects were seen at weeks 6 - 9 and were maintained until the end of the study. The mean treatment difference from placebo at week 12 was a decrease in pain score of approximately 1.0 point in both trials (using an 11-point scale). Maximum effect on CSBM frequency occurred within the first week, and for change from baseline in CSBM frequency at week 12, the difference between placebo and LINZESS was approximately 1.5 CSBMs per week in both trials.
In each trial, in addition to improvements in abdominal pain and CSBM frequency over the first 12 weeks of the treatment period, improvements were observed in the following when LINZESS was compared to placebo: SBM frequency [SBMs/week], stool consistency [as measured by the Bristol Stool Form Scale (BSFS)], and amount of straining with bowel movements [amount of time pushing or physical effort to pass stool].
During the 4-week randomized withdrawal period in Trial 1, patients who received LINZESS during the 12-week treatment period were re-randomized to receive placebo or continue treatment on LINZESS 290 mcg. In LINZESS-treated patients re-randomized to placebo, CSBM frequency and abdominal-pain severity returned toward baseline within 1 week and did not result in worsening compared to baseline. Patients who continued on LINZESS maintained their response to therapy over the additional 4 weeks. Patients on placebo who were allocated to LINZESS had an increase in CSBM frequency and a decrease in abdominal pain levels that were similar to the levels observed in patients taking LINZESS during the treatment period.
Trial 6 (NCT03573908) was a randomized, double-blind, placebo-controlled, parallel-group trial that evaluated the safety and efficacy of LINZESS in patients with IBS-C over a 12-week treatment period followed by a 4-week randomized withdrawal period. A total of 614 patients [mean age of 47 years (range 18 to 85 years), 81% female, 63% white, 24% black, and 27% Hispanic] received treatment with LINZESS 290 mcg or placebo once daily and all patients met Rome III criteria for IBS-C.
The efficacy of LINZESS was assessed using a primary endpoint based on the mean abdominal score (composite of abdominal bloating, abdominal discomfort, and abdominal pain) across 12 weeks. The secondary endpoint was a responder analysis based on at least a 2.5-point improvement in the abdominal score from baseline for at least 6 out of 12 weeks as shown in Table 5.
| Trial 6 | |||
| LINZESS
290 mcg Once Daily (N=306) | Placebo
(N=308) | Treatment Difference
[95% CI] |
|
| Baseline Abdominal Score | 6.4 | 6.5 | |
| Least Squares 12-week Mean Change from Baseline in Abdominal Score* | -1.9 | -1.2 | -0.7 [-1.0, -0.4] |
| Abdominal Score 6 of 12-Week Responder** | 34% | 18.5% | 15.5% [8.6%, 22.3%] |
| * Primary Endpoint, ** Secondary Endpoint Each abdominal symptom was rated on a 0-to-10-point numeric rating scale where 0=no [symptom] and 10=worst possible [symptom]. CI = Confidence Interval |
|||
14.2 Chronic Idiopathic Constipation (CIC) in Adults
The efficacy of LINZESS for the treatment of CIC was established in two double-blind, placebo-controlled, randomized, multicenter clinical trials in adult patients (Trials 3 and 4). A total of 642 patients in Trial 3 and 630 patients in Trial 4 [overall mean age of 48 years (range 18 to 85 years), 89% female, 76% white, 22% black, 10% Hispanic] received treatment with LINZESS 145 mcg, 290 mcg, or placebo once daily and were evaluated for efficacy. All patients met modified Rome II criteria for functional constipation. Modified Rome II criteria were less than 3 Spontaneous Bowel Movements (SBMs) per week and 1 of the following symptoms for at least 12 weeks, which need not be consecutive, in the preceding 12 months:
- Straining during greater than 25% of bowel movements
- Lumpy or hard stools during greater than 25% of bowel movements
- Sensation of incomplete evacuation during greater than 25% of bowel movements
Patients were also required to have less than 3 CSBMs per week and less than or equal to 6 SBMs per week during a 2-week baseline period. Patients were excluded if they met criteria for IBS-C or had fecal impaction that required emergency room treatment.
The trial designs were identical through the first 12 weeks. Trial 3 also included an additional 4-week randomized withdrawal (RW) period. During the trials, patients were allowed to continue stable doses of bulk laxatives or stool softeners but were not allowed to take laxatives, bismuth, prokinetic agents, or other drugs to treat chronic constipation.
The efficacy of LINZESS was assessed using a responder analysis and change-from-baseline endpoints. Results for endpoints were based on information provided daily by patients in diaries.
A CSBM responder in the CIC trials was defined as a patient who had at least 3 CSBMs and an increase of at least 1 CSBM from baseline in a given week for at least 9 weeks out of the 12-week treatment period. The CSBM responder rates are shown in Table 6. During the individual double-blind placebo-controlled trials, LINZESS 290 mcg did not consistently offer additional clinically meaningful treatment benefit over placebo than that observed with the LINZESS 145 mcg dose. Therefore, the 145 mcg dose is the recommended dose. Only the data for the approved 145 mcg dose of LINZESS are presented in Table 6.
In Trials 3 and 4, the proportion of patients who were CSBM responders was statistically significantly greater with the LINZESS 145 mcg dose than with placebo.
| Trial 3 | Trial 4 | |||||
| LINZESS
145 mcg Once Daily (N=217) | Placebo
(N=209) | Treatment Difference
[95% CI] | LINZESS
145 mcg Once Daily (N=213) | Placebo
(N=215) | Treatment Difference
[95% CI] |
|
| CSBM Responder*
(≥ 3 CSBMs and Increase ≥ 1 CSBM from Baseline) | 20% | 3% | 17% [11.0%, 22.8%] | 15% | 6% | 10% [4.2%, 15.7%] |
| *Primary Endpoint CI=Confidence Interval |
||||||
CSBM frequency reached maximum level during week 1 and was also demonstrated over the remainder of the 12-week treatment period in Trial 3 and Trial 4. For the mean change from baseline in CSBM frequency at week 12, the difference between placebo and LINZESS was approximately 1.5 CSBMs.
On average, patients who received LINZESS across the 2 trials had significantly greater improvements compared with patients receiving placebo in stool frequency (CSBMs/week and SBMs/week), and stool consistency (as measured by the BSFS).
In each trial, in addition to improvements in CSBM frequency over the first 12 weeks of the treatment period, improvements were observed in each of the following when LINZESS was compared to placebo: SBM frequency [SBMs/week], stool consistency [as measured by the BSFS], and amount of straining with bowel movements [amount of time pushing or physical effort to pass stool].
During the 4-week randomized withdrawal period in Trial 3, patients who received LINZESS during the 12-week treatment period were re-randomized to receive placebo or continue treatment on the same dose of LINZESS taken during the treatment period. In LINZESS-treated patients re-randomized to placebo, CSBM and SBM frequency returned toward baseline within 1 week and did not result in worsening compared to baseline. Patients who continued on LINZESS maintained their response to therapy over the additional 4 weeks. Patients on placebo who were allocated to LINZESS had an increase in CSBM and SBM frequency similar to the levels observed in patients taking LINZESS during the treatment period.
A 72 mcg dose of LINZESS was established in a randomized, double-blind, placebo-controlled, multicenter clinical trial in adult patients (Trial 5). A total of 1223 patients [overall mean age of 46 years (range 18 to 90 years), 77% female, 71% white, 24% black, 43% Hispanic] received treatment with LINZESS 72 mcg or placebo once daily and were evaluated for efficacy. All patients met modified Rome III criteria for functional constipation. Trial 5 was identical to Trials 3 and 4 through the first 12 weeks. The efficacy of the 72 mcg dose was assessed using a responder analysis where a CSBM responder was defined as a patient who had at least 3 CSBMs and an increase of at least 1 CSBM from baseline in a given week for at least 9 weeks out of the 12-week treatment period, which was the same as the one defined in Trials 3 and 4. The response rates for the CSBM responder endpoint were 13% for LINZESS 72 mcg and 5% for placebo. The difference between LINZESS 72 mcg and placebo was 9% (95% CI: 4.8%, 12.5%).
A separate analysis was performed using an alternate CSBM responder definition. In this analysis a CSBM responder was defined as a patient who had at least 3 CSBMs and an increase of at least 1 CSBM from baseline in a given week for at least 9 weeks out of the 12-week treatment period and at least 3 of the last 4 weeks of the treatment period. The response rates for the alternate CSBM responder endpoint were 12% for LINZESS 72 mcg and 5% for placebo. The difference between LINZESS 72 mcg and placebo was 8% (95% CI: 3.9%, 11.5%).
14.3 Functional Constipation (FC) in Pediatric Patients 6 to 17 Years of Age
The efficacy of LINZESS for the treatment of FC in pediatric patients 6 to 17 years of age was established in a 12-week double-blind, placebo-controlled, randomized, multicenter, clinical trial (Trial 7; NCT04026113). A total of 328 patients received treatment with LINZESS 72 mcg or placebo once daily and were evaluated for efficacy. Patients in the trial had a mean age of 11 years (range 6 to 17 years); 55% were female; 45% identified as Hispanic or Latino; 70% identified as White, 26% as Black or African American, 2% as Asian, and 2% identified as another racial group. For trial enrollment, Rome III criteria for child/adolescent FC were modified to require that patients have less than 3 Spontaneous Bowel Movements (SBMs) per week (defined as a BM that occurred in the absence of laxative, enema, or suppository use on the calendar day of or before the BM) and 1 or more of the following criteria at least once per week for at least 2 months before the screening visit:
- History of stool withholding or excessive voluntary stool retention
- History of painful or hard bowel movements (BMs)
- History of large diameter stools that may obstruct the toilet
- Presence of a large fecal mass in the rectum
- At least 1 episode of fecal incontinence per week
Patients were also required to have an average of less than 3 SBMs per week during the 2-week baseline period. Patients were excluded if they met criteria for pediatric IBS-C or had fecal impaction. Patients were allowed to continue previously stable doses of bulk laxatives, fiber, stool softeners, or probiotics. During the trial, patients could use bisacodyl or senna as needed, but were not allowed to take other laxatives, bismuth, prokinetic agents, or other drugs to treat functional constipation.
The efficacy of LINZESS in the treatment of FC in pediatric patients 6 to 17 years of age was assessed using change-from-baseline endpoints. The primary efficacy endpoint was the 12-week change from baseline in SBM frequency rate. The results demonstrated that patients who received LINZESS had statistically significant improvements compared with placebo as shown in Table 7.
| Trial 7 | |||
| LINZESS 72 mcg Once Daily
(N=164) | Placebo (N=164) | Treatment Difference [95% CI] | |
| Baseline SBM Frequency Rate
| 1.2 | 1.3 | |
| Least Squares 12-week Mean Change from Baseline in SBM Frequency Rate* | 2.6 | 1.3 | 1.3 [0.7, 1.8] |
| * Primary Endpoint CI = Confidence Interval |
|||
SBM frequency improved during week 1 and was maintained throughout the remainder of the 12-week treatment period.
Medication Guide
| MEDICATION GUIDE
LINZESS® (lin-ZESS) (linaclotide) capsules, for oral use |
What is the most important information I should know about LINZESS?
|
| What is LINZESS?
LINZESS is a prescription medicine used to treat:
|
Who should not take LINZESS?
|
Before you take LINZESS, tell your doctor about all of your medical conditions, including if you:
|
How should I take LINZESS?
Taking LINZESS in applesauce:
Gather the supplies you will need to take your LINZESS dose. Your doctor should tell you what size catheter tipped syringe you will need for your dose. Ask your doctor if you have any questions about how to give LINZESS the right way.
|
| What are the possible side effects of LINZESS?
LINZESS can cause serious side effects, including:
These are not all the possible side effects of LINZESS. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
How should I store LINZESS?
|
| General information about the safe and effective use of LINZESS.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use LINZESS for a condition for which it was not prescribed. Do not give LINZESS to other people, even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information about LINZESS that is written for health professionals. |
| What are the ingredients in LINZESS?
Active ingredient: linaclotide Inactive ingredients for the 145 mcg and 290 mcg capsules: calcium chloride dihydrate, hypromellose, L-leucine, and microcrystalline cellulose. Capsule shell: gelatin and titanium dioxide. Inactive ingredients for the 72 mcg capsules: calcium chloride dihydrate, L-histidine, microcrystalline cellulose, polyvinyl alcohol, and talc. Capsule shell: gelatin and titanium dioxide. LINZESS® is a registered trademark of Ironwood Pharmaceuticals, Inc. Marketed by: AbbVie, Inc. North Chicago, IL 60064 and Ironwood Pharmaceuticals, Inc. Boston, MA 02110 © 2023 AbbVie and Ironwood Pharmaceuticals, Inc. All rights reserved. For more information, go to www.LINZESS.com or call 1-800-678-1605. |
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: 6/2023
V4.0MG1201



| LINZESS
linaclotide capsule, gelatin coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| LINZESS
linaclotide capsule, gelatin coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| LINZESS
linaclotide capsule, gelatin coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Allergan, Inc. (144796497) |
