Drug Detail:Onureg (Azacitidine [ ay-za-sye-ti-deen ])
Drug Class: Miscellaneous antineoplastics
Highlights of Prescribing Information
ONUREG (azacitidine) tablets, for oral use
Initial U.S. Approval: 2004
Indications and Usage for Onureg
ONUREG is a nucleoside metabolic inhibitor indicated for continued treatment of adult patients with acute myeloid leukemia who achieved first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy (1).
Onureg Dosage and Administration
- •
- Do not substitute ONUREG for intravenous or subcutaneous azacitidine. The indications and dosing regimen for ONUREG differ from that of intravenous or subcutaneous azacitidine (2.1, 5.1).
- •
- Administer ONUREG 300 mg orally once daily on Days 1 through 14 of each 28-day cycle (2.2).
- •
- Administer an antiemetic before each dose for at least the first 2 cycles (2.2).
Dosage Forms and Strengths
Tablets: 200 mg and 300 mg (3).
Contraindications
History of severe hypersensitivity to azacitidine or its components (4).
Warnings and Precautions
- •
- Risks of Substitution with Other Azacitidine Products: Do not substitute ONUREG for intravenous or subcutaneous azacitidine (2.1, 5.1).
- •
- Myelosuppression: Monitor complete blood counts every other week for the first 2 cycles and prior to the start of each cycle thereafter. Increase monitoring to every other week for the 2 cycles after any dose reduction. Withhold and then resume at same or reduced dose or discontinue ONUREG based on severity (2.3, 5.2).
- •
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and use of effective contraception (5.4, 8.1, 8.3).
Adverse Reactions/Side Effects
The most common adverse reactions (≥ 10%) are nausea, vomiting, diarrhea, fatigue/asthenia, constipation, pneumonia, abdominal pain, arthralgia, decreased appetite, febrile neutropenia, dizziness, and pain in extremity (6).
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb Company at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
Lactation: Advise not to breastfeed (8.2).
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2022
Full Prescribing Information
1. Indications and Usage for Onureg
ONUREG is indicated for continued treatment of adult patients with acute myeloid leukemia who achieved first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy.
2. Onureg Dosage and Administration
2.1 Important Administration Information
Do not substitute ONUREG for intravenous or subcutaneous azacitidine. The indications and dosing regimen for ONUREG differ from that of intravenous or subcutaneous azacitidine [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
The recommended dosage of ONUREG is 300 mg orally once daily with or without food on Days 1 through 14 of each 28-day cycle. Continue ONUREG until disease progression or unacceptable toxicity.
Administer an antiemetic 30 minutes prior to each dose of ONUREG for the first 2 cycles. Antiemetic prophylaxis may be omitted after 2 cycles if there has been no nausea and vomiting.
If the absolute neutrophil count (ANC) is less than 0.5 Gi/L on Day 1 of a cycle, do not administer ONUREG. Delay the start of the cycle until the ANC is 0.5 Gi/L or more.
Instruct patients on the following:
- •
- Swallow tablets whole. Do not cut, crush, or chew the tablets.
- •
- Take a dose about the same time each day.
- •
- If a dose of ONUREG is missed, or not taken at the usual time, take the dose as soon as possible on the same day, and resume the normal schedule the following day. Do not take 2 doses on the same day.
- •
- If a dose is vomited, do not take another dose on the same day. Resume the normal schedule the following day.
ONUREG is a hazardous drug. Follow applicable special handling and disposal procedures.1
2.3 Monitoring and Dosage Modifications for Adverse Reactions
Monitor complete blood count every other week for the first 2 cycles and prior to the start of each cycle thereafter. Increase monitoring to every other week for the 2 cycles after any dose reduction for myelosuppression.
The recommended dosage modifications for adverse reactions are provided in Table 1.
| Adverse Reaction | Severity | Recommended Dosage Modification |
|---|---|---|
|
Myelosuppression [see Warnings and Precautions (5.2)] |
Neutrophils less than 0.5 Gi/L on Cycle Day 1 |
|
|
Neutrophils less than 1 Gi/L with fever at anytime |
First Occurrence
Occurrence in 2 Consecutive Cycles
|
|
|
Platelets less than 50 Gi/L with bleeding |
First Occurrence
Occurrence in 2 Consecutive Cycles
|
|
|
Gastrointestinal Toxicity [see Adverse Reactions (6.1)] |
Grade 3 or 4 Nausea or Vomiting |
|
|
Grade 3 or 4 Diarrhea |
|
|
|
Other Adverse Reactions [see Adverse Reactions (6.1)] |
Grade 3 or 4 |
|
3. Dosage Forms and Strengths
Tablets:
- •
- 200 mg, pink, oval, film-coated tablet with debossed "200" on one side and "ONU" on the other side.
- •
- 300 mg, brown, oval, film-coated tablet with debossed "300" on one side and "ONU" on the other side.
4. Contraindications
ONUREG is contraindicated in patients with known severe hypersensitivity to azacitidine or its components [see Adverse Reactions (6.2), Description (11)].
5. Warnings and Precautions
5.1 Risks of Substitution with Other Azacitidine Products
Due to substantial differences in the pharmacokinetic parameters [see Clinical Pharmacology (12.3)], the recommended dose and schedule for ONUREG are different from those for the intravenous or subcutaneous azacitidine products. Treatment of patients using intravenous or subcutaneous azacitidine at the recommended dosage of ONUREG may result in a fatal adverse reaction. Treatment of patients using ONUREG at the doses recommended for intravenous or subcutaneous azacitidine may not be effective.
Do not substitute ONUREG for intravenous or subcutaneous azacitidine [see Dosage and Administration (2.1)].
5.2 Myelosuppression
New or worsening Grade 3 or 4 neutropenia and thrombocytopenia occurred in 49% and 22% of patients who received ONUREG, respectively. Febrile neutropenia occurred in 12%. A dose reduction was required for 7% and 2% of patients due to neutropenia and thrombocytopenia, respectively. Less than 1% of patients discontinued ONUREG due to either neutropenia or thrombocytopenia.
Monitor complete blood counts and modify the dosage as recommended [see Dosage and Administration (2.2, 2.3)]. Provide standard supportive care, including hematopoietic growth factors, if myelosuppression occurs.
5.3 Increased Early Mortality in Patients with Myelodysplastic Syndromes
In AZA-MDS-003 (NCT01566695), 216 patients with red blood cell transfusion-dependent anemia and thrombocytopenia due to myelodysplastic syndromes were randomized to ONUREG or placebo. One-hundred and seven patients received a median of 5 cycles of ONUREG 300 mg daily for 21 days of a 28-day cycle. Enrollment was discontinued early due to a higher incidence of early fatal and/or serious adverse reactions in patients who received ONUREG compared with placebo. The most frequent fatal adverse reaction was sepsis. The safety and effectiveness of ONUREG for treatment of myelodysplastic syndromes have not been established. Treatment of patients with myelodysplastic syndromes with ONUREG is not recommended outside of controlled trials.
5.4 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, ONUREG can cause fetal harm when administered to a pregnant woman. Azacitidine administered to pregnant rats via a single intraperitoneal dose less than the recommended human daily dose of oral azacitidine on a mg/m2 basis caused fetal death and anomalies.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ONUREG and for at least 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with ONUREG and for at least 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Myelosuppression [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Acute Myeloid Leukemia
The safety of ONUREG was evaluated in QUAZAR [see Clinical Studies (14)]. Patients received ONUREG 300 mg (N=236) or placebo (N=233) orally once daily on Days 1 through 14 of each 28-day cycle. Among patients who received ONUREG, 71% were exposed for 6 months or longer, and 49% were exposed for greater than one year. The median duration of exposure to ONUREG was 11.6 months (range: 0.5 to 74.3 months) and the median number of cycles was 12 (range: 1 to 82 cycles).
Serious adverse reactions occurred in 15% of patients who received ONUREG. Serious adverse reactions in ≥ 2% of patients who received ONUREG were pneumonia (8%) and febrile neutropenia (7%). One fatal adverse reaction (sepsis) occurred in a patient who received ONUREG.
Permanent discontinuation of ONUREG due to an adverse reaction occurred in 8% of patients. Adverse reactions which resulted in permanent discontinuation of ONUREG in > 1% of patients included nausea (2.1%), diarrhea (1.7%), and vomiting (1.3%). Interruptions of ONUREG due to an adverse reaction occurred in 35% of patients. Adverse reactions which required an interruption of ONUREG in > 5% of patients included neutropenia (20%), thrombocytopenia (8%), and nausea (6%).
Dose reductions of ONUREG due to an adverse reaction occurred in 14% of patients. Adverse reactions which required a dose reduction in > 1% of patients included neutropenia (6%), diarrhea (3.4%), thrombocytopenia (1.7%), and nausea (1.7%).
The most common (≥ 10%) adverse reactions were nausea, vomiting, diarrhea, fatigue/asthenia, constipation, pneumonia, abdominal pain, arthralgia, decreased appetite, febrile neutropenia, dizziness, and pain in extremity.
Table 2 summarizes the adverse reactions in QUAZAR.
| Adverse Reaction | ONUREG
(N=236) | Placebo
(N=233) |
||
|---|---|---|---|---|
| All Grades
(%) | Grade 3 or 4
(%) | All Grades
(%) | Grade 3 or 4
(%) |
|
| a Grouped term includes abdominal pain, abdominal pain upper, abdominal discomfort, and gastrointestinal pain. b Grouped term includes fatigue and asthenia. c Broad scope term includes influenza, pneumonia, respiratory tract infection, respiratory tract infection viral, bronchopulmonary aspergillosis, lung infection, Staphylococcal infection, atypical pneumonia, lower respiratory tract infection, lung abscess, Pneumocystis jirovecii pneumonia, pneumonia bacterial, pneumonia fungal, Pseudomonas infection, hemoptysis, productive cough, pleural effusion, atelectasis, pleuritic pain, rales, Enterobacter test positive, and Hemophilus test positive. |
||||
|
Gastrointestinal disorders |
||||
|
Nausea |
65 |
3 |
24 |
< 1 |
|
Vomiting |
60 |
3 |
10 |
0 |
|
Diarrhea |
50 |
5 |
21 |
1 |
|
Constipation |
39 |
1 |
24 |
0 |
|
Abdominal paina |
22 |
2 |
13 |
< 1 |
|
General disorders and administration site conditions |
||||
|
Fatigue / astheniab |
44 |
4 |
25 |
1 |
|
Infections |
||||
|
Pneumoniac |
27 |
9 |
17 |
5 |
|
Musculoskeletal and connective tissue disorders |
||||
|
Arthralgia |
14 |
1 |
10 |
< 1 |
|
Pain in extremity |
11 |
< 1 |
5 |
0 |
|
Metabolism and nutrition disorders |
||||
|
Decreased appetite |
13 |
1 |
6 |
1 |
|
Blood and lymphatic disorders |
||||
|
Febrile neutropenia |
12 |
11 |
8 |
8 |
|
Nervous system disorders |
||||
|
Dizziness |
11 |
0 |
9 |
0 |
Clinically relevant adverse reactions that did not meet criteria for inclusion in Table 2 were weight decreased (4%) in patients who received ONUREG.
Neutropenia, thrombocytopenia, and anemia of any grade occurred in 74%, 65%, and 25% of patients who received ONUREG. Table 3 summarizes select Grades 3 or 4 hematological laboratory abnormalities in QUAZAR.
| ONUREG | Placebo | |||
|---|---|---|---|---|
| Laboratory Abnormality | Baseline
Grade 0-2 N | Post-Baseline
Grade 3 or 4 n (%) | Baseline
Grade 0-2 N | Post-Baseline
Grade 3 or 4 n (%) |
|
Neutropenia |
223 |
109 (49) |
217 |
50 (23) |
|
Thrombocytopenia |
222 |
46 (21) |
212 |
22 (10) |
|
Anemia |
229 |
10 (4) |
223 |
7 (3) |
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of intravenous or subcutaneous azacitidine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- •
- Hypersensitivity reaction
- •
- Interstitial lung disease
- •
- Tumor lysis syndrome
- •
- Sweet's syndrome (acute febrile neutrophilic dermatosis)
- •
- Necrotizing fasciitis (including fatal cases)
- •
- Differentiation syndrome
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action [see Clinical Pharmacology (12.1)] and findings in animals, ONUREG can cause fetal harm when administered to a pregnant woman. There are no available data on ONUREG use in pregnant women to evaluate for a drug-associated risk. Azacitidine was teratogenic and caused embryo-fetal lethality in animals at doses less than the recommended human daily dose of oral azacitidine on a mg/m2 basis (see Data). Advise pregnant women of the potential risk to the fetus.
The estimated background of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
No reproductive or developmental toxicity studies have been conducted with oral azacitidine.
Early embryotoxicity studies in mice revealed a 44% frequency of intrauterine embryonal death (increased resorption) after a single intraperitoneal injection of 6 mg/m2 azacitidine (at doses less than the recommended human daily dose of oral azacitidine on a mg/m2 basis) on gestation Day 10. Developmental abnormalities in the brain have been detected in mice given azacitidine on or before gestation Day 15 at doses of approximately 3 to 12 mg/m2 (at doses less than the recommended human daily dose on a mg/m2 basis).
In rats, azacitidine was clearly embryotoxic when given an intraperitoneal injection on gestation Days 4 to 8 (postimplantation) at a dose of 6 mg/m2 (at doses less than the recommended human daily dose on a mg/m2 basis), although treatment in the preimplantation period (on gestation Days 1 to 3) had no adverse effect on the embryos. Azacitidine caused multiple fetal abnormalities in rats after a single intraperitoneal dose of 3 to 12 mg/m2 (at doses less than the recommended human daily dose on a mg/m2 basis) given on gestation Days 9, 10, 11, or 12. In this study, azacitidine caused fetal death when administered at 3 to 12 mg/m2 on gestation Days 9 and 10; average live animals per litter was reduced to 9% of control at the highest dose on gestation Day 9. Fetal anomalies included: CNS anomalies (exencephaly/encephalocele), limb anomalies (micromelia, club foot, syndactyly, oligodactyly), and others (micrognathia, gastroschisis, edema, and rib abnormalities).
8.2 Lactation
Risk Summary
There are no data regarding the presence of azacitidine in human milk or the effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with ONUREG and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
ONUREG can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential before starting ONUREG.
8.4 Pediatric Use
The safety and effectiveness of ONUREG in pediatric patients have not been established.
8.5 Geriatric Use
Of the 238 patients in QUAZAR who received ONUREG, 72% were 65 years of age or older, while 12% were 75 years of age or older. No overall differences in safety or effectiveness of ONUREG were observed between these patients and younger patients.
8.6 Renal Impairment
Monitor patients with severe renal impairment (creatinine clearance [CLcr] 15 to 29 mL/min calculated by Cockcroft-Gault formula) more frequently for adverse reactions and modify the ONUREG dosage for adverse reactions [see Dosage and Administration (2.3)].
No dose adjustment of ONUREG is recommended for patients with mild to severe renal impairment (CLcr 15 to 89 mL/min) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
ONUREG has not been studied in patients with pre-existing severe hepatic impairment (total bilirubin > 3 × ULN).
A recommended dosage of ONUREG has not been established for patients with moderate hepatic impairment (total bilirubin > 1.5 to 3 × ULN).
No dose adjustment of ONUREG is recommended for patients with mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin 1 to 1.5 × ULN and any AST) [see Clinical Pharmacology (12.3)].
11. Onureg Description
Azacitidine is a nucleoside metabolic inhibitor with a molecular formula of C8H12N4O5 and a molecular weight of 244 g/mol. The chemical name is: 4-amino-1-β-D-ribofuranosyl-s-triazin-2(1H)-one and the chemical structural is:
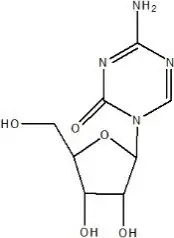
Azacitidine is a white to off-white solid. Azacitidine was found to be soluble in aqueous media across a pH range from 1.0 to 7.0.
ONUREG (azacitidine) is supplied as film-coated tablets containing 200 mg or 300 mg of azacitidine for oral use. Each core tablet contains the following inactive ingredients: croscarmellose sodium, magnesium stearate, mannitol, and silicified microcrystalline cellulose. The 200 and 300 mg tablet coating contains hypromellose, lactose monohydrate, polyethylene glycol, titanium dioxide, and triacetin. In addition, the 200 mg tablet coating contains iron oxide red and the 300 mg tablet coating contains black iron oxide, iron oxide red, and iron oxide yellow.
12. Onureg - Clinical Pharmacology
12.1 Mechanism of Action
Azacitidine is a pyrimidine nucleoside analog of cytidine that inhibits DNA/RNA methyltransferases. Azacitidine is incorporated into DNA and RNA following cellular uptake and enzymatic biotransformation to nucleotide triphosphates.
Incorporation of azacitidine into the DNA of cancer cells in vitro, including acute myeloid leukemia cells, inhibited DNA methyltransferases, reduced DNA methylation and altered gene expression, including re-expression of genes regulating tumor suppression and cell differentiation. Incorporation of azacitidine into the RNA of cancer cells, including leukemic cells, inhibited RNA methyltransferases, reduced RNA methylation, decreased RNA stability and decreased protein synthesis.
Antileukemic activity of azacitidine was demonstrated by reduction of cell viability and induction of apoptosis in AML cell lines in vitro. Azacitidine decreased tumor burden and increased survival in leukemic tumor models in vivo.
12.2 Pharmacodynamics
Greater reduction in global DNA methylation was observed with higher azacitidine plasma exposure in patients with AML administered ONUREG for 14 days of a 28-day cycle.
12.3 Pharmacokinetics
The systemic exposure of azacitidine is approximately dose proportional over the dose range of 120 mg to 600 mg once daily of ONUREG (0.4 to 2 times the recommended dosage). Following a single 300 mg dose of ONUREG, the mean (coefficient of variation [CV%]) Cmax of azacitidine was 145 ng/mL (64%) and the mean AUC of azacitidine was 242 ng h/mL (65%). No accumulation was observed following ONUREG 300 mg once daily.
Absorption
The mean oral bioavailability is approximately 11% relative to subcutaneous administration. The median time to peak plasma concentration of azacitidine is 1 hour.
Distribution
The mean (CV%) apparent volume of distribution (Vz/F) of azacitidine is 881 L (67%). The in vitro serum protein binding of azacitidine is approximately 6% to 12%. The blood-to-plasma ratio is approximately 0.3.
Elimination
The mean (CV%) terminal half-life is approximately 0.5 hours (27%) and the apparent clearance (CL/F) is 1240 L/hour (64%).
Specific Populations
Age (46 years to 93 years), sex, body weight (39.3 kg to 129 kg), mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin 1 to 1.5 × ULN and any AST), and mild to moderate renal impairment (CLcr 30 to 89 mL/min) have no clinically meaningful effect on the pharmacokinetics of oral azacitidine. The effects of race/ethnicity, moderate to severe hepatic impairment (total bilirubin > 1.5 × ULN and any AST), and severe renal impairment (CLcr 15 to 29 mL/min) on the pharmacokinetics of oral azacitidine is unknown.
Severe renal impairment increased azacitidine exposure by approximately 70% after a single or 41% after multiple subcutaneous daily administration.
Drug Interaction Studies
Effect of Gastric Acid Reducing Agents on Azacitidine:
Coadministration of omeprazole (a proton pump inhibitor) with ONUREG increased azacitidine AUC0-INF by 19% and had no effect on Cmax.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Azacitidine does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, or CYP2E1 at clinically relevant concentrations. Azacitidine is not an inducer of CYP1A2, CYP2C19, or CYP3A.
Transporter Systems: Azacitidine is not a substrate of P-glycoprotein (P-gp). Azacitidine does not inhibit P-gp, breast cancer resistance protein (BCRP), organic anion transporters (OAT) OAT1 and OAT3, organic anion transporting polypeptides (OATP) OATP1B1 and OATP1B3, or organic cation transporter (OCT) OCT2 at clinically relevant concentrations.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The potential carcinogenicity of azacitidine was evaluated in mice and rats. Azacitidine induced tumors of the hematopoietic system in female mice at 2.2 mg/kg (6.6 mg/m2, approximately 4% of the recommended human daily dose of oral azacitidine on a mg/m2 basis) administered intraperitoneal 3 times per week for 52 weeks. An increased incidence of tumors in the lymphoreticular system, lung, mammary gland, and skin was seen in mice treated with intraperitoneal azacitidine at 2 mg/kg (6 mg/m2, approximately 3% of the recommended human daily dose of oral azacitidine on a mg/m2 basis) once a week for 50 weeks. A tumorigenicity study in rats dosed twice weekly at 15 or 60 mg/m2 (approximately 8% to 32% of the recommended human daily dose of oral azacitidine on a mg/m2 basis) revealed an increased incidence of testicular tumors compared with controls.
The mutagenic and clastogenic potential of azacitidine was tested in in vitro bacterial systems Salmonella typhimurium strains TA100 and several strains of trpE8, Escherichia coli strains WP14 Pro, WP3103P, WP3104P, and CC103; in an in vitro forward gene mutation assay in mouse lymphoma cells and human lymphoblast cells; and in an in vitro micronucleus assay in mouse L5178Y lymphoma cells and Syrian hamster embryo cells. Azacitidine was mutagenic in bacterial and mammalian cell systems. The clastogenic effect of azacitidine was shown by the induction of micronuclei in L5178Y mouse cells and Syrian hamster embryo cells.
Administration of azacitidine by intraperitoneal injection to male mice at 9.9 mg/m2 (at doses less than the recommended human daily dose on a mg/m2 basis) daily for 3 days prior to mating with untreated female mice resulted in decreased fertility and loss of offspring during subsequent embryonic and postnatal development. Treatment of male rats 3 times per week for 11 or 16 weeks at doses of 15 to 30 mg/m2 (at doses less than the recommended human daily dose on a mg/m2 basis) resulted in decreased weight of the testes and epididymides, decreased sperm counts accompanied by decreased pregnancy rates, and increased loss of embryos in mated females. In a related study, male rats treated for 16 weeks at 24 mg/m2 resulted in an increase in abnormal embryos in mated females when examined on Day 2 of gestation.
14. Clinical Studies
The efficacy of ONUREG was evaluated in QUAZAR (NCT01757535), a multicenter, randomized, double-blind, placebo-controlled study. Eligible patients were ages 55 years or older, had AML, and were within 4 months of achieving first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) with intensive induction chemotherapy. Patients may have received consolidation (see Table 4). Patients were excluded if they were candidates for hematopoietic stem cell transplantation at the time of screening.
A total of 472 patients who completed induction with or without consolidation therapy were randomized 1:1 to receive ONUREG 300 mg (n=238) or placebo (n=234) orally on Days 1 through 14 of each 28-day cycle. Randomization was stratified by age at time of induction therapy (55 to 64 vs. ≥ 65 years), cytogenetic risk category at time of induction therapy (intermediate risk vs. poor risk), prior history of MDS/CMML (yes vs. no), and received consolidation therapy following induction therapy (yes vs. no). Baseline demographic and disease characteristics are shown in Table 4.
| Parameter | ONUREG
(N=238) | Placebo
(N=234) |
|---|---|---|
| AML=Acute Myeloid Leukemia, MDS=Myelodysplastic Syndrome, CMML=Chronic Myelomonocytic Leukemia, ECOG=Eastern Cooperative Oncology Group, CR=Morphologic Complete Remission, CRi=Morphologic complete remission with incomplete blood count recovery. Source for Intermediate and Poor Risk: National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology for Acute Myeloid Leukemia. National Comprehensive Cancer Network website. Available at http://www.nccn.org/professionals/physician_gls/PDF/aml.pdf. Accessed 01 Mar 2011. 1 Intermediate risk was defined as normal cytogenetics +8, t(9;11), or Other undefined. 2 Poor risk was defined as Complex (≥ 3 abnormalities): -5; 5q-; -7; 7q-; 11q23 - non t(9;11); inv(3); t(3;3); t(6;9); or t(9;22). |
||
|
Age (years) | ||
|
Median (Min, Max) |
68.0 (55, 86) |
68.0 (55, 82) |
|
Age Category, n (%) | ||
|
< 65 years |
66 (28) |
68 (29) |
|
65 years to < 75 years |
144 (61) |
142 (61) |
|
≥ 75 years |
28 (12) |
24 (10) |
|
Sex, n (%) | ||
|
Male |
118 (50) |
127 (54) |
|
Female |
120 (50) |
107 (46) |
|
Race, n (%) | ||
|
White |
216 (91) |
197 (84) |
|
Black or African American |
2 (1) |
6 (3) |
|
Asian |
6 (3) |
20 (9) |
|
Other |
12 (5) |
11 (5) |
|
Not Collected or Reported |
2 (1) |
0 (0) |
|
ECOG Performance Status, n (%) | ||
|
0 |
116 (49) |
111 (47) |
|
1 |
101 (42) |
106 (45) |
|
2 |
21 (9) |
15 (6) |
|
3 |
0 (0) |
2 (1) |
|
Cytogenetic Risk Status at Diagnosis, n (%) | ||
|
Intermediate Risk1 |
203 (85) |
203 (87) |
|
Poor Risk2 |
35 (15) |
31 (13) |
|
Initial AML Classification, n (%) | ||
|
AML with recurrent genetic abnormalities |
39 (16) |
46 (20) |
|
AML with myelodysplasia-related changes |
49 (21) |
42 (18) |
|
Therapy related myeloid neoplasms |
2 (1) |
0 (0) |
|
AML not otherwise specified |
148 (62) |
145 (62) |
|
Missing |
0 (0) |
1 (< 1) |
|
Type of AML, n (%) | ||
|
Primary (de novo) |
213 (89) |
216 (92) |
|
Secondary |
25 (11) |
18 (8) |
|
Induction Response | ||
|
CR |
187 (79) |
197 (84) |
|
CRi |
51 (21) |
37 (16) |
|
Consolidation following induction therapy | ||
|
None |
52 (22) |
42 (18) |
|
1 cycle |
110 (46) |
102 (44) |
|
2 cycles |
70 (29) |
77 (33) |
|
3 cycles |
6 (3) |
13 (6) |
|
Disease status at study baseline | ||
|
CR |
185 (78) |
181 (77) |
|
CRi |
44 (18) |
38 (16) |
|
Not in CR or CRi |
9 (4) |
13 (6) |
|
Not Reported |
0 |
2 (1) |
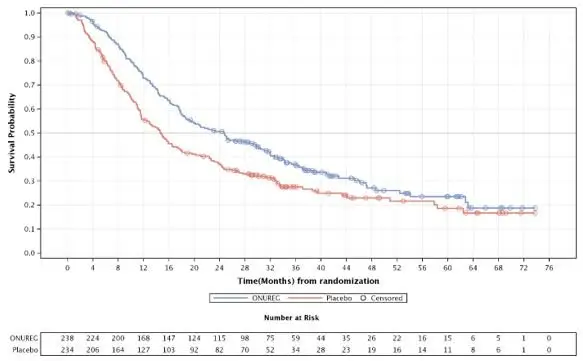
The efficacy of ONUREG was established on the basis of overall survival (OS). The trial demonstrated a statistically significant improvement in OS for patients randomized to ONUREG compared to placebo. A subgroup analysis showed consistency in the OS benefit for patients in either CR or CRi. The efficacy results are summarized in Table 5 and Figure 1.
| ONUREG
(N=238) | Placebo
(N=234) |
||||
|---|---|---|---|---|---|
| 1 The hazard ratio is from a Cox proportional hazards model stratified by age (55 to 64 vs. ≥65 years), cytogenetic risk category at time of induction therapy (intermediate risk vs. poor risk), and received consolidation therapy (yes vs. no). The p-value is two-sided from a log-rank test stratified by the same factors. | |||||
|
Overall Survival | |||||
|
OS Events, n (%) |
158 (66) |
171 (73) |
|||
|
Median OS (95% CI) Months |
24.7 (18.7, 30.5) |
14.8 (11.7, 17.6) |
|||
|
Hazard Ratio (95% CI)1 |
0.69 (0.55, 0.86) |
||||
|
p value1 |
0.0009 |
||||
Figure 1: Kaplan-Meier Curve for Overall Survival (ITT Population) in QUAZAR
16. How is Onureg supplied
How Supplied
ONUREG tablets are available as:
- •
- 200 mg: pink, oval, film-coated tablets with debossed "200" on one side and "ONU" on the other side.
- •
- 300 mg: brown, oval, film-coated tablets with debossed "300" on one side and "ONU" on the other side.
Table 6 lists the package configurations and strengths.
| Package Configuration | Tablet Strength | NDC Number |
|---|---|---|
|
Bottles of 14 with two desiccant cannisters |
200 mg |
59572-730-14 |
|
Bottles of 14 with two desiccant cannisters |
300 mg |
59572-740-14 |
|
One blister card containing 7 tablets |
200 mg |
59572-730-07 |
|
One blister card containing 7 tablets |
300 mg |
59572-740-07 |
Storage
- •
- Store bottles at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Keep bottle tightly closed.
Store and dispense in the original bottle (with two desiccant canisters). - •
- Store blisters at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Store in the original aluminum-aluminum blisters.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Myelosuppression
Advise patients of the risk of myelosuppression with ONUREG and of the need to monitor complete blood counts before and during treatment [see Warnings and Precautions (5.2)].
Gastrointestinal Toxicity
Advise patients of the risk of gastrointestinal toxicity with ONUREG and of the potential need to use anti-emetic or anti-diarrheal medications during treatment [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with ONUREG and for at least 6 months after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with ONUREG and for at least 3 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with ONUREG and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Administration
Advise patients to take ONUREG with or without food at about the same time each day and how to make up a missed or vomited dose. Advise patients to swallow tablets whole. Advise patients not to cut, crush, or chew the tablets [see Dosage and Administration (2.2)].
Storage Instructions
Advise patients to keep ONUREG in the original container (bottles or blisters). If bottles are dispensed, advise patients to keep the container tightly closed with both desiccant canisters inside and to not eat the desiccant canisters [see How Supplied/Storage and Handling (16)].
Marketed by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
ONUREG® is a trademark of Celgene Corporation, a Bristol-Myers Squibb company.
ONUPI.004
| This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: October 2022 |
||
|
Patient Information
|
||
|
What is ONUREG? |
||
|
ONUREG is a prescription medicine used for continued treatment of adults with acute myeloid leukemia (AML) who: |
||
|
||
|
It is not known if ONUREG is safe and effective in children under 18 years of age. |
||
|
Do not take ONUREG if you: |
||
|
||
|
Before taking ONUREG, tell your healthcare provider about all of your medical conditions, including if you: |
||
|
||
|
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
||
|
How should I take ONUREG? |
||
|
||
|
What are the possible side effects of ONUREG? |
||
|
ONUREG can cause serious side effects, including: |
||
|
||
|
|
|
|
||
|
ONUREG may cause fertility problems in males and females, which may affect your ability to have children. Talk with your healthcare provider if you have concerns about fertility. |
||
|
The most common side effects of ONUREG include: |
||
|
|
|
|
These are not all of the possible side effects of ONUREG. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
|
How should I store ONUREG? |
||
|
||
|
Keep ONUREG and all medicines out of the reach of children |
||
|
General information about the safe and effective use of ONUREG. |
||
|
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ONUREG for a condition for which it was not prescribed. Do not give ONUREG to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ONUREG that is written for health professionals. |
||
|
What are the ingredients in ONUREG? |
||
|
Active ingredient: azacitidine |
||
|
Inactive ingredients: |
||
|
Each core tablet contains: croscarmellose sodium, magnesium stearate, mannitol, and silicified microcrystalline cellulose. |
||
|
The pink 200 mg tablet coating contains: hypromellose, iron oxide red, lactose monohydrate, polyethylene glycol, titanium dioxide, and triacetin. |
||
|
The brown 300 mg tablet coating contains: black iron oxide, hypromellose, iron oxide red, iron oxide yellow, lactose monohydrate, polyethylene glycol, titanium dioxide, and triacetin. |
||
|
Marketed by: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA |
||
PRINCIPAL DISPLAY PANEL - 200 mg Bottle Label
NDC 59572-730-14
ONUREG™
(azacitidine) tablets
200 mg
Swallow tablets whole.
Do not cut, crush, or chew the tablets.
Rx only
14 Tablets
CAUTION: Hazardous Agent

PRINCIPAL DISPLAY PANEL - 200 mg Blister Pack Carton
NDC 59572-730-07
Rx only
ONUREG™
(azacitidine) tablets
200 mg
Each tablet contains 200 mg of azacitidine.
Swallow tablets whole. Do not cut, crush, or chew the tablets.
One Blister Card
Containing 7 Tablets
Bristol Myers Squibb
CAUTION: Hazardous Agent
PRINCIPAL DISPLAY PANEL - 300 mg Blister Pack Carton
NDC 59572-740-07
Rx only
ONUREG™
(azacitidine) tablets
300 mg
Each tablet contains 200 mg of azacitidine.
Swallow tablets whole. Do not cut, crush, or chew the tablets.
One Blister Card
Containing 7 Tablets
Bristol Myers Squibb
CAUTION: Hazardous Agent
| ONUREG
azacitidine tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ONUREG
azacitidine tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Celgene Corporation (174201137) |
