Drug Detail:Orilissa (Elagolix [ el-a-goe-lix ])
Drug Class: Gonadotropin-releasing hormone antagonists
Highlights of Prescribing Information
ORILISSA® (elagolix) tablets, for oral use
Initial U.S. Approval: 2018
Indications and Usage for Orilissa
ORILISSA is a gonadotropin-releasing hormone (GnRH) receptor antagonist indicated for the management of moderate to severe pain associated with endometriosis. (1)
Limitations of Use:
- Limit the duration of use based on the dose and coexisting condition (see Table 1). (1)
Orilissa Dosage and Administration
Normal liver function or mild hepatic impairment: 150 mg once daily for up to 24 months or 200 mg twice daily for up to 6 months. (2.1)
Moderate hepatic impairment: 150 mg once daily for up to 6 months. (2.1)
Dosage Forms and Strengths
Oral tablets: 150 mg and 200 mg (3)
Contraindications
- Pregnancy (4)
- Known osteoporosis (4)
- Severe hepatic impairment (4)
- Organic anion transporting polypeptide (OATP) 1B1 inhibitors that significantly increase elagolix plasma concentrations (4)
- Hypersensitivity reactions (4, 6.2)
Warnings and Precautions
-
Bone Loss: Dose- and duration-dependent decreases in bone mineral density (BMD) that may not be completely reversible. Assess BMD in women with additional risk factors for bone loss (5.1)
-
Reduced Ability to Recognize Pregnancy: ORILISSA may alter menstrual bleeding, which may reduce the ability to recognize pregnancy. Perform testing if pregnancy is suspected. Discontinue if pregnancy is confirmed (5.2)
-
Suicidal Ideation and Mood Disorders: Advise patients to seek medical attention for suicidal ideation, suicidal behavior, new onset or worsening depression, anxiety, or other mood changes (5.3)
-
Hepatic Transaminase Elevations: Dose-dependent elevations in serum alanine aminotransferase (ALT). Counsel patients on signs and symptoms of liver injury (5.4)
- Interactions with Hormonal Contraceptives: Use non-hormonal contraception during treatment and for 28 days after discontinuing ORILISSA. Coadministration of ORILISSA 200 mg twice daily with an estrogen-containing contraceptive is not recommended because of the potential for increased estrogen-associated risks. Coadministration of ORILISSA with an estrogen-containing contraceptive may reduce the efficacy of ORILISSA. Coadministration with progestin-containing oral contraceptives may reduce the efficacy of the contraceptive. (5.5)
Adverse Reactions/Side Effects
Most common adverse reactions (>5%) in clinical trials included hot flushes and night sweats, headache, nausea, insomnia, amenorrhea, anxiety, arthralgia, depression-related adverse reactions and mood changes (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Drug Interactions
See full prescribing information for a list of clinically important drug interactions (7).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Orilissa
ORILISSA is indicated for the management of moderate to severe pain associated with endometriosis.
Limitations of Use:
Limit the duration of use based on the dose and coexisting condition (see Table 1) [see Dosage and Administration (2.1) and Warnings and Precautions (5.1)].
2. Orilissa Dosage and Administration
2.1 Important Dosing Information
- Exclude pregnancy before starting ORILISSA or start ORILISSA within 7 days from the onset of menses.
- Take ORILISSA at approximately the same time each day, with or without food.
- Use the lowest effective dose, taking into account the severity of symptoms and treatment objectives [see Warnings and Precautions (5.1, 5.3, 5.4) and Clinical Studies (14)].
- Limit the duration of use because of bone loss (Table 1) [see Warnings and Precautions (5.1)].
| Dosing Regimen | Maximum
Treatment Duration | Coexisting Condition |
| Initiate treatment with ORILISSA 150 mg once daily | 24 months | None |
| Consider initiating treatment with ORILISSA 200 mg twice daily | 6 months | Dyspareunia |
| Initiate treatment with ORILISSA 150 mg once daily. Use of 200 mg twice daily is not recommended. | 6 months | Moderate hepatic impairment (Child-Pugh Class B) |
2.2 Hepatic Impairment
No dosage adjustment of ORILISSA is required in women with mild hepatic impairment (Child-Pugh A).
Compared to women with normal liver function, those with moderate hepatic impairment had approximately 3-fold higher elagolix exposures and those with severe hepatic impairment had approximately 7-fold higher elagolix exposures. Because of these increased exposures and risk for bone loss:
- ORILISSA 150 mg once daily is recommended for women with moderate hepatic impairment (Child-Pugh B) with the duration of treatment limited to 6 months. Use of ORILISSA 200 mg twice daily is not recommended for women with moderate hepatic impairment [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
- ORILISSA is contraindicated in women with severe hepatic impairment (Child-Pugh C) [see Contraindications (4), Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
The 150 mg tablets are light pink, oblong, film-coated tablets with “EL 150” debossed on one side. Each tablet contains 155.2 mg of elagolix sodium equivalent to 150 mg of elagolix.
The 200 mg tablets are light orange, oblong, film-coated tablets with “EL 200” debossed on one side. Each tablet contains 207.0 mg of elagolix sodium equivalent to 200 mg of elagolix.
4. Contraindications
ORILISSA is contraindicated in women:
● Who are pregnant [see Use in Specific Populations (8.1)]. Exposure to ORILISSA early in pregnancy may increase the risk of early pregnancy loss.
● With known osteoporosis because of the risk of further bone loss [see Warnings and Precautions (5.1)]
● With severe hepatic impairment [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)]
● Taking inhibitors of organic anion transporting polypeptide (OATP)1B1 (a hepatic uptake transporter) that are known or expected to significantly increase elagolix plasma concentrations [see Drug Interactions (7.2)]
● With known hypersensitivity reaction to ORILISSA or any of its inactive components. Reactions have included anaphylaxis and angioedema [see Adverse Reactions (6.2)].
5. Warnings and Precautions
5.1 Bone Loss
ORILISSA causes a dose-dependent decrease in bone mineral density (BMD). BMD loss is greater with increasing duration of use and may not be completely reversible after stopping treatment [see Adverse Reactions (6.1)]. The impact of these BMD decreases on long-term bone health and future fracture risk are unknown.
ORILISSA is contraindicated in women with known osteoporosis [see Contraindications (4)]. Consider assessment of BMD in patients with a history of a low-trauma fracture or other risk factors for osteoporosis or bone loss. Limit the duration of use to reduce the extent of bone loss [see Dosage and Administration (2.2)]. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients.
5.2 Change in Menstrual Bleeding Pattern and Reduced Ability to Recognize Pregnancy
Women who take ORILISSA may experience a reduction in the amount, intensity or duration of menstrual bleeding, which may reduce the ability to recognize the occurrence of a pregnancy in a timely manner [see Adverse Reactions (6.1)]. Perform pregnancy testing if pregnancy is suspected, and discontinue ORILISSA if pregnancy is confirmed.
5.3 Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders
Suicidal ideation and behavior, including one completed suicide, occurred in subjects treated with ORILISSA in the endometriosis clinical trials. ORILISSA subjects had a higher incidence of depression and mood changes compared to placebo, and ORILISSA subjects with a history of suicidality or depression had a higher incidence of depression compared to subjects without such a history [see Adverse Reactions (6.1)]. Promptly evaluate patients with depressive symptoms to determine whether the risks of continued therapy outweigh the benefits [see Adverse Reactions (6.1)]. Patients with new or worsening depression, anxiety or other mood changes should be referred to a mental health professional, as appropriate. Advise patients to seek immediate medical attention for suicidal ideation and behavior. Reevaluate the benefits and risks of continuing ORILISSA if such events occur.
5.4 Hepatic Transaminase Elevations
In clinical trials, dose-dependent elevations of serum alanine aminotransferase (ALT) at least 3-times the upper limit of the reference range occurred with ORILISSA. Use the lowest effective dose of ORILISSA and instruct patients to promptly seek medical attention in case of symptoms or signs that may reflect liver injury, such as jaundice. Promptly evaluate patients with elevations in liver tests to determine whether the benefits of continued therapy outweigh the risks [see Adverse Reactions (6.1)].
5.5 Interactions with Hormonal Contraceptives
Advise women to use effective non-hormonal contraceptives during treatment with ORILISSA and for 28 days after discontinuing ORILISSA [see Use in Specific Populations (8.1, 8.3), Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Increase in Estrogen Exposure and Potential Associated Increased Risks When ORILISSA 200 mg Twice Daily is Taken With Combined Hormonal Contraceptives
Co-administration of a combined oral contraceptive (COC) (containing 20 mcg ethinyl estradiol/0.1 mg levonorgestrel) following administration of ORILISSA 200 mg twice daily for 14 days increases the plasma ethinyl estradiol concentration by 2.2-fold compared to this COC alone. ORILISSA 200 mg twice daily co-administered with a COC containing ethinyl estradiol may lead to increased risk of ethinyl estradiol-related adverse events including thromboembolic disorders and vascular events and is not recommended [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Potential for Reduced Efficacy of Progestin-Containing Hormonal Contraceptives
Co-administration of ORILISSA 200 mg twice daily and a COC containing 0.1 mg levonorgestrel decreases the plasma concentrations of levonorgestrel by 27%, potentially affecting contraceptive efficacy. Co-administration of ORILISSA with COCs containing norethindrone acetate did not show reduction in plasma concentrations of norethindrone [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Co-administration of ORILISSA with progestin-containing intrauterine contraceptive systems has not been studied.
Reduced efficacy of ORILISSA
Based on the mechanism of action of ORILISSA, estrogen-containing contraceptives are expected to reduce the efficacy of ORILISSA. The effect of progestin-only contraceptives on the efficacy of ORILISSA is unknown.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed elsewhere in labeling:
- Bone loss [see Warnings and Precautions (5.1)]
- Change in menstrual bleeding pattern and reduced ability to recognize pregnancy [see Warnings and Precautions (5.2)]
- Suicidal ideation, suicidal behavior, and exacerbation of mood disorders [see Warnings and Precautions (5.3)]
- Hepatic transaminase elevations [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of ORILISSA was evaluated in two six-month, randomized, double-blind, placebo-controlled clinical trials [EM-1 (NCT01620528) and EM-2 (NCT01931670)] in which a total of 952 adult women with moderate to severe pain associated with endometriosis were treated with ORILISSA (475 with 150 mg once daily and 477 with 200 mg twice daily) and 734 were treated with placebo. The population age range was 18-49 years old. Women who completed six months of treatment and met eligibility criteria continued treatment in two uncontrolled, blinded six-month extension trials [EM-3 (NCT01760954) and EM-4 (NCT02143713)], for a total treatment duration of up to 12 months.
Serious Adverse Events
Overall, the most common serious adverse events reported for subjects treated with ORILISSA in the two placebo-controlled clinical trials (Studies EM-1 and EM-2) included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%). In these trials, 0.2% of subjects treated with ORILISSA 150 mg once daily and 0.2% of subjects treated with ORILISSA 200 mg twice daily discontinued therapy due to serious adverse reactions compared to 0.5% of those given placebo.
Adverse Reactions Leading to Study Discontinuation
In the two placebo-controlled clinical trials (Studies EM-1 and EM-2), 5.5% of subjects treated with ORILISSA 150 mg once daily and 9.6% of subjects treated with ORILISSA 200 mg twice daily discontinued therapy due to adverse reactions compared to 6.0% of those given placebo. Discontinuations were most commonly due to hot flushes or night sweats (1.1% with 150 mg once daily and 2.5% with 200 mg twice daily) and nausea (0.8% with 150 mg once daily and 1.5% with 200 mg twice daily) and were dose-related. The majority of discontinuations due to hot flushes or night sweats (10 of 17, 59%) and nausea (7 of 11, 64%) occurred within the first 2 months of therapy.
In the two extension trials (Studies EM-3 and EM-4), discontinuations were most commonly due to decreased BMD and were dose-related. In these trials, 0.3% of subjects treated with ORILISSA 150 mg once daily and 3.6% of subjects treated with ORILISSA 200 mg twice daily discontinued therapy due to decreased BMD.
Common Adverse Reactions:
Adverse reactions reported in ≥ 5% of women in the two placebo-controlled trials in either ORILISSA dose group and at a greater frequency than placebo are noted in the following table.
| ORILISSA
150 mg Once Daily N=475 | ORILISSA
200 mg Twice Daily N=477 | Placebo
N=734 |
|
| % | % | % | |
| Hot Flush | 24 | 46 | 9 |
| Headache | 17 | 20 | 12 |
| Nausea | 11 | 16 | 13 |
| Insomnia | 6 | 9 | 3 |
| Mood altered, mood swings | 6 | 5 | 3 |
| Amenorrhea | 4 | 7 | <1 |
| Depressed mood, depression, depressive symptoms and/or tearfulness | 3 | 6 | 2 |
| Anxiety | 3 | 5 | 3 |
| Arthralgia | 3 | 5 | 3 |
The most commonly reported adverse reactions in the extension trials (EM-3 and EM-4) were similar to those in the placebo-controlled trials.
Less Common Adverse Reactions:
In Study EM-1 and Study EM-2, adverse reactions reported in ≥ 3% and < 5% in either ORILISSA dose group and greater than placebo included: decreased libido, diarrhea, abdominal pain, weight gain, dizziness, constipation and irritability.
Bone Loss
The effect of ORILISSA on BMD was assessed by dual-energy X-ray absorptiometry (DXA).
In Studies EM-1 and EM-2, there was a dose-dependent decrease in BMD in ORILISSA-treated subjects compared to an increase in placebo-treated subjects.
In Study EM-1, compared to placebo, the mean change from baseline in lumbar spine BMD at 6 months was -0.9% (95% CI: -1.3, -0.4) with ORILISSA 150 mg once daily and -3.1% (95% CI: -3.6, -2.6) with ORILISSA 200 mg twice daily (Table 3). The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the placebo-controlled treatment period was 2% with ORILISSA 150 mg once daily, 7% with ORILISSA 200 mg twice daily and < 1% with placebo. In the blinded extension Study EM-3, continued bone loss was observed with 12 months of continuous treatment with ORILISSA. The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the extension treatment period was 8% with continuous ORILISSA 150 mg once daily and 21% with continuous ORILISSA 200 mg twice daily.
In Study EM-2, compared to placebo, the mean change from baseline in lumbar spine BMD at 6 months was -1.3% (95% CI: -1.8, -0.8) with ORILISSA 150 mg once daily and -3.0% (95% CI: -3.5, -2.6) with ORILISSA 200 mg twice daily (Table 3). The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the placebo-controlled treatment period was < 1% with ORILISSA 150 mg once daily, 6% with ORILISSA 200 mg twice daily and 0% with placebo. In the blinded extension Study EM-4, continued bone loss was observed with 12 months of continuous treatment with ORILISSA. The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the extension treatment period was 2% with continuous ORILISSA 150 mg once daily and 21% with continuous ORILISSA 200 mg twice daily.
| ORILISSA
150 mg Once Daily | ORILISSA
200 mg Twice Daily | Placebo | |
| EM-1 | |||
| N | 183 | 180 | 277 |
| Percent Change from Baseline, % | -0.3 | -2.6 | 0.5 |
| Treatment Difference, % (95% CI) | -0.9 (-1.3, -0.4) | -3.1 (-3.6, -2.6) | |
| EM-2 | |||
| N | 174 | 183 | 271 |
| Percent Change from Baseline, % | -0.7 | -2.5 | 0.6 |
| Treatment Difference, % (95% CI) | -1.3 (-1.8, -0.8) | -3.0 (-3.5, -2.6) | |
To assess for recovery, the change in lumbar spine BMD over time was analyzed for subjects who received continuous treatment with ORILISSA 150 mg once daily or ORILISSA 200 mg twice daily for up to 12 months and who were then followed after cessation of therapy for an additional 6 months. Partial recovery of BMD was seen in these subjects (Figure 1).
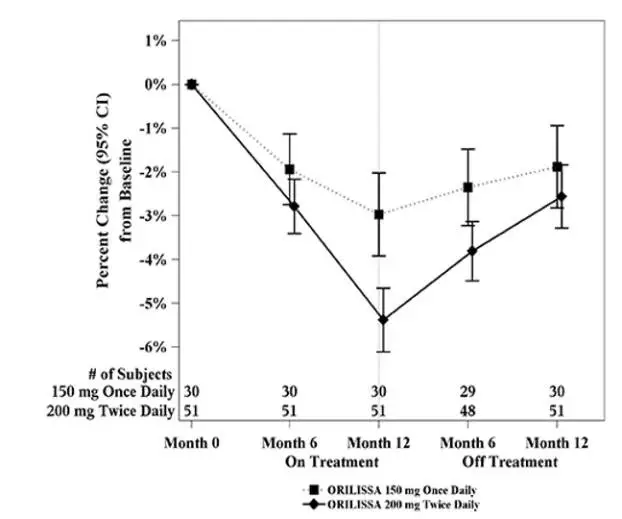
In Study EM-3, if a subject had BMD loss of more than 1.5% at the lumbar spine or more than 2.5% at the total hip at the end of treatment, follow-up DXA was required after 6 months off-treatment. In Study EM-4, all subjects were required to have a follow-up DXA 6 months off treatment regardless of change in BMD and if a subject had BMD loss of more than 1.5% at the lumbar spine or more than 2.5% at the total hip after 6 months off treatment, follow-up DXA was required after 12 months off-treatment. Figure 2 shows the change in lumbar spine BMD for the subjects in Study EM-2/EM-4 who completed 12 months of treatment with ORILISSA and who had a follow-up DXA 12-months off treatment.
Figure 1. Percent Change from Baseline in Lumbar Spine BMD in Subjects Who Received 12 Months of ORILISSA and Had Follow-up BMD 6 Months off Therapy in Studies EM-2/EM-4
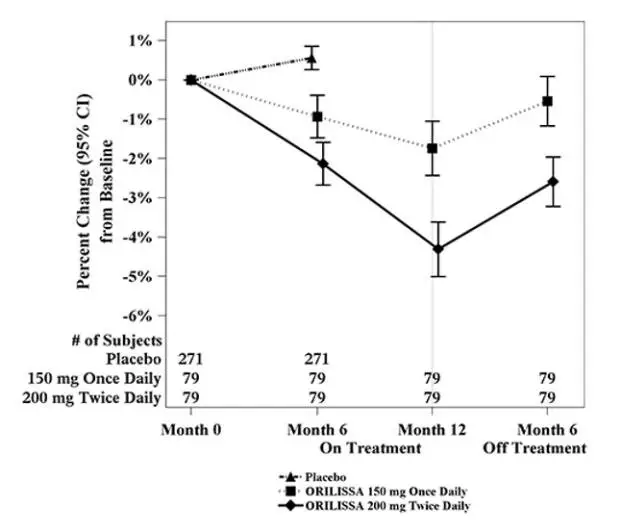
Figure 2. Percent Change from Baseline in Lumbar Spine BMD in Subjects Who Received 12 Months of ORILISSA and Had Follow-up BMD 12 Months off Therapy in Studies EM-2/EM-4
Suicidal Ideation, Suicidal Behavior and Exacerbation of Mood Disorders
In the placebo-controlled trials (Studies EM-1 and EM-2), ORILISSA was associated with adverse mood changes (see Table 2 and Table 4), particularly in those with a history of depression.
| ORILISSA | |||
|
Adverse Reactions | 150 mg
Once Daily (N=475) n (%) | 200 mg
Twice Daily (N=477) n (%) | Placebo
(N=734) n (%) |
| Completed suicide | 1 (0.2) | 0 | 0 |
| Suicidal ideation | 1 (0.2) | 1 (0.2) | 0 |
A 44-year-old woman received 31 days of ORILISSA 150 mg once daily then completed suicide 2 days after ORILISSA discontinuation. She had no relevant past medical history; life stressors were noted.
Among the 2090 subjects exposed to ORILISSA in the endometriosis Phase 2 and Phase 3 studies, there were four reports of suicidal ideation. In addition to the two subjects in Table 4, there were two additional reports of suicidal ideation: one subject in EM-3 (150 mg once daily) and one in a Phase 2 study (75 mg once daily, an unapproved dose). Three of these subjects had a history of depression. Two subjects discontinued ORILISSA and two completed the clinical trial treatment periods.
Hepatic Transaminase Elevations
In the placebo-controlled clinical trials (Studies EM-1 and EM-2), dose-dependent asymptomatic elevations of serum ALT to at least 3-times the upper limit of the reference range occurred during treatment with ORILISSA (150 mg once daily – 1/450, 0.2%; 200 mg twice daily – 5/443, 1.1%; placebo – 1/696, 0.1%). Similar increases were seen in the extension trials (Studies EM-3 and EM-4).
Changes in Lipid Parameters
Dose-dependent increases in total cholesterol, low-density lipoprotein cholesterol (LDL-C), high density lipoprotein cholesterol (HDL-C), and serum triglycerides were noted during ORILISSA treatment in EM-1 and EM-2. In EM-1 and EM-2, 12% and 1% of subjects with mildly elevated LDL-C (130-159 mg/dL) at baseline had an increase in LDL-C concentrations to 190 mg/dL or higher during treatment with ORILISSA and placebo, respectively. In EM-1 and EM-2, 4% and 1% of subjects with mildly elevated serum triglycerides (150-300 mg/dL) at baseline had an increase in serum triglycerides to at least 500 mg/dL during treatment with ORILISSA and placebo, respectively. The highest measured serum triglyceride concentration during treatment with ORILISSA was 982 mg/dL.
| ORILISSA
150 mg Once Daily N=475 | ORILISSA
200 mg Twice Daily N=477 | Placebo
N=734 |
|
| LDL-C (mg/dL) | |||
| Mean change at Month 6 | 5 | 13 | -3 |
| Maximum increase during Treatment Period | 137 | 107 | 122 |
| HDL-C (mg/dL) | |||
| Mean change at Month 6 | 2 | 4 | 1 |
| Maximum increase during Treatment Period | 43 | 52 | 45 |
| Triglycerides (mg/dL) | |||
| Mean change at Month 6 | <1 | 11 | -3 |
| Maximum increase during Treatment Period | 624 | 484 | 440 |
Lipid increases occurred within 1 to 2 months after the start of ORILISSA and remained stable thereafter over 12 months.
Hypersensitivity Reactions
In Studies EM-1 and EM-2, non-serious hypersensitivity reactions including rash occurred in 5.8% of ORILISSA treated-subjects and 6.1% of placebo-treated subjects. These events led to study drug discontinuation in 0.4% of ORILISSA-treated subjects and 0.5% of placebo-treated subjects.
Effects on Menstrual Bleeding Patterns
The effects of ORILISSA on menstrual bleeding were evaluated for up to 12 months using an electronic daily diary where subjects classified their flow of menstrual bleeding (if present in the last 24 hours) as spotting, light, medium, or heavy. ORILISSA led to a dose-dependent reduction in mean number of bleeding and spotting days and bleeding intensity in those subjects who reported menstrual bleeding.
| ORILISSA
150mg Once Daily | ORILISSA
200mg Twice Daily | Placebo | ||||
| Baseline | Month 3 | Baseline | Month 3 | Baseline | Month 3 | |
| Mean bleeding/ spotting days in prior 28 days | 5.3 | 2.8 | 5.7 | 0.8 | 5.4 | 4.6 |
| Mean Intensity scorea | 2.6 | 2.2 | 2.5 | 2.0 | 2.6 | 2.4 |
| aIntensity for subjects who reported at least 1 day of bleeding or spotting during 28 day interval. Scale ranges from 1 to 4, 1 = spotting, 2 = light, 3 = medium, 4 = heavy | ||||||
ORILISSA also demonstrated a dose-dependent increase in the percentage of women with amenorrhea (defined as no bleeding or spotting in a 56-day interval) over the treatment period. The incidence of amenorrhea during the first six months of treatment ranged from 6-17% for ORILISSA 150 mg once daily, 13-52% for ORILISSA 200 mg twice daily and less than 1% for placebo. During the second 6 months of treatment, the incidence of amenorrhea ranged from 11-15% for ORILISSA 150 mg once daily and 46-57% for ORILISSA 200 mg twice daily.
After 6 months of therapy with ORILISSA 150 mg once daily, resumption of menses after stopping treatment was reported by 59%, 87% and 95% of women within 1, 2, and 6 months, respectively. After 6 months of therapy with ORILISSA 200 mg twice daily, resumption of menses after stopping treatment was reported by 60%, 88%, and 97% of women within 1, 2, and 6 months, respectively.
After 12 months of therapy with ORILISSA 150 mg once daily resumption of menses after stopping treatment was reported by 77%, 95% and 98% of women within 1, 2, and 6 months respectively. After 12 months of therapy with ORILISSA 200 mg twice daily resumption of menses after stopping treatment was reported by 55%, 91% and 96% of women within 1, 2, and 6 months respectively.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ORILISSA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune system disorders: hypersensitivity reactions (including anaphylaxis, angioedema, and urticaria).
7. Drug Interactions
7.1 Potential for ORILISSA to Affect Other Drugs
Elagolix is:
- A weak to moderate inducer of cytochrome P450 (CYP) 3A. Co-administration with ORILISSA may decrease plasma concentrations of drugs that are substrates of CYP3A (see Table 7).
- A weak inhibitor of CYP 2C19. Co-administration with ORILISSA may increase plasma concentrations of drugs that are substrates of CYP2C19 (see Table 7).
- An inhibitor of efflux transporter P-glycoprotein (P-gp). Co-administration with ORILISSA may increase plasma concentrations of drugs that are substrates of P-gp (see Table 7).
The effects of co-administration of ORILISSA on concentrations of concomitant drugs and the clinical recommendations for these drug interactions are summarized in Table 7.
| Concomitant
Drug Class: Drug Name | Effect on Plasma
Exposure of Concomitant Drug | Clinical Recommendations |
| Cardiac glycosides: digoxin | ↑ digoxin | Increase monitoring of digoxin concentrations and potential signs and symptoms of clinical toxicity when initiating ORILISSA in patients who are taking digoxin. If ORILISSA is discontinued, increase monitoring of digoxin concentrations. |
| Benzodiazepines: oral midazolam | ↓ midazolam | Consider increasing the dose of midazolam by no more than 2-fold and individualize midazolam therapy based on the patient’s response. |
| Statins: rosuvastatin | ↓ rosuvastatin | Monitor lipid levels and adjust the dose of rosuvastatin, if necessary. |
| Proton pump inhibitors: omeprazole | ↑ omeprazole | No dose adjustment needed for omeprazole 40 mg once daily when co-administered with ORILISSA. When ORILISSA is used concomitantly with higher doses of omeprazole, consider dosage reduction of omeprazole. |
| Combined hormonal contraceptives: oral ethinyl estradiol/levonorgestrel | ↑ethinyl estradiol ↓levonorgestrel | Advise women to use effective non-hormonal contraception during treatment with ORILISSA and for 28 days after discontinuing ORILISSA. |
| See Tables 10 and 11 [see Clinical Pharmacology (12.3)]. The direction of the arrow indicates the direction of the change in the area under the curve (AUC) (↑= increase, ↓ = decrease). |
||
7.2 Potential for Other Drugs to Affect ORILISSA
Elagolix is a substrate of CYP3A, P-gp, and OATP1B1.
Concomitant use of ORILISSA 200 mg twice daily and strong CYP3A inhibitors for more than 1 month is not recommended. Limit concomitant use of ORILISSA 150 mg once daily and strong CYP3A inhibitors to 6 months.
Co-administration of ORILISSA with strong CYP3A inducers may decrease elagolix plasma concentrations and may result in a decrease of the therapeutic effects of ORILISSA.
Concomitant use of ORILISSA 200 mg twice daily and rifampin is not recommended. Limit concomitant use of ORILISSA 150 mg once daily and rifampin to 6 months.
The effect of concomitant use of P-gp inhibitors or inducers on the pharmacokinetics of ORILISSA is unknown. OATP1B1 inhibitors that are known or expected to significantly increase elagolix plasma concentrations are contraindicated due to increased risk of elagolix-associated adverse reactions [see Contraindications (4)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy registry that monitors pregnancy outcomes in women exposed to ORILISSA during pregnancy. Healthcare providers are encouraged to register patients, or pregnant women may enroll themselves in the registry by calling 1-833-782-7241 or visiting https://www.bloompregnancyregistry.com.
Risk Summary
Use of ORILISSA is contraindicated in pregnant women. Exposure to ORILISSA early in pregnancy may increase the risk of early pregnancy loss. Discontinue ORILISSA if pregnancy occurs during treatment.
The limited human data with the use of ORILISSA in pregnant women are insufficient to determine whether there is a risk for major birth defects or miscarriage. Although two cases of congenital malformations were reported in clinical trials with ORILISSA, no pattern was identified and miscarriages were reported at a similar incidence across treatment groups (see Data).
When pregnant rats and rabbits were orally dosed with elagolix during the period of organogenesis, postimplantation loss was observed in pregnant rats at doses 20 times the maximum recommended human dose (MRHD). Spontaneous abortion and total litter loss was observed in rabbits at doses 7 and 12 times the MRHD. There were no structural abnormalities in the fetuses at exposures up to 40 and 12 times the MRHD for the rat and rabbit, respectively (see Data).
Data
Human Data
There were 49 pregnancies reported in clinical trials of more than 3,500 women (of whom more than 2,000 had endometriosis) treated with ORILISSA for up to 12 months. These pregnancies occurred while the women were receiving ORILISSA or within 30 days after stopping ORILISSA. Among these 49 pregnancies, two major congenital malformations were reported. In one case of infant cleft palate, the mother was treated with ORILISSA 150 mg daily and the estimated fetal exposure to ORILISSA occurred during the first 30 days of pregnancy. In one case of infant tracheoesophageal fistula, the mother was treated with ORILISSA 150 mg daily and the estimated fetal exposure to ORILISSA occurred during the first 15 days of pregnancy.
Among these 49 pregnancies, there were five cases of spontaneous abortion (miscarriage) compared to five cases among the 20 pregnancies that occurred in more than 1100 women treated with placebo. Although the duration of fetal exposure was limited in ORILISSA clinical trials, there were no apparent decreases in birth weights associated with ORILISSA in comparison to placebo.
Animal Data
Embryofetal development studies were conducted in the rat and rabbit. Elagolix was administered by oral gavage to pregnant rats (25 animals/dose) at doses of 0, 300, 600 and 1200 mg/kg/day and to rabbits (20 animals/dose) at doses of 0, 100, 150, and 200 mg/kg/day, during the period of organogenesis (gestation day 6-17 in the rat and gestation day 7-20 in the rabbit).
In rats, maternal toxicity was present at all doses and included six deaths and decreases in body weight gain and food consumption. Increased postimplantation losses were present in the mid dose group, which was 20 times the MRHD based on AUC. In rabbits, three spontaneous abortions and a single total litter loss were observed at the highest, maternally toxic dose, which was 12 times the MRHD based on AUC. A single total litter loss occurred at a lower non-maternally toxic dose of 150 mg/kg/day, which was 7 times the MRHD.
No fetal malformations were present at any dose level tested in either species even in the presence of maternal toxicity. At the highest doses tested, the exposure margins were 40 and 12 times the MRHD for the rat and rabbit, respectively. However, because elagolix binds poorly to the rat gonadotropin-releasing hormone (GnRH) receptor (~1000 fold less than to the human GnRH receptor), the rat study is unlikely to identify pharmacologically mediated effects of elagolix on embryofetal development. The rat study is still expected to provide information on potential non-target-related effects of elagolix.
In a pre- and postnatal development study in rats, elagolix was given in the diet to achieve doses of 0, 100 and 300 mg/kg/day (25 per dose group) from gestation day 6 to lactation day 20. There was no evidence of maternal toxicity. At the highest dose, two dams had total litter loss, and one failed to deliver. Pup survival was decreased from birth to postnatal day 4. Pups had lower birth weights and lower body weight gains were observed throughout the pre-weaning period at 300 mg/kg/day. Smaller body size and effect on startle response were associated with lower pup weights at 300 mg/kg/day. Post-weaning growth, development and behavioral endpoints were unaffected.
Maternal plasma concentrations in rats on lactation day 21 at 100 and 300 mg/kg/day (47 and 125 ng/mL) were 0.06-fold and 0.16-fold the maximal elagolix concentration (Cmax) in humans at the MRHD. Because the exposures achieved in rats were much lower than the human MRHD, this study is not predictive of potentially higher lactational exposure in humans.
8.3 Females and Males of Reproductive Potential
Based on the mechanism of action, there is a risk of early pregnancy loss if ORILISSA is administered to a pregnant woman [see Use in Specific Populations (8.1), Clinical Pharmacology (12.1)].
Pregnancy Testing
ORILISSA may delay the ability to recognize the occurrence of a pregnancy because it may reduce the intensity, duration, and amount of menstrual bleeding. Exclude pregnancy before initiating treatment with ORILISSA. Perform pregnancy testing if pregnancy is suspected during treatment with ORILISSA and discontinue treatment if pregnancy is confirmed [see Contraindications (4) and Warnings and Precautions (5.2)].
Contraception
Advise women to use effective non-hormonal contraception during treatment with ORILISSA and for 28 days after discontinuing ORILISSA [see Warnings and Precautions (5.5)].
8.7 Hepatic Impairment
No dosage adjustment of ORILISSA is required for women with mild hepatic impairment (Child-Pugh A). Only the 150 mg once daily regimen is recommended for women with moderate hepatic impairment (Child-Pugh B) and the duration of treatment should be limited to 6 months.
ORILISSA is contraindicated in women with severe hepatic impairment (Child-Pugh C) [see Contraindications (4), and Clinical Pharmacology (12.3)].
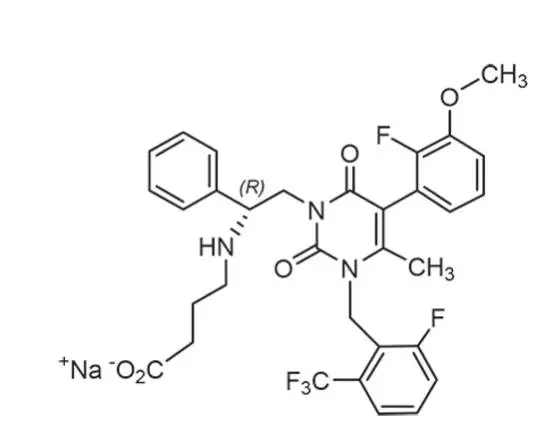
12. Orilissa - Clinical Pharmacology
12.1 Mechanism of Action
ORILISSA is a GnRH receptor antagonist that inhibits endogenous GnRH signaling by binding competitively to GnRH receptors in the pituitary gland. Administration of ORILISSA results in dose-dependent suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), leading to decreased blood concentrations of the ovarian sex hormones, estradiol and progesterone.
12.3 Pharmacokinetics
The pharmacokinetic properties of ORILISSA in healthy subjects are summarized in Table 8. The steady state pharmacokinetic parameters under fasting conditions are summarized in Table 9.
| Absorption | |
| Tmax (h) | 1.0 |
| Effect of high-fat meal (relative to fasting) | AUC: ↓24%, Cmax: ↓36% |
| Distribution | |
| % Bound to human plasma proteins | 80 |
| Blood-to-plasma ratio | 0.6 |
| Metabolism | |
| Metabolism | CYP3A (major) Minor pathways include: CYP2D6, CYP2C8, and uridine glucuronosyl transferases (UGTs) |
| Elimination | |
| Major route of elimination | Hepatic metabolism |
| Terminal phase elimination half-life (t1/2) (h) | 4-6 |
| % of dose excreted in urine | <3 |
| % of dose excreted in feces | 90 |
| Pharmacokinetic Parameter
(Units) | 150 mg Once Daily
N = 6 | 200 mg Twice Daily
N = 7 |
| Cmax (ng/mL) | 574 (29) | 774 (68) |
| AUCτ (ng●hr/mL) | 1292 (31) | 1725 (57) |
| CL/F (L/hr) | 123 (21) | 144 (43) |
| Vdss/F | 1674 (94) | 881 (38) |
| Rac | 0.98 (7) | 0.89 (19) |
| CV: Coefficient of variation Cmax: peak concentration AUCτ: area under the plasma concentration-time curve during the dosing interval (τ) i.e., 12 hours for twice daily regimen, 24 hours for once daily regimen. CL/F: oral clearance Vdss/F: apparent volume of distribution at steady state Rac: drug accumulation ratio |
||
Specific Populations
Patients with Renal Impairment
Elagolix exposures (Cmax and AUC) are not altered by renal impairment. The mean exposures are similar for women with moderate to severe or end stage renal disease (including women on dialysis) compared to women with normal renal function.
Patients with Hepatic Impairment
Elagolix exposures (Cmax and AUC) are similar between women with normal hepatic function and women with mild hepatic impairment. Elagolix exposures in women with moderate and severe hepatic impairment are approximately 3-fold and 7-fold, respectively, higher than exposures from women with normal hepatic function [see Use in Specific Populations (8.7)].
Racial or Ethnic Groups
No clinically meaningful difference in the pharmacokinetics of ORILISSA between White and Black subjects or between Hispanics and others was observed. There is no clinically meaningful difference in the pharmacokinetics of ORILISSA between Japanese and Han Chinese subjects.
Body weight/Body mass index
Body weight or body mass index does not affect the pharmacokinetics of ORILISSA.
Drug Interaction Studies
Drug interaction studies were performed with ORILISSA and other drugs that are likely to be co-administered and with drugs commonly used as probes for pharmacokinetic interactions. Tables 10 and 11 summarize the pharmacokinetic effects when elagolix was co-administered with these drugs.
| Co-
administered Drug | Regimen
of Co- administered Drug | Regimen
of Elagolix | N | Ratio (90% CI)* | |
| Ketoconazole | 400 mg once daily | 150 mg single dose | 11 | Cmax | AUC |
| 1.77 (1.48 – 2.12) | 2.20 (1.98 – 2.44) |
||||
| Rifampin# | 600 mg single dose | 150 mg single dose | 12 | 4.37 (3.62 – 5.28) | 5.58 (4.88 – 6.37) |
| 600 mg once daily | 2.00 (1.66 – 2.41) | 1.65 (1.45 – 1.89) |
|||
| CI: Confidence interval *ratios for Cmax and AUC compare co-administration of the medication with elagolix vs. administration of elagolix alone. # A single dose of 600 mg rifampin inhibits OATP1B1; 600 mg once daily dose of rifampin inhibits OATP1B1 and induces CYP3A. |
|||||
No clinically significant changes in elagolix exposures were observed when co-administered with rosuvastatin (20 mg once daily), sertraline (25 mg once daily) or fluconazole (200 mg single dose).
| Co-
administered Drug | Regimen
of Co- administered Drug | Regimen
of Elagolix | N | Ratio (90% CI)* | |
| Digoxin | 0.5 mg single dose | 200 mg twice daily x 10 days | 11 | Cmax | AUC |
| 1.71 (1.53 – 1.91) | 1.26 (1.17 – 1.35) |
||||
| Rosuvastatin | 20 mg once daily | 300 mg twice daily x 7 days | 10 | 0.99 (0.73 – 1.35) | 0.60 (0.50 – 0.71) |
| Midazolam | 2 mg single dose | 300 mg twice daily x 11 days | 20 | 0.56 (0.51 – 0.62) | 0.46 (0.41 – 0.50) |
| 150 mg once daily x 13 days | 11 | 0.81 (0.74 – 0.89) | 0.65 (0.58 – 0.72) |
||
| Norethindrone | 0.35 mg once daily x 112 days | 150 mg once daily x 56 days | 32 | 0.95 (0.86 – 1.05) | 0.88 (0.79 – 0.99) |
| Ethinyl Estradiol | Ethinyl estradiol 35 mcg and triphasic norgestimate 0.18/0.215/0.25 mg once daily | 150 mg once daily | 21 | 1.15 (1.07 – 1.25) | 1.30 (1.19 – 1.42) |
| Norelgestromina | 0.87 (0.78 – 0.97) | 0.85 (0.78 – 0.92) |
|||
| Norgestrela | 0.89 (0.78 – 1.00) | 0.92 (0.84 – 1.01) |
|||
| Ethinyl Estradiol | Ethinyl estradiol 20 mcg/Levonorgestrel 0.1 mg single dose | 200 mg twice daily x 15 days | 20 | 1.36 (1.27 – 1.45) | 2.18 (1.99 – 2.39) |
| Levonorgestrel | 0.97 (0.88 – 1.07) | 0.73 (0.64 – 0.82) |
|||
| Omeprazole | 40 mg single dose | 300 mg twice daily x 9 days | 20 | 1.95 (1.50 – 2.53) | 1.78 (1.39 – 2.27) |
| CI: Confidence interval *ratios for Cmax and AUC compare co-administration of the medication with elagolix vs. administration of the medication alone. a metabolite of norgestimate |
|||||
No clinically significant changes were observed in exposures of sertraline, fluconazole, or bupropion when co-administered with elagolix 300 mg twice daily.
14. Clinical Studies
The efficacy of ORILISSA 150 mg once daily and 200 mg twice daily for the management of moderate to severe pain associated with endometriosis was demonstrated in two multinational double-blind, placebo-controlled trials in 1686 premenopausal women [Study EM-1 (NCT01620528) and Study EM-2 (NCT01931670)]. The median age of women in the trials was 32 years; 88% were White, 9% were Black or African American and 3% were other races. Each placebo-controlled trial assessed the reduction in endometriosis-associated pain over 6 months of treatment.
Moderate to severe pain associated with endometriosis was required for entry into the trials and was assessed during screening using the composite pelvic signs and symptoms score (CPSSS) and other baseline criteria.
The CPSSS is based on a modified Biberoglu and Behrman scale with five elements: three responses reported by study subjects (dysmenorrhea, dyspareunia, and non-menstrual pelvic pain) and two findings based on investigator assessment during physical examination (rating of pelvic tenderness and induration). Each element is scored from 0 (absent) to 3 (severe) for a maximum total score of 15. A total score of at least 6, with a score of at least 2 for dysmenorrhea and at least 2 for non-menstrual pelvic pain was required to qualify for randomization. Subjects were also required to have non-menstrual pelvic pain for at least four days in the preceding calendar month, defined as 35 days. Other criteria to determine eligibility for randomization included subject responses in a daily electronic diary (Endometriosis Daily Pain Impact Scale, described below) for both dysmenorrhea and non-menstrual pelvic pain in the 35 days prior to randomization.
Dysmenorrhea and Non-Menstrual Pelvic Pain
The co-primary efficacy endpoints were (1) the proportion of subjects whose dysmenorrhea responded to treatment at Month 3 and (2) the proportion of subjects whose pelvic pain not related to menses (also known as non-menstrual pelvic pain) responded to treatment at Month 3. Dysmenorrhea and non-menstrual pelvic pain were evaluated daily using the Endometriosis Daily Pain Impact Scale that asked subjects to rate their pain severity and its impact on daily activities during the prior 24 hours as none, mild, moderate or severe (correlating with a score of 0 to 3, respectively, where higher scores indicated greater severity). Scores at baseline and at each month were averaged over a 35-day interval.
Women were defined as responders if they experienced a reduction in dysmenorrhea and non-menstrual pelvic pain as defined in Table 12 with no increase in analgesic use (nonsteroidal anti-inflammatory drug or opioid) for endometriosis-associated pain. The threshold for defining responders was based on a receiver operating characteristic (ROC) analysis using the patient global impression of change as an anchor. A higher proportion of women treated with ORILISSA 150 mg once daily or 200 mg twice daily were responders for dysmenorrhea and non-menstrual pelvic pain compared to placebo in a dose-dependent manner at Month 3 [see Table 12].
| Study EM-1 | Study EM-2 | |||||
| ORILISSA | Placebo | ORILISSA | Placebo | |||
| 150 mg
Once Daily N=248 | 200 mg
Twice Daily N=244 | N=373 | 150 mg
Once Daily N=221 | 200 mg
Twice Daily N=225 | N=353 | |
| Dysmenorrhea Difference from placebo | 46% 27%** | 76% 56%** | 20% | 43% 21%** | 72% 50%** | 23% |
| Non-Menstrual Pelvic Pain Difference from placebo | 50% 14%** | 55% 18%** | 36% | 50% 13%* | 58% 21%** | 37% |
| † Study EM-1-Dysmenorrhea responder threshold: at least 0.81 point decrease from baseline in dysmenorrhea score; Non-Menstrual Pelvic Pain responder threshold: at least 0.36 point decrease from baseline in Non-Menstrual Pelvic Pain score Study EM-2 - Dysmenorrhea responder threshold: at least 0.85 point decrease from baseline in dysmenorrhea score; Non-Menstrual Pelvic Pain responder threshold: at least 0.43 point decrease from baseline in Non-Menstrual Pelvic Pain score *p ≤0.01 for test of difference from placebo **p≤0.001 for test of difference from placebo |
||||||
Women in these studies also provided a daily self-assessment of their endometriosis pain using a numeric rating scale (NRS) that asked subjects to rate their endometriosis pain at its worst over the last 24 hours on a scale from 0 (no pain) to 10 (worst pain ever). In Study EM-1, baseline NRS scores were 5.7 for ORILISSA 150 mg once daily, 5.5 for ORILISSA 200 mg twice daily and 5.6 for placebo. In Study EM-2, baseline NRS scores were 5.7 for ORILISSA 150 mg once daily, 5.3 for ORILISSA 200 mg twice daily and 5.6 for placebo. Women taking ORILISSA 150 mg once daily and 200 mg twice daily reported a statistically (p <0.001) significant reduction from baseline in NRS scores compared to placebo at Month 3 in both Studies EM-1 and EM-2 (Study EM-1: 0.7 points for ORILISSA 150 mg once daily and 1.3 points for ORILISSA 200 mg twice daily; Study EM-2: 0.6 points for ORILISSA 150 mg once daily and 1.2 points for ORILISSA 200 mg twice daily).
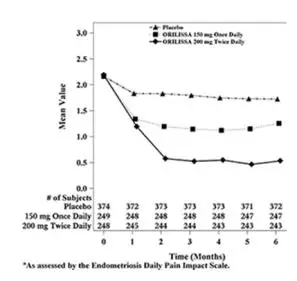
In addition, both ORILISSA treatment groups showed statistically significantly greater mean decreases from baseline compared to placebo in dysmenorrhea and non-menstrual pelvic pain scores at Month 6. Figures 3 through 6 show the mean scores for dysmenorrhea and non-menstrual pelvic pain over time for Study EM-1 and EM-2.
Figure 3. Mean Dysmenorrhea Pain Scoresa in Study EM-1 Over 6 Months
 | Figure 4. Mean Dysmenorrhea Pain Scoresa in Study EM-2 Over 6 Months
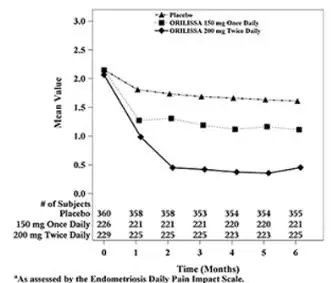 |
Figure 5. Mean Non-Menstrual Pelvic Paina Scores in Study EM-1 Over 6 Months
 | Figure 6. Mean Non-Menstrual Pelvic Paina Scores in Study EM-2 Over 6 Months
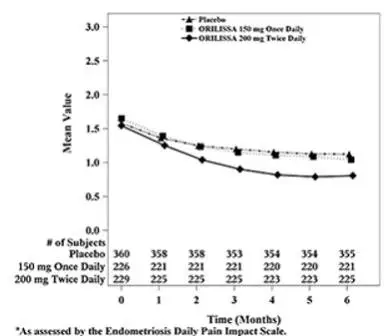 |
Dyspareunia
Dyspareunia associated with endometriosis was evaluated as a secondary endpoint using the Endometriosis Daily Pain Impact Scale that asked subjects to rate their pain during sexual intercourse in the prior 24 hours as none, mild, moderate, severe (correlating with a score of 0 to 3, respectively, where higher scores indicated greater severity), or not applicable. In both Studies EM-1 and EM-2, women treated with ORILISSA 200 mg twice daily showed statistically significantly greater reduction in dyspareunia from baseline to Month 3 than women given placebo (Study EM-1: 0.2; Study EM-2: 0.3). Figures 7 and 8 show the mean scores over time for Study EM-1 and EM-2.
Figure 7. Mean Dyspareunia Scoresa in Study EM-1 Over 3 Months
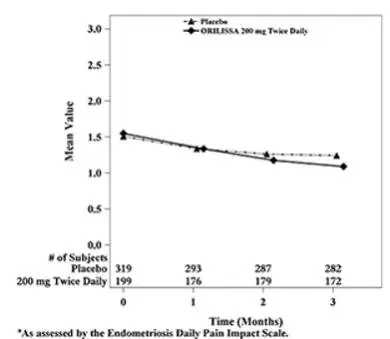 | Figure 8. Mean Dyspareunia Scoresa in Study EM-2 Over 3 Months
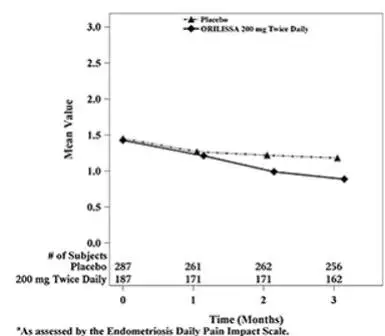 |
Use of rescue pain medication
In EM-1 and EM-2, 59% and 60% of patients used an opioid rescue analgesic for pain at baseline. The opioid rescue analgesics used at baseline were predominantly hydrocodone/acetaminophen (HC/APAP) and codeine/APAP at strengths of 5/300-325 mg and 30/300-500 mg. In EM-1, of all patients on an opioid at baseline, 98% and 2% were on HC/APAP and codeine/APAP, respectively. In EM-2, of all patients on an opioid at baseline, 50% were on HC/APAP and 16% were on codeine/APAP.
Other data related to opioid rescue analgesic use are summarized in Table 13.
| Study EM-1 | Study EM-2 | |||||
| ORILISSA
150 mg Once Daily | ORILISSA
200 mg Twice Daily | Placebo | ORILISSA
150 mg Once Daily | ORILISSA
200 mg Twice Daily | Placebo | |
| Tablets per month at baseline (mean±SD) | 15 ±24 | 15 ±25 | 13 ±21 | 13 ±29 | 12 ±26 | 12 ±21 |
| Tablets per month at baseline [Median (Min, Max)] | 4 (0, 184) | 4 (0, 195) | 4 (0, 146) | 4 (0, 236) | 3 (0, 214) | 4 (0, 152) |
| Tablets per month at Month 3 (mean±SD) | 12 ±29 | 7 ±18 | 10 ±17 | 8 ±22 | 5 ±14 | 8 ±15 |
| Tablets per month at Month 3 [Median (Min, Max)] | 0 (0, 251) | 0 (0, 162) | 2 (0, 144) | 0 (0, 168) | 0 (0, 136) | 2 (0, 142) |
| Tablets per month at Month 6 (mean±SD) | 11 ±26 | 7 ±17 | 11 ±19 | 7 ±19 | 5 ±14 | 8 ±15 |
| Tablets per month at Month 6 [Median (Min, Max)] | 0 (0, 224) | 0 (0, 157) | 3 (0, 185) | 0 (0, 185) | 0 (0, 157) | 2 (0, 142) |
| Number and % of patients on any dose of opioid rescue at baseline who were off opioid at Month 3* | 46/150 (31%) | 59/151 (39%) | 36/211 (17%) | 44/124 (35%) | 68/134 (51%) | 54/220 (25%) |
| Number and % of patients on any dose of opioid rescue at baseline who were off opioid at Month 6* | 43/149 (29%) | 66/150 (44%) | 36/211 (17%) | 50/124 (40%) | 78/134 (58%) | 70/222 (32%) |
| Number and % of patients not on opioid rescue at baseline who were on any opioid at Month 3^ | 9/98 (9%) | 6/93 (6%) | 17/162 (10%) | 10/97 (10%) | 10/91 (11%) | 29/133 (22%) |
| Number and % of patients not on opioid rescue at baseline who were on any opioid at Month 6^ | 16/98 (16%) | 6/93 (6%) | 32/161 (20%) | 13/97 (13%) | 6/91 (7%) | 32/133 (24%) |
| Min = minimum; Max = maximum; SD = standard deviation Monthly calculations are based on a 35-day interval. *Denominator is the number of subjects on opioid rescue at baseline. ^Denominator is the number of subjects not on opioid rescue at baseline. |
||||||
The clinical relevance of these data has not been demonstrated.
Medication Guide
| MEDICATION GUIDE
ORILISSA® (awr-ah-lih-sah) (elagolix) tablets, for oral use |
| What is the most important information I should know about ORILISSA?
ORILISSA may cause serious side effects, including:
|
| What is ORILISSA?
ORILISSA is a prescription medicine used to treat moderate to severe pain associated with endometriosis. It is not known if ORILISSA is safe and effective in children. |
Do not take ORILISSA if you:
|
Before you take ORILISSA, tell your healthcare provider about all of your medical conditions, including if you:
Especially tell your healthcare provider if you take:
|
How should I take ORILISSA?
|
| What are the possible side effects of ORILISSA?
ORILISSA may cause serious side effects including:
These are not all the possible side effects of ORILISSA. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
How should I store ORILISSA?
|
| General information about the safe and effective use of ORILISSA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ORILISSA for a condition for which it was not prescribed. Do not give ORILISSA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ORILISSA that is written for health professionals. |
| What are the ingredients in ORILISSA?
Active ingredient: elagolix Inactive ingredients 150 mg tablets: mannitol, sodium carbonate monohydrate, pregelatinized starch, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and carmine high tint. Inactive ingredients 200 mg tablets: mannitol, sodium carbonate monohydrate, pregelatinized starch, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide red. Manufactured by AbbVie Inc. North Chicago, IL 60064 20075867 For more information, go to www.orilissa.com or call 1-844-674-3676. |
This Medication Guide has been approved by the U.S. Food and Drug Administration
Revised: June 2023

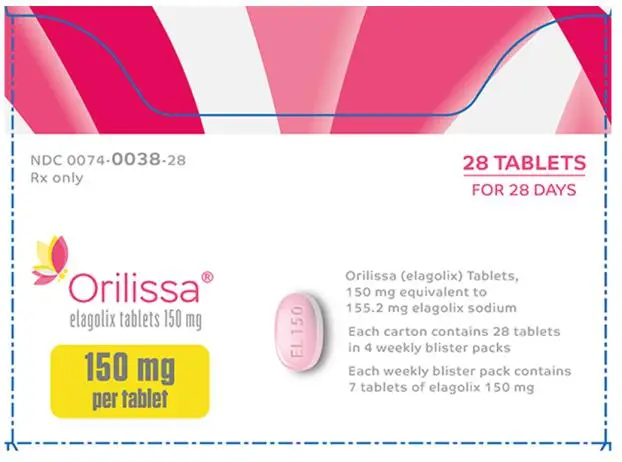

| ORILISSA
elagolix tablet, film coated |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| ORILISSA
elagolix tablet, film coated |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - AbbVie Inc. (078458370) |
| Registrant - AbbVie Inc. (078458370) |
