Drug Detail:Roctavian (Valoctocogene roxaparvovec-rvox)
Drug Class: Miscellaneous coagulation modifiers
Highlights of Prescribing Information
ROCTAVIAN (valoctocogene roxaparvovec-rvox) suspension for intravenous infusion
Initial U.S. Approval: 2023
Indications and Usage for Roctavian
ROCTAVIAN is an adeno-associated virus vector-based gene therapy indicated for the treatment of adults with severe hemophilia A (congenital factor VIII deficiency with factor VIII activity < 1 IU/dL) without pre-existing antibodies to adeno-associated virus serotype 5 detected by an FDA-approved test. (1)
Roctavian Dosage and Administration
For one-time single-dose intravenous use only. (2)
- Perform baseline testing to select patients, including testing for pre-existing antibodies to adeno-associated virus serotype 5 (AAV5), factor VIII inhibitor presence, and liver health assessments. (2)
- The recommended dose of ROCTAVIAN is 6 × 1013 vector genomes (vg) per kg of body weight. (2.1)
- Start the infusion at 1 mL/min. If tolerated, the rate may be increased every 30 minutes by 1 mL/min up to a maximum rate of 4 mL/min. (2.1)
Dosage Forms and Strengths
- ROCTAVIAN is a suspension for intravenous infusion. (3)
- ROCTAVIAN has a nominal concentration of 2 × 1013 vg valoctocogene roxaparvovec-rvox per mL, each vial contains an extractable volume of not less than 8 mL (16 × 1013 vg). (3)
Contraindications
- Active infections, either acute or uncontrolled chronic. (4)
- Known significant hepatic fibrosis (stage 3 or 4), or cirrhosis. (4)
- Known hypersensitivity to mannitol. (4)
Warnings and Precautions
- Infusion-related reactions: Infusion reactions, including hypersensitivity reactions and anaphylaxis, have occurred. Monitor during and for at least 3 hours after ROCTAVIAN administration. If symptoms occur, slow or interrupt administration and give appropriate treatment. Restart infusion at slower rate once symptoms resolve. Discontinue infusion for anaphylaxis. (2.3, 5.1)
- Hepatotoxicity: Monitor alanine aminotransferase (ALT) weekly for at least 26 weeks and institute corticosteroid treatment in response to ALT elevations as required. Continue to monitor ALT until it returns to baseline. Monitor factor VIII activity levels since ALT elevation may be accompanied by a decrease in factor VIII activity. Monitor for and manage adverse reactions from corticosteroid use. (5.2)
- Thromboembolic events: Thromboembolic events may occur in the setting of elevated factor VIII activity above the upper limit of normal (ULN). Factor VIII activity above ULN has been reported following ROCTAVIAN infusion. Evaluate for risk factors for thrombosis including cardiovascular risk factors prior to and after ROCTAVIAN use and advise patients accordingly. (5.3)
- Monitoring laboratory tests: Monitor for factor VIII activity and factor VIII inhibitors. (5.4)
- Malignancy: Monitor for hepatocellular malignancy in patients with risk factors for hepatocellular carcinoma (e.g., hepatitis B or C, non-alcoholic fatty liver disease, chronic alcohol consumption, non-alcoholic steatohepatitis, advanced age). Perform regular liver ultrasound (e.g., annually) and alpha-fetoprotein testing following administration. In the event that any malignancy occurs after treatment with ROCTAVIAN, contact BioMarin Pharmaceutical Inc. at 1-866-906-6100. (5.5)
Adverse Reactions/Side Effects
- Most common adverse reactions (incidence ≥ 5%) were nausea, fatigue, headache, infusion-related reactions, vomiting, and abdominal pain. (6)
- Most common laboratory abnormalities (incidence ≥ 10%) were ALT, aspartate aminotransferase (AST), lactate dehydrogenase (LDH), creatine phosphokinase (CPK), factor VIII activity levels, gamma-glutamyl transferase (GGT) and bilirubin > ULN. (6)
To report SUSPECTED ADVERSE REACTIONS, contact BioMarin Pharmaceutical Inc. at 1-866-906-6100, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- For 6 months after administration, men must not donate semen, and men and their female partners must prevent or postpone pregnancy. (8.3)
- There is limited information on the safety and effectiveness of ROCTAVIAN in patients with HIV infection. (8.6)
- The safety and effectiveness of ROCTAVIAN in patients with prior or active factor VIII inhibitors have not been established. (8.7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Roctavian
ROCTAVIAN is an adeno-associated virus vector-based gene therapy indicated for the treatment of adults with severe hemophilia A (congenital factor VIII deficiency with factor VIII activity < 1 IU/dL) without antibodies to adeno-associated virus serotype 5 (AAV5) detected by an FDA-approved test.
2. Roctavian Dosage and Administration
For one-time single-dose intravenous use only.
Treatment with ROCTAVIAN should be under the supervision of a physician experienced in the treatment of hemophilia and/or bleeding disorders.
2.1 Dose
The recommended dose of ROCTAVIAN is 6 × 1013 vector genomes per kilogram (vg/kg) body weight, administered as a single intravenous infusion. ROCTAVIAN is administered using an infusion pump at a rate of 1 mL/min, which can be increased every 30 minutes by 1 mL/min up to a maximum rate of 4 mL/min.
Calculating Dose in Milliliters (mL) and Number of Vials Required
- Patient dose volume in mL:
- Body weight in kg multiplied by 3 = dose in mL.
- Number of ROCTAVIAN vials to be thawed:
- Patient dose volume (mL) divided by 8 = number of vials to be thawed (round up to next whole number of vials).
| Patient Weight | Patient Dose by Volume (mL) (body weight multiplied by 3) | Number of Vials to be Thawed (dose volume divided by 8, then rounded up) |
|---|---|---|
| 70 kg | 210 mL | 27 vials (rounded up from 26.25) |
ROCTAVIAN can be administered only once.
2.2 Preparation for Administration
Required Equipment and Materials
- Flow rate-controlled syringe pump
- Syringes for ROCTAVIAN administration (the number of syringes will depend on the patient's dose volume and the syringe pump used and should be prepared prior to administration of ROCTAVIAN)
- 18- to 21-gauge sharp needles
- High-volume, in-line, low protein binding infusion filter with a pore size of 0.22 microns and maximum operating pressure adequate for the syringe pump or pump settings. Ensure availability of a sufficient number of replacement filters, according to the specifications for maximum filtered fluid volume.
- Syringe of 0.9% Sodium Chloride Injection, USP sodium chloride 9 mg/mL (0.9%) solution for priming and flushing the infusion line
- When assembling the infusion system, refer to the compatible materials with ROCTAVIAN suspension listed in Table 2.
| Component | Compatible Materials |
|---|---|
|
|
| Syringes | Polypropylene barrel with a synthetic rubber plunger tip |
| Syringe cap | Polypropylene |
| Infusion tubing* | Polyethylene |
| 0.22 micron in-line filter | Polyvinylidene fluoride filter with polyvinyl chloride body |
| Infusion catheter | Polyurethane based polymer |
| Stopcocks | Polycarbonate |
| 18 to 21-gauge sharp needles for extraction from vials† | Stainless steel |
2.3 Administration
- Administer ROCTAVIAN in a setting where personnel and equipment are immediately available to treat infusion-related reactions [see Warnings and Precautions (5.1)].
- Infuse the suspension through a suitable peripheral vein, using an infusion catheter with in-line filter and a programmable syringe pump.
- Start the infusion at a rate of 1 mL/min. If tolerated, the rate may be increased every 30 minutes by 1 mL/min up to a maximum rate of 4 mL/min. The infusion time depends on infusion volume, rate and patient response and can be, for example, 2 to 5 hours or longer for a patient weighing 100 kg.
- In the event of an infusion-related reaction during administration [see Warnings and Precautions (5.1)],
- Decrease the infusion rate or stop the infusion.
- Administer treatment as needed to manage infusion reaction.
- If the infusion is stopped, restart the infusion at a rate of 1 mL/min and consider maintaining it at a previously tolerated level for the remainder of the infusion. If the infusion needs to be restarted, the infusion should be completed within 10 hours of initial drug product thaw.
- Discontinue infusion for anaphylaxis.
- DO NOT administer ROCTAVIAN as an intravenous push or bolus.
- DO NOT infuse ROCTAVIAN in the same intravenous line with any other products.
- DO NOT use a central line or port.
- To ensure the patient receives the complete dose, after the content of the last ROCTAVIAN-containing syringe is infused, flush the infusion line with a sufficient volume of 0.9% Sodium Chloride Injection, USP, through the same tubing and filter, and at the same infusion rate.
- Maintain venous access during the subsequent observation period [see Warnings and Precautions (5.1)].
Monitoring Post-Administration
Conduct the following tests after ROCTAVIAN administration [see Warnings and Precautions (5.2, 5.4, 5.5)].
- Perform regular ALT testing to monitor for elevations. Elevated liver enzymes, especially elevated ALT, may indicate immune-mediated hepatotoxicity and may be associated with decline in factor VIII activity.
The monitoring schedule for ALT, and recommendations for corticosteroid use (initiation and taper) are based on the clinical efficacy and safety experience of 112 patients in a clinical study with ROCTAVIAN.- Monitor ALT weekly for at least 26 weeks following administration of ROCTAVIAN. See Table 3 for monitoring schedule. Monitor AST and creatine phosphokinase (CPK) as needed to help rule out alternative causes for ALT elevations (including potentially hepatotoxic medications or agents, alcohol consumption, or strenuous exercise). Consider repeating ALT testing within 24 to 48 hours to confirm ALT elevation prior to initiation of corticosteroid treatment and using the same laboratory to measure ALT activity at baseline and over time to minimize the impact of inter-laboratory variability on test results.
- If ALT ≥ 1.5 × baseline or above ULN consider corticosteroid treatment. For patients who need corticosteroid therapy, the recommended starting dose is 60 mg with a subsequent taper upon return of ALT levels to baseline (see Table 4 below for recommendation on corticosteroid treatment).
- Monitor ALT weekly, and as clinically indicated, during corticosteroid therapy. Continue to monitor ALT until its return to baseline.
- Monitor for and manage adverse reactions secondary to corticosteroid use. Refer to the corticosteroid prescribing information for risks and required precautions.
| Timeframe | Monitoring Frequency† |
|---|---|
|
|
| First 26 weeks | Weekly |
| Weeks 26 to 52 (Year 1) | Every 1 to 2 weeks |
| Year 2 | Every 3 months |
| After Year 2 | Every 6 months |
| Corticosteroid Regimen (Prednisone or Equivalent Dose of Another Corticosteroid) |
|
|---|---|
|
|
| Starting Dose* | 60 mg daily for 2 weeks |
| Tapering† | 40 mg daily for 3 weeks 30 mg daily for 1 week 20 mg daily for 1 week 10 mg daily for 1 week |
There is limited information on the benefit of starting a corticosteroid course after the first year of ROCTAVIAN administration.
Other immunosuppressive therapies (e.g., tacrolimus, mycophenolate mofetil) may be considered if corticosteroids are contraindicated, ineffective or there are adverse reactions secondary to corticosteroid use necessitating discontinuation.
-
Monitor Factor VIII Activity
- Monitor factor VIII activity using the same schedule for ALT monitoring in Table 3 [see Warnings and Precautions (5.4)].
- Consider more frequent monitoring in patients with factor VIII activity levels ≤ 5 IU/dL and evidence of bleeding, taking into account the stability of factor VIII levels since the previous measurement.
- It may take several weeks after ROCTAVIAN infusion before ROCTAVIAN-derived factor VIII activity rises to a level sufficient for prevention of spontaneous bleeding episodes. Therefore, continued routine prophylaxis support with exogenous factor VIII or other hemostatic products used in the management of hemophilia A may be needed during the first few weeks after ROCTAVIAN infusion [see Clinical Pharmacology (12.3)]. Exogenous factor VIII or other hemostatic products may also be required in case of surgery, invasive procedures, trauma, or bleeds in the event that ROCTAVIAN-derived factor VIII activity is deemed insufficient for adequate hemostasis in such situations.
- The use of different assays may impact test results; therefore, use the same assay and reagents to monitor patients over time, if feasible [see Warnings and Precautions (5.4)].
- Use of exogenous factor VIII products before and after ROCTAVIAN administration may impede assessment of ROCTAVIAN-derived factor VIII activity.
- Monitor patients for factor VIII inhibitors (neutralizing antibodies to factor VIII). Test for factor VIII inhibitors especially if bleeding is not controlled, or plasma factor VIII activity levels decrease [see Warnings and Precautions (5.4) and Clinical Pharmacology (12.6)].
- Perform regular liver ultrasound (e.g., annually) and alpha-fetoprotein (AFP) testing in patients with risk factors of hepatocellular carcinoma (e.g., hepatitis B or C, non-alcoholic fatty liver disease, chronic alcohol consumption, non-alcoholic steatohepatitis, advanced age) [see Warnings and Precautions (5.5)].
3. Dosage Forms and Strengths
ROCTAVIAN is a clear, colorless to pale yellow suspension for intravenous infusion containing 2 × 1013 vg valoctocogene roxaparvovec-rvox per mL.
ROCTAVIAN is provided in vials containing an extractable volume of not less than 8 mL.
Dose volume is based on body weight, with a recommended dose of 6 × 1013 vg/kg.
4. Contraindications
Administration of ROCTAVIAN is contraindicated in:
- patients with active infections, either acute (such as acute respiratory infections or acute hepatitis) or uncontrolled chronic (such as chronic active hepatitis B).
- patients with known significant hepatic fibrosis (stage 3 or 4 on the Batts-Ludwig scale or equivalent), or cirrhosis [see Dosage and Administration (2)].
- patients with known hypersensitivity to mannitol.
5. Warnings and Precautions
5.1 Infusion-Related Reactions
Infusion-related reactions, including hypersensitivity reactions and anaphylaxis, have occurred during and/or following ROCTAVIAN administration. Symptoms included one or more of the following: urticaria, pruritus, rash, sneezing, coughing, dyspnea, rhinorrhea, watery eyes, tingling throat, nausea, diarrhea, hypotension, tachycardia, presyncope, pyrexia, rigors, and chills.
Monitor patients during and for at least 3 hours after completion of ROCTAVIAN infusion. Do not infuse the product faster than 4 mL/min [see Dosage and Administration (2.3)].
In the event of an infusion reaction, administration of ROCTAVIAN should be slowed or stopped. Restart at a lower rate after the infusion reaction has resolved. Discontinue infusion for anaphylaxis. Consider treatment with a corticosteroid, antihistamine, and other measures for management of an infusion reaction [see Dosage and Administration (2.3)].
5.2 Hepatotoxicity
Intravenous administration of a liver-directed AAV vector could lead to liver enzyme elevations (transaminitis), especially ALT elevation. Transaminitis is presumed to occur due to immune-mediated injury of transduced hepatocytes and may reduce the therapeutic efficacy of AAV-vector based gene therapy.
Majority of patients treated with ROCTAVIAN experienced ALT elevations. Most ALT elevations occurred within the first year following ROCTAVIAN administration, especially within the first 26 weeks, were low-grade, and resolved. The median time (range) to the first ALT elevation (defined as ALT ≥ 1.5 × baseline or above ULN) was 7 weeks (0.4, 159 weeks) and the median duration (range) was 4 weeks (0.1, 135 weeks). Some ALT elevations were associated with a decline in factor VIII activity.
The majority of the 112 patients in the clinical trial of ROCTAVIAN required corticosteroids for ALT elevation [see Adverse Reactions (6.1) and Clinical Studies (14)]. The median duration (range) of corticosteroid use was 35 weeks (3, 120 weeks). The median duration (range) of alternate immunosuppressive medications use was 26 weeks (6, 118 weeks). In 20 (18%) patients the duration of immunosuppression was > 1 year.
Monitor ALT and institute corticosteroid treatment in response to ALT elevations, as required. Monitor ALT and factor VIII activity levels weekly and, as clinically indicated, during corticosteroid therapy [see Dosage and Administration (2.3)]. Monitor for and manage adverse reactions secondary to corticosteroid therapy.
Since some ALT elevations have been attributed to alcohol consumption in clinical studies, patients should abstain from alcohol consumption for at least a year following ROCTAVIAN infusion and limit alcohol use thereafter. Concomitant medications may cause hepatotoxicity, or decrease factor VIII activity, or change plasma corticosteroid levels which may impact liver enzyme elevation and/or factor VIII activity [see Drug Interactions (7.0, 7.1, 7.2)]. Closely monitor concomitant medication use including herbal products and nutritional supplements and consider alternative medications in case of potential drug interactions.
5.3 Thromboembolic Events
Elevated factor VIII activity level above the ULN as measured by the chromogenic substrate assays (CSA), or one-stage clotting assays (OSA), or both assays has occurred following ROCTAVIAN administration. Thirty-eight (28%) patients experienced elevations of factor VIII above ULN with a median time to first occurrence of 14 weeks and a median total duration above ULN of 12 weeks.
An increase in factor VIII activity may increase the risk for venous and arterial thromboembolic events. There are no data in patients with a history of venous or arterial thromboembolism or known history of thrombophilia since such patients were excluded from clinical trials of ROCTAVIAN.
Evaluate patients for risk of thrombosis including general cardiovascular risk factors before and after administration of ROCTAVIAN. Advise patients on their individual risk of thrombosis in relation to their factor VIII activity levels above ULN and consider prophylactic anticoagulation. Advise patients to seek immediate medical attention for signs or symptoms indicative of a thrombotic event.
5.5 Malignancy
The integration of liver-targeting AAV vector DNA into the genome may carry the theoretical risk of hepatocellular carcinoma development.
ROCTAVIAN is composed of a non-replicating AAV5 vector whose DNA persists largely in episomal form. Low levels of vector integration were found following evaluation of liver samples from 5 patients and parotid gland tissue sample from 1 patient in clinical studies and liver samples from 12 nonhuman primates [see Clinical Pharmacology (12.2) and Nonclinical Toxicology (13.1)]. ROCTAVIAN can also insert into the DNA of other human body cells. No malignancies assessed as being likely related to ROCTAVIAN were observed in clinical studies.
Monitor patients with risk factors for hepatocellular carcinoma (e.g., hepatitis B or C, non-alcoholic fatty liver disease, chronic alcohol consumption, non-alcoholic steatohepatitis, advanced age) with regular liver ultrasound (e.g., annually) and alpha-fetoprotein testing for 5 years following ROCTAVIAN administration [see Dosage and Administration (2.3)].
In the event that a malignancy occurs, contact BioMarin Pharmaceutical Inc. at 1-866-906-6100 to obtain instructions on collecting patient samples for testing.
6. Adverse Reactions/Side Effects
The following adverse reactions are also discussed in other sections of the label:
- Infusion-Related Reactions [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described in this section reflect the exposure of 134 adult patients with severe hemophilia A (defined as residual factor VIII activity ≤ 1 IU/dL) to a single dose of 6 × 1013 vg/kg of body weight of ROCTAVIAN. Patients with detectable pre-existing antibodies to AAV5, active infections, history of thrombosis, immunosuppressive disorders, and liver dysfunction were excluded. All patients had a median follow-up of 162 weeks (range: 66 to 255 weeks) [see Clinical Studies (14)].
The most common adverse reactions (≥ 5%) to ROCTAVIAN were nausea, fatigue, headache, infusion-related reactions, vomiting, and abdominal pain.
The most common laboratory abnormalities (≥ 10%) to ROCTAVIAN were ALT, AST, LDH, CPK, factor VIII activity levels, GGT, and bilirubin > ULN.
Non-laboratory adverse reactions (≥ 5%) to ROCTAVIAN are listed in Table 5. There were 6 serious adverse reactions related to ROCTAVIAN treatment including ALT elevation, presyncope, maculopapular rash, anaphylaxis, and hypersensitivity reaction.
| Adverse Reactions | Number of Patients (%) N = 134 |
|
|---|---|---|
| All Grades | ≥ Grade 3 | |
|
||
| Nervous system disorders | ||
| Headache | 9 (7%) | 0 (0%) |
| Gastrointestinal disorders | ||
| Nausea | 42 (31%) | 0 (0%) |
| Vomiting | 8 (6%) | 0 (0%) |
| Abdominal pain† | 8 (6%) | 0 (0%) |
| General disorders and administration site conditions | ||
| Fatigue‡ | 21 (16%) | 0 (0%) |
| Infusion-related reactions§ | 9 (7%) | 2 (1%) |
Table 6 lists laboratory abnormalities in patients treated with ROCTAVIAN.
| Laboratory Abnormalities | Number of Patients (%) N = 134 |
|---|---|
|
|
| ALT increases > ULN | 109 (81%) |
| AST increases > ULN | 92 (69%) |
| LDH increases > ULN | 77 (57%) |
| CPK increases > ULN | 60 (45%) |
| Factor VIII activity levels > ULN* | 38 (28%) |
| GGT increases > ULN | 24 (18%) |
| Bilirubin increases > ULN | 18 (13%) |
Twelve (9%), 9 (7%) and 1 (1%) of patients experienced > 5-20 × ULN ALT, AST and GGT elevations, respectively. Seven (5%) patients and 5 (4%) patients experienced > 5-10 × ULN and > 10 × ULN CPK increases, respectively.
Infusion-related reactions were observed in 9 patients (7%), including hypersensitivity reactions (4%) and anaphylaxis (1%), and have occurred during and/or following ROCTAVIAN administration.
Hepatotoxicity as defined by ALT ≥ 1.5 × baseline or ALT > ULN occurred in 107 of 112 (96%) patients. Nine (8%) patients had ALT between > 5-20 × ULN. One hundred (89%) patients had ALT elevations that occurred within the first 26 weeks, 74 (66%) patients had ALT elevations that occurred between weeks 27 to 52, and 72 (64%) patients had ALT elevations that occurred beyond 52 weeks after administration. Thirty-four of 112 (30%) patients had ALT elevations that were associated with a decline in factor VIII activity of ≥ 30%.
The majority of patients (82%, 92/112) required corticosteroids for one or more episodes of ALT elevation while 35% (39/112) required alternate immunosuppression. Seventy-six percent, 5%, and 1% of patients used corticosteroids within the first 26 weeks, weeks 27 to 52, and beyond 52 weeks respectively following ROCTAVIAN administration.
The most common (≥ 10%) adverse reactions from corticosteroid use (N = 92) included acne (34%), insomnia (27%), mood disorders (20%), cushingoid (20%), rash (18%), weight gain (16%), hypertension (12%), folliculitis (11%), abdominal pain (10%), and vision disorders (10%). Other clinically meaningful adverse events while on corticosteroid therapy included bone fracture (5%), impaired glucose tolerance (5%), herpes zoster (3%), oral candidiasis (3%), and adrenal insufficiency (1%). The most common adverse reactions from alternate immunosuppressant use (N = 39) included hypomagnesemia (15%) and diarrhea (10%). Infections requiring intravenous antimicrobial therapy occurred in 3 (3%) patients while on corticosteroid or other immunosuppressant therapy (N = 97).
One case of autoimmune hepatitis was reported during third year follow-up in a patient with history of hepatitis C and steatohepatitis.
7. Drug Interactions
Prior to ROCTAVIAN administration, the patient's existing medications should be reviewed to determine if they should be modified to prevent anticipated interactions described in this section.
Concomitant medications should be monitored after ROCTAVIAN administration, and the need to change concomitant medications based on patient's hepatic status and risk should be evaluated. When a new medication is started, close monitoring of ALT and factor VIII activity levels (e.g., weekly to every 2 weeks for the first month) is recommended to assess potential effects on both levels.
No in vivo interaction studies have been performed.
7.1 Isotretinoin
In one patient, decreased factor VIII activity without ALT elevation was detected after starting treatment with systemic isotretinoin following ROCTAVIAN infusion. Factor VIII activity was 75 IU/dL at Week 60 and transiently decreased to < 3 IU/dL at Week 64, after initiating isotretinoin. After discontinuing isotretinoin at Week 72, factor VIII activity recovered to 46 IU/dL at Week 122. An in vitro study in human primary hepatocytes indicated that isotretinoin suppressed factor VIII transcription independent of hepatotoxicity, without impact on ALT, and expression was partially restored upon cessation of isotretinoin treatment. Isotretinoin is not recommended in patients who are benefiting from ROCTAVIAN.
7.2 Efavirenz
One HIV positive patient treated with ROCTAVIAN at a dose of 4 × 1013 vg/kg ROCTAVIAN while on an antiretroviral therapy regimen consisting of efavirenz, lamivudine, and tenofovir experienced asymptomatic elevations of ALT, AST, and GGT (> 5.0 × ULN) and serum bilirubin (> ULN and up to 1.5 × ULN) at Week 4 [see Use in Specific Populations (8.6)]. The reaction resolved after the antiretroviral therapy regimen was changed to a regimen without efavirenz. The patient later reverted to prophylactic use of factor VIII concentrates. An in vitro study in human primary hepatocytes indicated that efavirenz suppressed factor VIII transcription independent of hepatotoxicity, and expression was not restored upon discontinuation of efavirenz. Efavirenz is not recommended in patients treated with ROCTAVIAN.
7.3 Interactions with Agents that May Reduce or Increase Plasma Concentrations of Corticosteroids
Agents that may reduce or increase the plasma concentration of corticosteroids (e.g., agents that induce or inhibit cytochrome P450 3A4) can decrease the efficacy of the corticosteroid regimen or increase their side effects [see Dosage and Administration (2.3)].
7.4 Vaccinations
Prior to ROCTAVIAN infusion, ensure up to date vaccinations. Individual vaccination schedules may need to be adjusted to accommodate concomitant immunosuppressive therapy [see Dosage and Administration (2.3)]. Live vaccines should not be administered to patients while on immunosuppressive therapy.
8. Use In Specific Populations
8.3 Females and Males of Reproductive Potential
ROCTAVIAN is not intended for administration in women.
8.4 Pediatric Use
The safety and effectiveness of ROCTAVIAN in pediatric patients have not been established.
8.5 Geriatric Use
A single patient ≥ 65 years of age was treated with ROCATVIAN in clinical studies. Clinical studies of ROCTAVIAN did not include sufficient numbers of patients aged 65 and over to determine whether the efficacy or safety differs compared to younger patients.
8.6 Human Immunodeficiency Virus (HIV) Positive Patients
In clinical studies, 3 HIV infected patients have been treated with ROCTAVIAN. Clinical studies of ROCTAVIAN did not include sufficient numbers of patients with HIV to determine whether the efficacy and safety differs compared to patients without HIV infection.
A single HIV infected patient treated with ROCTAVIAN developed hepatocellular injury that subsequently resolved and was attributed to concomitant administration with antiretroviral drug efavirenz [see Drug Interactions (7.2)].
8.7 Factor VIII Inhibitors
The safety and effectiveness of ROCTAVIAN in patients with prior or active factor VIII inhibitors have not been established [see Clinical Pharmacology (12.6)]. Patients with active factor VIII inhibitors should not take ROCTAVIAN.
After administration of ROCTAVIAN, patients should be monitored for the development of factor VIII inhibitors by appropriate clinical observations and laboratory tests [see Warnings and Precautions (5.4)].
8.8 Hepatic Impairment
The safety and effectiveness of ROCTAVIAN in patients with hepatic impairment has not been established. Clinical studies excluded patients with known hepatic cirrhosis, significant fibrosis (stage 3 or 4 on the Batts-Ludwig scale or equivalent), current hepatitis B or C, or history of hepatic malignancy. No dose adjustments can be recommended for patients with hepatic impairment.
11. Roctavian Description
ROCTAVIAN (valoctocogene roxaparvovec-rvox) is an adeno-associated virus (AAV) vector-based gene therapy product. ROCTAVIAN is replication-incompetent and consists of an AAV serotype 5 capsid containing a DNA sequence encoding the B-domain deleted SQ form of the human coagulation factor VIII (hFVIII-SQ). ROCTAVIAN is derived from naturally occurring adeno-associated virus and is produced using Sf9 insect cells and recombinant baculovirus technology.
ROCTAVIAN is a sterile suspension for intravenous infusion. When thawed, the suspension is clear and colorless to pale yellow.
Each vial of ROCTAVIAN contains an extractable volume of 8 mL of valoctocogene roxaparvovec-rvox at a concentration of 2 × 1013 vector genomes (vg) per mL, and the following excipients: mannitol (20 mg/mL), poloxamer 188 (2.0 mg/mL), sodium chloride (8.2 mg/mL), sodium phosphate monobasic dihydrate (0.23 mg/mL), sodium phosphate dibasic dodecahydrate (3.05 mg/mL) and Water for Injection, USP. The pH of ROCTAVIAN is 6.9 to 7.8. The osmolarity of ROCTAVIAN is 364 to 445 mOsm/L.
The product contains no preservative.
12. Roctavian - Clinical Pharmacology
12.1 Mechanism of Action
Valoctocogene roxaparvovec-rvox is an adeno-associated virus serotype 5 (AAV5) based gene therapy vector, designed to introduce a functional copy of a transgene encoding the B-domain deleted SQ form of human coagulation factor VIII (hFVIII-SQ). Transcription of this transgene occurs within the liver, using a liver-specific promoter, which results in the expression of hFVIII-SQ. The expressed hFVIII-SQ replaces the missing coagulation factor VIII needed for effective hemostasis.
12.2 Pharmacodynamics
Following ROCTAVIAN infusion, vector DNA is processed in vivo to form full-length, episomal transgenes that increase circulating hFVIII-SQ up to 5 years.
Liver samples from 5 patients in clinical studies collected 0.5-4.1 years post-dose were analyzed. Vector integration into human genomic DNA was observed in all samples. No preferential integration of the vector and no clonal outgrowth of cells as a result of vector integration was observed. ROCTAVIAN can also insert into DNA of other human body cells; vector insertion into parotid gland DNA samples without associated clinical manifestations was observed in one patient treated with ROCTAVIAN.
Factor VIII Activity
The pharmacodynamic effect of ROCTAVIAN was assessed by measuring circulating factor VIII activity levels.
Factor VIII activity levels (IU/dL) over time post-ROCTAVIAN infusion in ITT population are reported by both the CSA and OSA. The mean factor VIII activity levels at Month 36 was 18.2 IU/dL (95% CI: 12.9, 23.4) using the CSA, a statistically significant (p < 0.0001) improvement from 1 IU/dL at baseline.
Table 7 shows factor VIII activity levels (IU/dL) over time post-ROCTAVIAN infusion in patients rolled over from a non-interventional study prospectively collecting patients' baseline annualized bleeding rate (ABR) and factor VIII usage data.
| Timepoint | Rollover Population N = 112 | Directly Enrolled Population N = 22 |
||
|---|---|---|---|---|
| CSA | OSA | CSA | OSA | |
| Month 3 | N = 111 | N = 111 | N = 22 | N = 22 |
| Mean (SD) | 34.9 (40.4) | 54.6 (60.8) | 31.4 (25.7) | 48.3 (36.0) |
| Median (Q1, Q3) | 20.7 (10.3, 40.5) | 31.3 (15.3, 71.7) | 20.9 (12.6, 45.7) | 36.0 (22.4, 63.9) |
| Min, Max | 0, 249.5 | 1.5, 335.8 | 0, 85.8 | 4.5, 126.0 |
| Month 6 | N = 111 | N = 111 | N = 22 | N = 22 |
| Mean (SD) | 55.4 (57.5) | 84.9 (83.1) | 40.0 (37.9) | 63.0 (57.2) |
| Median (Q1, Q3) | 38.8 (16.8, 76.5) | 62.0 (28.0, 115.2) | 33.2 (14.7, 46.3) | 53.5 (23.7, 78.2) |
| Min, Max | 0, 367.3 | 1.9, 483.9 | 0, 169.4 | 1.8, 261.9 |
| Month 10 | N = 111 | N = 111 | N = 20 | N = 20 |
| Mean (SD) | 49.4 (49.5) | 73.6 (70.5) | 44.2 (49.6) | 70.2 (70.9) |
| Median (Q1, Q3) | 31.7 (17.1, 64.5) | 51.3 (25.1, 96.2) | 30.9 (14.1, 68.6) | 55.4 (24.8, 101.4) |
| Min, Max | 0, 265.3 | 1.2, 375.6 | 0, 223.6 | 2.4, 313.7 |
| Month 12 | N = 111 | N = 111 | N = 21 | N = 21 |
| Mean (SD) | 43.6 (45.5) | 64.7 (64.6) | 38.2 (46.3) | 59.7 (67.0) |
| Median (Q1, Q3) | 24.0 (12.5, 63.7) | 40.0 (20.4, 87.5) | 23.9 (11.2, 52.8) | 40.5 (17.4, 82.6) |
| Min, Max | 0, 231.2 | 0, 311.1 | 1.6, 207.4 | 4.4, 294.1 |
| Month 18 | N = 99 | N = 99 | N = 18 | N = 18 |
| Mean (SD) | 27.7 (32.3) | 40.6 (45.9) | 28.5 (28.9) | 44.5 (43.9) |
| Median (Q1, Q3) | 13.5 (6.9, 36.8) | 22.5 (10.9, 55.30) | 15.3 (10.8, 43.9) | 24.4 (17.7, 60.4) |
| Min, Max | 0, 167.9 | 0, 232.2 | 3.3, 117.0 | 4.2, 173.7 |
| Month 24 | N = 98 | N = 99 | N = 19 | N = 18 |
| Mean (SD) | 25.0 (35.5) | 38.9 (50.7) | 22.0 (28.7) | 36.0 (40.8) |
| Median (Q1, Q3) | 12.7 (5.1, 26.5) | 22.7 (7.9, 45.7) | 8.9 (5.8, 25.9) | 19.5 (7.9, 37.7) |
| Min, Max | 0, 187.1 | 0, 271.3 | 0, 110.6 | 2.4, 146.7 |
| Month 36 | N = 96 | N = 97 | N = 15 | N = 15 |
| Mean (SD) | 21.0 (34.0) | 33.8 (47.6) | 20.8 (24.4) | 32.2 (33.1) |
| Median (Q1, Q3) | 10.0 (4.3, 19.8) | 17.7 (7.2, 35.1) | 9.4 (6.6, 31.7) | 20.6 (8.5, 46.7) |
| Min, Max | 0, 217.7 | 0, 291.4 | 0, 74.5 | 1.9, 104.2 |
The proportion of patients achieving factor VIII activity level thresholds by year are presented in Table 8 by both the CSA and OSA. The majority (95%) of patients who reach factor VIII activity levels of ≥ 5 IU/dL using the CSA do so within 5 months post-infusion.
| Rollover Population (N = 112) | |||
| Factor VIII Activity Threshold Achieved by Assay | Year 1
N = 111 n (%) | Year 2
N = 98 n (%) | Year 3
N = 96 n (%) |
| CSA | |||
| > 150 IU/dL | 6 (5%) | 2 (2%) | 2 (2%) |
| 40 - ≤ 150 IU/dL | 37 (33%) | 14 (14%) | 9 (9%) |
| 15 - < 40 IU/dL | 37 (33%) | 27 (28%) | 23 (24%) |
| 5 - < 15 IU/dL | 18 (16%) | 33 (34%) | 35 (36%) |
| 3 - < 5 IU/dL | 3 (3%) | 10 (10%) | 8 (8%) |
| < 3 IU/dL | 10 (9%) | 12 (12%) | 19 (20%) |
| Year 1
N = 111 n (%) | Year 2
N = 99 n (%) | Year 3
N = 97 n (%) |
|
| OSA | |||
| > 150 IU/dL | 12 (11%) | 5 (5%) | 4 (4%) |
| 40 - ≤ 150 IU/dL | 44 (40%) | 25 (25%) | 17 (18%) |
| 15 - < 40 IU/dL | 37 (33%) | 36 (36%) | 36 (37%) |
| 5 - < 15 IU/dL | 10 (9%) | 20 (20%) | 26 (27%) |
| 1 - < 5 IU/dL | 6 (5%) | 11 (11%) | 12 (12%) |
| < 1 IU/dL | 2 (2%) | 2 (2%) | 2 (2%) |
| Directly Enrolled Population (N = 22) | |||
| Factor VIII Activity Threshold Achieved by Assay | Year 1
N = 21 n (%) | Year 2
N = 19 n (%) | Year 3
N = 15 n (%) |
| CSA | |||
| > 150 IU/dL | 1 (5%) | 0 (0%) | 0 (0%) |
| 40 - ≤ 150 IU/dL | 5 (24%) | 3 (16%) | 3 (20%) |
| 15 - < 40 IU/dL | 8 (38%) | 4 (21%) | 2 (13%) |
| 5 - < 15 IU/dL | 5 (24%) | 8 (42%) | 7 (47%) |
| 3 - < 5 IU/dL | 0 (0%) | 1 (5%) | 1 (7%) |
| < 3 IU/dL | 2 (10%) | 3 (16%) | 2 (13%) |
| Year 1
N = 21 n (%) | Year 2
N = 18 n (%) | Year 3
N = 15 n (%) |
|
| OSA | |||
| > 150 IU/dL | 1 (5%) | 0 (0%) | 0 (0%) |
| 40 - ≤ 150 IU/dL | 10 (48%) | 4 (22%) | 4 (27%) |
| 15 - < 40 IU/dL | 6 (29%) | 6 (33%) | 6 (40%) |
| 5 - < 15 IU/dL | 3 (14%) | 6 (33%) | 3 (20%) |
| 1 - < 5 IU/dL | 1 (5%) | 2 (11%) | 2 (13%) |
| < 1 IU/dL | 0 (0%) | 0 (0%) | 0 (0%) |
12.3 Pharmacokinetics
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of ROCTAVIAN or of other adeno-associated virus-based gene therapy products.
In clinical studies, all patients receiving treatment were required to screen negative for anti-AAV5 antibodies and negative (< 0.6 BU) for factor VIII inhibitors in a Nijmegen modified Bethesda assay following a lifetime minimum of 150 exposure days to factor VIII replacement therapy [see Use in Specific Populations (8.7)]. Following infusion of ROCTAVIAN, all patients remained negative for factor VIII inhibitors.
All patients seroconverted to anti-AAV5 antibody positive within 8 weeks of administration. Anti-AAV5 total antibody titers peaked by 36 weeks after administration with mean (SD) values of 12,528,983 (32,427,817), and remained stable until the last time point tested, Week 168 with mean (SD) values of 3,673,038 (3,344,713).
ROCTAVIAN-treated patients were tested for cellular immune responses against the AAV5 capsid and the factor VIII transgene product using an IFN-γ ELISpot assay. AAV5 capsid-specific cellular immune responses were detected beginning at Week 2 following dose administration and often declined or reverted to negative over the first 52 weeks in the majority of patients with available data. Incidence peaked at Week 2 with 67 of 96 patients (70%) testing positive in the IFN-γ ELISpot assay. This declined to 17 of 74 patients (23%) at Week 26, and 10 of 60 patients (17%) at Week 52.
Factor VIII-specific cellular responses were detected in 81 of 123 (65.9%) patients, often sporadically at a single time point and reverting to negative in most patients.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been conducted to evaluate the effects of ROCTAVIAN on carcinogenesis.
To evaluate vector integration, host genomic DNA was isolated from liver tissues obtained from nonhuman primates at 13 and 26 weeks following intravenous administration of ROCTAVIAN at dose levels of up to 5.4 × 1013 vg/kg. Vector DNA was mostly detected in the form of episomal DNA that was not integrated into the host genome. Vector integration events were observed at low frequencies and distributed throughout the host genome with no indication of clonal enrichment or preference to particular chromosomal locations, including genes associated with oncogenesis in humans [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.2)].
Since ROCTAVIAN vector DNA was detected in testes of adult male mice for at least 6 months following a single intravenous administration at dose level of 2.1 × 1014 vg/kg [see Clinical Pharmacology (12.3)], the potential for vertical transmission to offspring was studied in Rag2-/- immunodeficient mice. A single intravenous administration of ROCTAVIAN at a dose level of 6.7 × 1013 vg/kg into male immunodeficient mice that were mated 37 days later with naïve female mice did not result in adverse effects on fertility rates, pregnancy rates, and litter sizes. Quantitative polymerase chain reaction (qPCR) evaluation of liver tissues from the F1 pups did not detect the vector DNA.
13.2 Animal Toxicology and/or Pharmacology
A single intravenous administration of up to 2.1 × 1014 vg/kg of ROCTAVIAN in immunocompetent male mice with intact coagulation (CD1 mice), followed by an observation period of up to 26 weeks, showed dose-dependent toxicities in the heart, lung, thymus, and epididymis. At dose levels of 6.5 × 1013 vg/kg and higher, histopathologic findings in the heart included minimal to moderate epicardial hemorrhage, myocardial necrosis, fibrosis, inflammation, and vascular/perivascular necrosis, minimal to mild mesothelial hypertrophy/hyperplasia, fibroplasia, myocardial atrophy, and vascular/perivascular mixed cell infiltrates. Lung findings included moderate to marked hemorrhage, minimal to mild edema, vascular/perivascular necrosis, and mesothelial hypertrophy. Additional ROCTAVIAN-related findings included mild fibrosis, hemorrhage, and pigmented macrophages in the epididymis, and moderate to marked hemorrhage and hypocellularity of the thymus. ROCTAVIAN-related mortality, adverse clinical observations, and changes in gross pathology were observed at dose levels of 6.5 × 1013 vg/kg and higher, and were associated with fibrosis, hemorrhage, and necrosis in the heart.
In a toxicology study conducted in adult male nonhuman primates with a duration of 3 months, single intravenous administration of ROCTAVIAN at dose levels of 1.6 × 1013 vg/kg and 5.4 × 1013 vg/kg resulted in a dose-dependent prolongation of activated partial thromboplastin time (APTT). The transient APTT prolongation in a subset of nonhuman primates was temporally associated with an immune response to hFVIII-SQ protein. At 3 months post-administration, histopathology findings included minimal to mild mononuclear/mixed infiltration in the lungs.
14. Clinical Studies
The efficacy of ROCTAVIAN was evaluated in a prospective, phase 3, open-label, single-dose, single-arm, multinational study in 134 adult males (18 years of age and older) with severe hemophilia A, who received a single intravenous dose of 6 × 1013 vg/kg body weight of ROCTAVIAN and entered a follow-up period of 5 years. Patients previously treated with prophylactic factor VIII replacement therapy, but not emicizumab, were enrolled in the study. The study population was 72% White, 14% Asian, and 11% Black with a median age of 30 (range: 18 to 70) years. Twenty patients had a history of hepatitis B and 41 patients had a history of hepatitis C. All except 2 patients were HIV-negative.
Only patients without detectable, pre-existing antibodies to AAV5 capsid were eligible for therapy. Presence of pre-existing antibodies to AAV5 capsid was identified during screening using the ARUP Laboratories AAV5 DetectCDx™ total antibody assay, which is the FDA-approved test for selection of patients for ROCTAVIAN therapy. Other key exclusion criteria included active infection, chronic or active hepatitis B or C, immunosuppressive disorder including HIV, current or prior history of factor VIII inhibitor, stage 3 or 4 liver fibrosis, cirrhosis, liver function test abnormalities, history of thrombosis or thrombophilia, serum creatinine ≥ 1.4 mg/dL, and active malignancy.
Of the 134 patients who received ROCTAVIAN in the clinical trial, 112 patients had baseline annualized bleeding rate (ABR) data prospectively collected during a period of at least six months on factor VIII prophylaxis prior to receiving ROCTAVIAN (rollover population). The remaining 22 patients had baseline ABR collected retrospectively (directly enrolled population). All patients were followed for at least 3 years.
The primary efficacy outcome was a non-inferiority (NI) test of the difference in ABR in the efficacy evaluation period (EEP) following ROCTAVIAN administration compared with ABR during the baseline period in the rollover population. The NI margin was 3.5 bleeds per year. All bleeding episodes, regardless of treatment, were counted towards ABR. The EEP started from Study Day 33 (Week 5) or the end of factor VIII prophylaxis including a washout period after ROCTAVIAN treatment, whichever was later, and ended when a patient completed the study, had the last visit, or withdrew or was lost to follow-up from the study, whichever was the earliest.
Table 9 summarizes the NI comparison between the mean ABR after ROCTAVIAN treatment and the mean baseline ABR while patients were on factor VIII prophylaxis in the rollover population (N = 112). The mean EEP ABR was 2.6 bleeds/year, compared to a mean baseline ABR of 5.4 bleeds/year. The mean difference in ABR was -2.8 (95% confidence interval: -4.3, -1.2) bleeds/year. The NI analysis met the pre-specified NI margin, indicating the effectiveness of ROCTAVIAN.
A majority of patients treated with ROCTAVIAN received immunosuppressive medications, including steroids, to control elevations in transaminases and to prevent loss of transgene expression [see Dosage and Administration (2.3), Warnings and Precautions (5.2), and Adverse Reactions (6.1)].
| ABR and Bleeding Events | Baseline | Post-ROCTAVIAN Efficacy Evaluation Period |
|---|---|---|
| Min: Minimum; Max: Maximum; SD: Standard Deviation | ||
|
||
| Median (range) follow-up duration in years | 0.6 (0.5, 1.3) | 3.0 (1.7, 3.7) |
| Follow-up duration in person-years | 78.3 | 342.8 |
| Mean (SD) ABR in bleeds/year | 5.4 (6.9) | 2.6 (6.2)* |
| Median (min, max) ABR in bleeds/year | 3.3 (0, 34.6) | 0.3 (0, 35.0)* |
| Observed spontaneous bleed count (proportion of total bleeds) | 176 (42%) | 179 (41%) |
| Observed joint bleed count (proportion of total bleeds) | 240 (57%) | 195 (45%) |
In the rollover population, a total of 5 patients (4%) did not respond and 17 patients (15%) lost response to ROCTAVIAN treatment over a median time of 2.3 (range: 1.0 to 3.3) years. In the directly enrolled population with a longer follow-up, a total of 1 patient (5%) did not respond and 6 patients (27%) lost response to ROCTAVIAN treatment over a median time of 3.6 (range: 1.2 to 4.3) years.
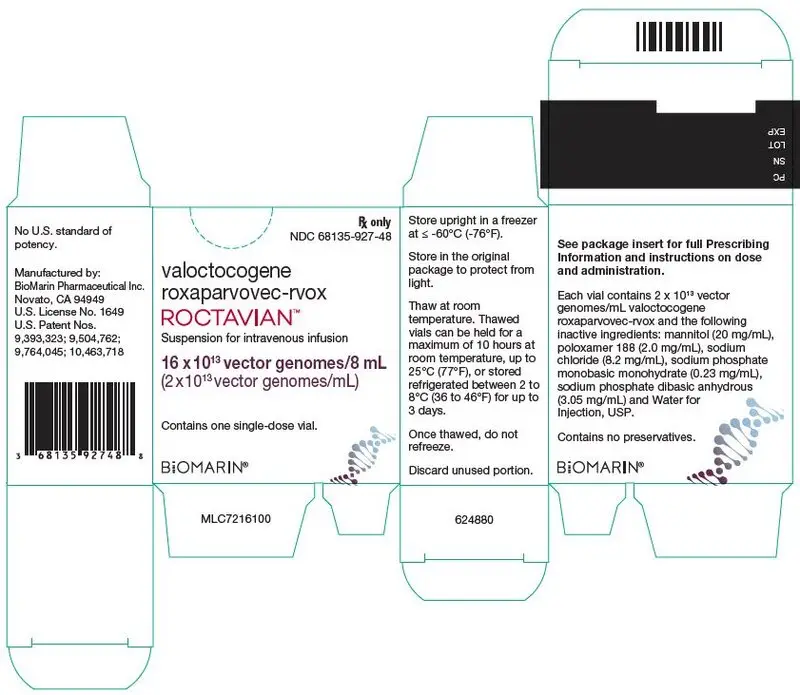
16. How is Roctavian supplied
16.1 How Supplied
ROCTAVIAN (valoctocogene roxaparvovec-rvox) injection is supplied as a sterile, preservative-free, clear and colorless to pale yellow suspension for intravenous infusion. ROCTAVIAN contains 2 × 1013 vector genomes (vg) per mL.
Each carton of ROCTAVIAN (NDC 68135-927-48) contains one single-dose vial (NDC 68135-927-01) with an extractable volume of not less than 8 mL, containing 16 × 1013 vector genomes (vg).

| ROCTAVIAN
valoctocogene roxaparvovec-rvox injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - BioMarin Pharmaceutical Inc. (079722386) |
