Drug Detail:Nilandron (Nilutamide [ nih-lute-ah-mide ])
Drug Class: Antiandrogens Hormones / antineoplastics
Interstitial Pneumonitis
Interstitial pneumonitis has been reported in 2% of patients in controlled clinical trials in patients exposed to nilutamide. A small study in Japanese subjects showed that 8 of 47 patients (17%) developed interstitial pneumonitis. Reports of interstitial changes including pulmonary fibrosis that led to hospitalization and death have been reported rarely post-marketing. Symptoms included exertional dyspnea, cough, chest pain, and fever. X-rays showed interstitial or alveolo-interstitial changes, and pulmonary function tests revealed a restrictive pattern with decreased DLco. Most cases occurred within the first 3 months of treatment with NILANDRON, and most reversed with discontinuation of therapy.
A routine chest X-ray should be performed prior to initiating treatment with NILANDRON. Baseline pulmonary function tests may be considered. Patients should be instructed to report any new or worsening shortness of breath that they experience while on NILANDRON.
If symptoms occur, NILANDRON should be immediately discontinued until it can be determined if the symptoms are drug related.
Nilandron Description
NILANDRON ® tablets contain nilutamide, a nonsteroidal, orally active antiandrogen having the chemical name 5,5-dimethyl-3-[4-nitro-3-(trifluoromethyl)phenyl]-2,4-imidazolidinedione with the following structural formula:
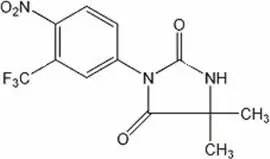
Nilutamide is a microcrystalline, white to practically white powder with a molecular weight of 317.25. Its molecular formula is C 12H 10F 3N 3O 4.
It is freely soluble in ethyl acetate, acetone, chloroform, ethyl alcohol, dichloromethane, and methanol. It is slightly soluble in water [<0.1% W/V at 25°C (77°F)]. It melts between 153°C and 156°C (307.4°F and 312.8°F).
Each NILANDRON tablet contains 150 mg of nilutamide. Other ingredients in NILANDRON tablets are corn starch, lactose, povidone, docusate sodium, magnesium stearate, and talc.
Nilandron - Clinical Pharmacology
Mechanism of Action
Prostate cancer is known to be androgen sensitive and responds to androgen ablation. In animal studies, nilutamide has demonstrated antiandrogenic activity without other hormonal (estrogen, progesterone, mineralocorticoid, and glucocorticoid) effects. In vitro, nilutamide blocks the effects of testosterone at the androgen receptor level. In vivo, nilutamide interacts with the androgen receptor and prevents the normal androgenic response.
Pharmacokinetics
Absorption:
Analysis of blood, urine, and feces samples following a single oral 150-mg dose of [ 14C]-nilutamide in patients with metastatic prostate cancer showed that the drug is rapidly and completely absorbed and that it yields high and persistent plasma concentrations.
Distribution:
After absorption of the drug, there is a detectable distribution phase. There is moderate binding of the drug to plasma proteins and low binding to erythrocytes. The binding is nonsaturable except in the case of alpha-1-glycoprotein, which makes a minor contribution to the total concentration of proteins in the plasma. The results of binding studies do not indicate any effects that would cause nonlinear pharmacokinetics.
Metabolism:
The results of a human metabolism study using 14C-radiolabelled tablets show that nilutamide is extensively metabolized and less than 2% of the drug is excreted unchanged in urine after 5 days. Five metabolites have been isolated from human urine. Two metabolites display an asymmetric center, due to oxidation of a methyl group, resulting in the formation of D- and L-isomers. One of the metabolites was shown, in vitro, to possess 25 to 50% of the pharmacological activity of the parent drug, and the D-isomer of the active metabolite showed equal or greater potency compared to the L-isomer. However, the pharmacokinetics and the pharmacodynamics of the metabolites have not been fully investigated.
Elimination:
The majority (62%) of orally administered [ 14C]-nilutamide is eliminated in the urine during the first 120 hours after a single 150-mg dose. Fecal elimination is negligible, ranging from 1.4% to 7% of the dose after 4 to 5 days. Excretion of radioactivity in urine likely continues beyond 5 days. The mean elimination half-life of nilutamide determined in studies in which subjects received a single dose of 100–300 mg ranged from 38.0 to 59.1 hours with most values between 41 and 49 hours. The elimination of at least one metabolite is generally longer than that of unchanged nilutamide (59–126 hours). During multiple dosing of 150 mg nilutamide (given as 3 × 50 mg) twice a day, steady state was reached within 2 to 4 weeks for most patients, and mean steady state AUC 0–12 was 110% higher than the AUC 0–∞ obtained from the first 150 mg dose. These data and in vitro metabolism data suggest that, upon multiple dosing, metabolic enzyme inhibition may occur for this drug.
Clinical Studies
Nilutamide through its antiandrogenic activity can complement surgical castration, which suppresses only testicular androgens. The effects of the combined therapy were studied in patients with previously untreated metastatic prostate cancer.
In a double-blind, randomized, multicenter study that enrolled 457 patients (225 treated with orchiectomy and NILANDRON, 232 treated with orchiectomy and placebo), the NILANDRON group showed a statistically significant benefit in time to progression and time to death. The results are summarized below.
| NILANDRON
| PLACEBO
|
|
| Median Survival (months)
| 27.3
| 23.6
|
| Progression-Free Survival (months)
| 21.1
| 14.9
|
| Complete or Partial Regression
| 41%
| 24%
|
| Improvement in Bone Pain
| 54%
| 37%
|
Contraindications
NILANDRON tablets are contraindicated:
- in patients with severe hepatic impairment (baseline hepatic enzymes should be evaluated prior to treatment)
- in patients with severe respiratory insufficiency
- in patients with hypersensitivity to nilutamide or any component of this preparation.
Warnings
Hepatitis
Rare cases of death or hospitalization due to severe liver injury have been reported post-marketing in association with the use of NILANDRON. Hepatotoxicity in these reports generally occurred within the first 3 to 4 months of treatment. Hepatitis or marked increases in liver enzymes leading to drug discontinuation occurred in 1% of NILANDRON patients in controlled clinical trials.
Serum transaminase levels should be measured prior to starting treatment with NILANDRON, at regular intervals for the first 4 months of treatment, and periodically thereafter. Liver function tests should also be obtained at the first sign or symptom suggestive of liver dysfunction, e.g. nausea, vomiting, abdominal pain, fatigue, anorexia, "flu-like" symptoms, dark urine, jaundice or right upper quadrant tenderness. If at any time, a patient has jaundice or their ALT rises above 2 times the upper limit of normal, NILANDRON should be immediately discontinued with close followup of liver function tests until resolution.
Precautions
Information For Patients
Patients should be informed that NILANDRON tablets should be started on the day of or on the day after, surgical castration. They should also be informed that they should not interrupt their dosing of NILANDRON or stop taking this medication without consulting their physician.
Because of the possibility of interstitial pneumonitis, patients should also be told to report immediately any dyspnea or aggravation of pre-existing dyspnea.
Because of the possibility of hepatitis, patients should be told to consult with their physician should nausea, vomiting, abdominal pain or jaundice occur.
Because of the possibility of an intolerance to alcohol (facial flushes, malaise, hypotension) following ingestion of NILANDRON, it is recommended that intake of alcoholic beverages be avoided by patients who experience this reaction. This effect has been reported in about 5% of patients treated with NILANDRON.
In clinical trials, 13% to 57% of patients receiving NILANDRON reported a delay in adaptation to dark, ranging from seconds to a few minutes, when passing from a lighted area to a dark area. This effect sometimes does not abate as drug treatment is continued. Patients who experience this effect should be cautioned about driving at night or through tunnels. This effect can be alleviated by the wearing of tinted glasses.
Drug Interactions
In vitro, nilutamide has been shown to inhibit the activity of liver cytochrome P-450 isoenzymes and, therefore, may reduce the metabolism of compounds requiring these systems.
Consequently, drugs with a low therapeutic margin, such as vitamin K antagonists, phenytoin, and theophylline, could have a delayed elimination and increases in their serum half-life leading to a toxic level. The dosage of these drugs or others with a similar metabolism may need to be modified if they are administered concomitantly with nilutamide. For example, when vitamin K antagonists are administered concomitantly with nilutamide, prothrombin time should be carefully monitored and, if necessary, the dosage of vitamin K antagonists should be reduced.
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Administration of nilutamide to rats for 18 months at doses of 0, 5, 15, or 45 mg/kg/day produced benign Leydig cell tumors in 35% of the high-dose male rats (AUC exposures in high-dose rats were approximately 1–2 times human AUC exposures with therapeutic doses). The increased incidence of Leydig cell tumors is secondary to elevated luteinizing hormone (LH) concentrations resulting from loss of feedback inhibition at the pituitary. Elevated LH and testosterone concentrations are not observed in castrated men receiving NILANDRON. Nilutamide had no effect on the incidence, size or time of onset of any spontaneous tumor in rats.
Nilutamide displayed no mutagenic effects in a variety of in vitro and in vivo tests (Ames test, mouse micronucleus test, and two chromosomal aberration tests).
In reproduction studies in rats, nilutamide had no effect on the reproductive function of males and females, and no lethal, teratogenic or growth-suppressive effects on fetuses were found. The maximal dose at which nilutamide did not affect reproductive function in either sex or have an effect on fetuses was estimated to be 45 mg/kg orally (AUC exposures in rats approximately 1–2 times human therapeutic AUC exposures).
Pregnancy
Animal reproduction studies have not been conducted with nilutamide. It is also not known whether nilutamide can cause fetal harm when administered to a pregnant woman or can affect reproductive capacity. Nilutamide should be given to a pregnant woman only if clearly needed.
Animal Pharmacology and Toxicology
Administration of NILANDRON to beagle dogs resulted in drug-related deaths at dose levels that produce AUC exposures in dogs much lower than the AUC exposures of men receiving the therapeutic doses of 150 and 300 mg/day. Nilutamide-induced toxicity in dogs was cumulative with progressively lower doses producing death when given for longer durations. Nilutamide given to dogs at 60 mg/kg/day (1–2 times human AUC exposure) for 1 month produced 100% mortality. Administration of 20 and 30 mg/kg/day nilutamide (1/2–1 times human AUC exposure) for 6 months resulted in 20% and 70% mortality in treated dogs. Administration to dogs of 3, 6, and 12 mg/kg/day nilutamide (1/10–1/2 human AUC exposure) for 1 year resulted in 8%, 33%, and 50% mortality, respectively. A "no-effect level" for nilutamide-induced mortality in dogs was not identified. Pathology data from the one-year oral toxicity study suggest that the deaths in dogs were secondary to liver toxicity. Marked-to-massive hepatocellular swelling and vacuolization were observed in affected dogs. Liver toxicity in dogs was not consistently associated with elevations of liver enzymes.
Administration of nilutamide to rats at a dose level of 45 mg/kg/day (AUC exposure in rats 1–2 times human therapeutic AUC exposures) for 18 months increased the incidence of lung pathology (granulomatous inflammation and chronic alveolitis).
The hepatic and pulmonary adverse effects observed in nilutamide-treated animals and men are similar to effects observed with another nitroaromatic compound, nitrofurantoin. Nilutamide and nitrofurantoin are both metabolized in vitro to nitroanion free-radicals by microsomal NADPH-cytochrome P450 reductase in the lungs and liver of rats and humans.
Adverse Reactions/Side Effects
Clinical Trial Experience
The following adverse experiences were reported during a multicenter clinical trial comparing NILANDRON + surgical castration versus placebo + surgical castration. The most frequently reported (greater than 5%) adverse experiences during treatment with NILANDRON tablets in combination with surgical castration are listed below. For comparison, adverse experiences seen with surgical castration and placebo are also listed.
| Adverse Experience | NILANDRON + surgical castration (N=225) % All | Placebo + surgical castration
(N=232) % All |
| Cardiovascular System
| ||
| Hypertension
|
5.3 |
2.6 |
| Digestive System
| ||
| Nausea
|
9.8 |
6.0 |
| Constipation
|
7.1 |
3.9 |
| Endocrine System
| ||
| Hot flushes
|
28.4 |
22.4 |
| Metabolic and Nutritional System
| ||
| Increased AST
|
8.0 |
3.9 |
| Increased ALT
|
7.6 |
4.3 |
| Nervous System
| ||
| Dizziness
|
7.1 |
3.4 |
| Respiratory System
| ||
| Dyspnea
|
6.2 |
7.3 |
| Special Senses
| ||
| Impaired adaptation to dark
|
12.9 |
1.3 |
| Abnormal vision
|
6.7 |
1.7 |
| Urogenital System
| ||
| Urinary tract infection
|
8.0 |
9.1 |
The overall incidence of adverse experiences was 86% (194/225) for the NILANDRON group and 81% (188/232) for the placebo group.
The following adverse experiences were reported during a multicenter clinical trial comparing NILANDRON + leuprolide versus placebo + leuprolide. The most frequently reported (greater than 5%) adverse experiences during treatment with NILANDRON tablets in combination with leuprolide are listed below. For comparison, adverse experiences seen with leuprolide and placebo are also listed.
| Adverse Experience | NILANDRON + leuprolide
(N=209) % All | Placebo + leuprolide
(N=202) % All |
| Body as a Whole
| ||
| Pain
|
26.8 |
27.7 |
| Headache
|
13.9 |
10.4 |
| Asthenia
|
19.1 |
20.8 |
| Back pain
|
11.5 |
16.8 |
| Abdominal pain
|
10.0 |
5.4 |
| Chest pain
|
7.2 |
4.5 |
| Flu syndrome
|
7.2 |
3.0 |
| Fever
|
5.3 |
6.4 |
| Cardiovascular System
| ||
| Hypertension
|
9.1 |
9.9 |
| Digestive System
| ||
| Nausea
|
23.9 |
8.4 |
| Constipation
|
19.6 |
16.8 |
| Anorexia
|
11.0 |
6.4 |
| Dyspepsia
|
6.7 |
4.5 |
| Vomiting
|
5.7 |
4.0 |
| Endocrine System
| ||
| Hot flushes
|
66.5 |
59.4 |
| Impotence
|
11.0 |
12.9 |
| Libido decreased
|
11.0 |
4.5 |
| Hemic and Lymphatic System
| ||
| Anemia
|
7.2 |
6.4 |
| Metabolic and Nutritional System
| ||
| Increased AST
|
12.9 |
13.9 |
| Peripheral edema
|
12.4 |
17.3 |
| Increased ALT
|
9.1 |
8.9 |
| Musculoskeletal System
| ||
| Bone Pain
|
6.2 |
5.0 |
| Nervous System
| ||
| Insomnia
|
16.3 |
15.8 |
| Dizziness
|
10.0 |
11.4 |
| Depression
|
8.6 |
7.4 |
| Hypesthesia
|
5.3 |
2.0 |
| Respiratory System
| ||
| Dyspnea
|
10.5 |
7.4 |
| Upper respiratory infection
|
8.1 |
10.9 |
| Pneumonia
|
5.3 |
3.5 |
| Skin and Appendages
| ||
| Sweating
|
6.2 |
3.0 |
| Body hair loss
|
5.7 |
0.5 |
| Dry skin
|
5.3 |
2.5 |
| Rash
|
5.3 |
4.0 |
| Special Senses
| ||
| Impaired adaptation to dark
|
56.9 |
5.4 |
| Chromatopsia
|
8.6 |
0.0 |
| Impaired adaptation to light
|
7.7 |
1.0 |
| Abnormal vision
|
6.2 |
4.5 |
| Urogenital System
| ||
| Testicular atrophy
|
16.3 |
12.4 |
| Gynecomastia
|
10.5 |
11.9 |
| Urinary tract infection
|
8.6 |
21.3 |
| Hematuria
|
8.1 |
7.9 |
| Urinary tract disorder
|
7.2 |
10.4 |
| Nocturia
|
6.7 |
6.4 |
The overall incidence of adverse experiences is 99.5% (208/209) for the NILANDRON group and 98.5% (199/202) for the placebo group.
Some frequently occurring adverse experiences, for example hot flushes, impotence, and decreased libido, are known to be associated with low serum androgen levels and known to occur with medical or surgical castration alone. Notable was the higher incidence of visual disturbances (variously described as impaired adaptation to darkness, abnormal vision, and colored vision), which led to treatment discontinuation in 1% to 2% of patients.
Interstitial pneumonitis occurred in one (<1%) patient receiving NILANDRON in combination with surgical castration and in seven patients (3%) receiving NILANDRON in combination with leuprolide and one patient receiving placebo in combination with leuprolide. Overall, it has been reported in 2% of patients receiving NILANDRON. This included a report of interstitial pneumonitis in 8 of 47 patients (17%) in a small study performed in Japan.
In addition, the following adverse experiences were reported in 2 to 5% of patients treated with NILANDRON in combination with leuprolide or orchiectomy.
Body as a Whole: Malaise (2%).
Cardiovascular System: Angina (2%), heart failure (3%), syncope (2%).
Digestive System: Diarrhea (2%), gastrointestinal disorder (2%), gastrointestinal hemorrhage (2%), melena (2%).
Metabolic and Nutritional System: Alcohol intolerance (5%), edema (2%), weight loss (2%).
Musculoskeletal System: Arthritis (2%).
Nervous System: Dry mouth (2%), nervousness (2%), paresthesia (3%).
Respiratory System: Cough increased (2%), interstitial lung disease (2%), lung disorder (4%), rhinitis (2%).
Skin and Appendages: Pruritus (2%).
Special Senses: Cataract (2%), photophobia (2%).
Laboratory Values: Haptoglobin increased (2%), leukopenia (3%), alkaline phosphatase increased (3%), BUN increased (2%), creatinine increased (2%), hyperglycemia (4%).
To report SUSPECTED ADVERSE REACTIONS, contact Concordia Pharmaceuticals at 1-877-370-1142 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Overdosage
One case of massive overdosage has been published. A 79-year-old man attempted suicide by ingesting 13 g of nilutamide (i.e., 43 times the maximum recommended dose). Despite immediate gastric lavage and oral administration of activated charcoal, plasma nilutamide levels peaked at 6 times the normal range 2 hours after ingestion. There were no clinical signs or symptoms or changes in parameters such as transaminases or chest X-ray. Maintenance treatment (150 mg/day) was resumed 30 days later.
In repeated-dose tolerance studies, doses of 600 mg/day and 900 mg/day were administered to 9 and 4 patients, respectively. The ingestion of these doses was associated with gastrointestinal disorders, including nausea and vomiting, malaise, headache, and dizziness. In addition, a transient elevation in hepatic enzyme levels was noted in one patient.
Since nilutamide is protein bound, dialysis may not be useful as treatment for overdose. As in the management of overdosage with any drug, it should be borne in mind that multiple agents may have been taken. If vomiting does not occur spontaneously, it should be induced if the patient is alert. General supportive care, including frequent monitoring of the vital signs and close observation of the patient, is indicated.
Nilandron Dosage and Administration
The recommended dosage is 300 mg once a day for 30 days, followed thereafter by 150 mg once a day. NILANDRON tablets can be taken with or without food.
How is Nilandron supplied
NILANDRON 150 mg tablets are supplied in boxes of 30 tablets. Each box contains 3 child-resistant, PVC, aluminum foil-backed blisters of 10 tablets (NDC 59212-111-14). Each white, biconvex, cylindrical (10 mm in diameter) tablet has a triangular logo on one side and an internal reference number (168D) on the other.
Store at 25°C (77°F); excursions permitted between 15-30°C (59-86°F) [see USP Controlled Room Temperature]. Protect from light. Keep out of reach of children.
Rev. 09/19
Mfd. for:
Concordia Pharmaceuticals
Distributed by:
Amdipharm Limited
17 Northwood House
Dublin 9, Ireland
© 2015 All rights reserved.
| NILANDRON
nilutamide tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - CENEXI HSC (268155718) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| CENEXI | 263382610 | manufacture(82454-0111) | |
