Drug Detail:Ruconest (C1 esterase inhibitor recombinant injection (conestat alfa))
Drug Class: Hereditary angioedema agents
Highlights of Prescribing Information
RUCONEST (C1 esterase inhibitor [recombinant])
For Intravenous Use, Lyophilized Powder For Reconstitution
Initial U.S. Approval: 2014
Indications and Usage for Ruconest
RUCONEST is a C1 esterase inhibitor [recombinant] indicated for the treatment of acute attacks in adult and adolescent patients with hereditary angioedema (HAE). ( 1)
Limitation of Use: Effectiveness was not established in HAE patients with laryngeal attacks.
Ruconest Dosage and Administration
For Intravenous Use Only.
- Reconstitute each vial (2100 U) by adding 14 mL sterile Water for Injection per vial to obtain a solution of 150 U per mL. ( 2.3)
- Administer the reconstituted solution at room temperature as a slow intravenous injection over approximately 5 minutes. ( 2.4)
- Appropriately trained patients may self-administer upon recognition of an HAE attack. ( 2.4)
Recommended dose of RUCONEST for an acute attack
|
Body weight |
RUCONEST Dose for Intravenous Injection |
Volume (mL) of Reconstituted Solution (150 U/mL) to be Administered |
|
< 84 kg |
50 U per kg |
Body weight in kg divided by 3 |
|
≥ 84 kg |
4200 U (2 vials) |
28 mL |
If the attack symptoms persist, an additional (second) dose can be administered at the recommended dose level. Do not exceed 4200 U per dose. No more than two doses should be administered within a 24 hour period.
Dosage Forms and Strengths
2100 U Lyophilized powder for reconstitution for injection in a single-use vial. ( 3)
Contraindications
- Known or suspected allergy to rabbits and rabbit-derived products. ( 4)
- History of immediate hypersensitivity reactions, including anaphylaxis, to C1 esterase inhibitor preparations. ( 4)
Warnings and Precautions
- Hypersensitivity reactions, including anaphylaxis may occur. Should symptoms occur, discontinue RUCONEST and administer appropriate treatment. ( 5.1)
- Serious arterial and venous thromboembolic (TE) events have been reported at the recommended dose of plasma derived C1 esterase inhibitor products in patients with risk factors. Monitor patients with known risk factors for TE events during and after RUCONEST administration. ( 5.2)
Adverse Reactions/Side Effects
- The serious adverse reaction reported in clinical trials was anaphylactic reaction. ( 6)
- The common adverse reactions (≥ 2%) reported in clinical trials were headache, nausea, and diarrhea. ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact Pharming Healthcare Inc. at 1-800-930-5221 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
Pregnancy: Limited animal data. No human data. Use only if clearly needed. ( 8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2018
Full Prescribing Information
1. Indications and Usage for Ruconest
RUCONEST is a C1 esterase inhibitor [recombinant] indicated for the treatment of acute attacks in adult and adolescent patients with hereditary angioedema (HAE).
Limitation of Use: Effectiveness was not established in HAE patients with laryngeal attacks.
2. Ruconest Dosage and Administration
For intravenous use after reconstitution only.
2.1 Recommended Dosing
- Initiate treatment with RUCONEST under the supervision of a qualified healthcare professional experienced in the treatment of HAE.
- Appropriately trained patients may self-administer upon recognition of an HAE attack.
- The recommended dose of RUCONEST is 50 U per kg with a maximum of 4200 U to be administered as a slow intravenous injection over approximately 5 minutes.
If the attack symptoms persist, an additional (second) dose can be administered at the recommended dose level. Do not exceed 4200 U per dose. No more than two doses should be administered within a 24 hour period.
|
Body Weight |
RUCONEST Dose for Intravenous Injection |
Volume (mL) of Reconstituted Solution (150 U/mL) to be Administered |
|
< 84 kg |
50 U per kg |
Body weight in kg divided by 3 |
|
≥ 84 kg |
4200 U (2 vials) |
28 mL |
2.2 Preparation and Handling
- Store RUCONEST in the original carton and protect from light prior to reconstitution.
- Do not use after expiration date on the product vial label.
- Water for Injection is not included in the RUCONEST package.
- Use aseptic technique to reconstitute, mix the solution, and to combine the reconstituted solution from more than one vial ( see Reconstitution [ 2.3] and Administration [ 2.4]).
- Do not mix or administer RUCONEST with other medicinal products or solutions.
- Discard all partially used vials after treatment.
2.3 Reconstitution
Each package contains one single-use vial of RUCONEST. To reconstitute, the following are also required:
- Sterile Water for Injection (diluent) - At least 14 mL per vial of RUCONEST requiring reconstitution.
- Antiseptic wipe
- Syringe
- Commercially available vial adapter and syringe luer lock or large bore needle. If using a syringe with vial adapter, use a new vial adapter for each vial of RUCONEST and diluent.
The procedures below are provided as general guidelines for the reconstitution and administration of RUCONEST.
- Ensure that the RUCONEST vial and diluent vial are at room temperature.
- Remove the flip caps from the RUCONEST and diluent vials. Treat the vial stoppers with the antiseptic wipe and allow to dry.
- Using the syringe/needle or syringe/vial adapter, withdraw 14 mL of sterile water for injection from the diluent vial.
- Remove the syringe and transfer the diluent to the RUCONEST vial. Add the diluent slowly to avoid forceful impact on the powder. Swirl the vial slowly to mix and avoid foaming.
- Repeat this procedure using another 14 mL of diluent and a second vial of RUCONEST.
- If the same patient is to receive more than one vial, the contents of multiple vials may be pooled into a single administration device (i.e., syringe).
- Inspect RUCONEST visually for particulate matter and discoloration after reconstitution and prior to administration. The reconstituted solution should be colorless, clear, and free from visible particles. Do not use if the solution is cloudy, colored, or contains particulates.
- RUCONEST vial is for single-use only.
- Use the reconstituted product immediately, or within 8 hours stored at 36ºF - 46ºF (2º - 8ºC). Discard partially used vials.
- Do not freeze the reconstituted solution.
2.4 Administration
- Do not mix RUCONEST with other medicinal products. Administer RUCONEST by a separate infusion line.
- Use aseptic technique when administering RUCONEST.
- Follow recommended venipuncture guidelines for initiating intravenous therapy.
- Administer RUCONEST by slow intravenous injection over approximately 5 minutes.
- For self-administration, provide the patient with instructions and training for intravenous injection outside of a clinic setting so patients may self-administer RUCONEST upon recognition of symptoms of an HAE attack ( see Patient Counseling Information [ 17]).
- After administration, immediately discard any unused product and all used disposable supplies in accordance with local requirements.
3. Dosage Forms and Strengths
- RUCONEST is available as a lyophilized powder for reconstitution for injection in a single-use 25 mL glass vial. Each vial contains 2100 U of rhC1INH.
4. Contraindications
- RUCONEST is contraindicated in patients with a history of allergy to rabbits or rabbit-derived products.
- RUCONEST is contraindicated in patients with a history of life-threatening immediate hypersensitivity reactions to C1 esterase inhibitor preparations, including anaphylaxis.
5. Warnings and Precautions
5.1 Hypersensitivity
Severe hypersensitivity reactions may occur [see Patient Counseling Information (17)]. The signs and symptoms of hypersensitivity reactions may include hives, generalized urticaria, tightness of the chest, wheezing, hypotension, and/or anaphylaxis during or after injection of RUCONEST. Should symptoms occur, discontinue RUCONEST and institute appropriate treatment. Because hypersensitivity reactions may have symptoms similar to HAE attacks, treatment methods should be carefully considered.
5.2 Thromboembolic Events
Serious arterial and venous thromboembolic (TE) events have been reported at the recommended dose of plasma derived C1 esterase inhibitor products in patients with risk factors. Risk factors may include the presence of an indwelling venous catheter/access device, prior history of thrombosis, underlying atherosclerosis, use of oral contraceptives or certain androgens, morbid obesity, and immobility. Monitor patients with known risk factors for TE events during and after RUCONEST administration.
6. Adverse Reactions/Side Effects
The serious adverse reaction in clinical studies of RUCONEST was anaphylaxis.
The most common adverse reactions (≥ 2%) reported in all clinical trials were headache, nausea, and diarrhea.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The RUCONEST clinical development program evaluated a combined total of 940 administrations in 236 subjects (symptomatic and non-symptomatic). In clinical studies, a total of 205 symptomatic HAE patients received treatment with RUCONEST for a combined total of 650 acute angioedema attacks. Among these HAE patients, 83 were treated for a single HAE attack and 122 were treated for multiple attacks.
Three randomized, placebo-controlled clinical trials (RCTs) were conducted in which 137 patients experiencing acute HAE attacks received RUCONEST (either an initial 50 U/kg or 100 U/kg body weight dose) or placebo (saline solution).
Table 2 shows all adverse reactions (ARs) in the RCTs, compared with the placebo group.
Table 2. Adverse reactions occurring In ≥ 2% of subjects in the three RCT studies
| MedDRA Preferred Term |
RUCONEST 100 U/kg (N=29) n (%) |
RUCONEST
50 U/kg*
|
Placebo**
(N=47)
|
| Total number of patients with adverse reactions | 4 (14%) | 6 (9%) | 13 (28%) |
| Headache | 3 (10%) | 0 | 2 (4%) |
| Sneezing | 0 | 1 (2%) | 0 |
| Angioedema | 1 (3%) | 0 | 0 |
| Erythema marginatum | 0 | 1 (2%) | 0 |
| Skin burning sensation | 0 | 1 (2%) | 0 |
| Back pain | 0 | 2 (3%) | 0 |
| C-reactive protein increased | 0 | 1 (2%) | 0 |
| Fibrin D-dimer increased | 0 | 1 (2%) | 0 |
| Vertigo | 1 (3%) | 0 | 0 |
| Procedural headache | 0 | 1 (2%) | 0 |
| Lipoma | 0 | 1 (2%) | 0 |
* Includes 5 patients who received an additional 50 U/kg dose
MedDRA: Medical Dictionary for Regulatory Activities, version 15.0.
** Events only occurring in placebo patients are not listed.
Integrated RCT and Open-Label Extension (OLE) Studies
In a total of seven RCT and OLE studies, 205 patients experiencing acute HAE attacks were treated with RUCONEST for a total of 650 HAE attacks. Included in this population were 124 patients who were treated at the 50 U/kg dosage strength for one or more attacks.
Table 3 shows adverse reactions in ≥ 2% of patients in any RUCONEST group for the integrated dataset combining all seven RCT and OLE studies in patients experiencing acute HAE attacks.
Table 3. Adverse reactions in the seven RCT and OLE studies occurring ≥ 2% of RUCONEST-treated patients (all doses), irrespective of causality
| MedDRA Preferred Term | All RUCONEST doses* N=205 n (%) |
| Headache | 19 (9%) |
| Nausea | 5 (2%) |
| Diarrhea | 5 (2%) |
* RUCONEST doses: doses up to 100 U/kg
MedDRA: Medical Dictionary for Regulatory Activities, version 15.0.
Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. Pre- and post-exposure samples from 205 HAE patients treated for 650 acute attacks with RUCONEST were tested for the antibodies against plasma-derived C1INH or rhC1INH and for antibodies against host-related impurities (HRI). Testing was performed prior to and after treatment of a first attack and subsequent repeated attacks at 7, 22 or 28, and 90 days after RUCONEST treatment.
Prior to the first exposure to RUCONEST, the frequency of anti-C1INH antibodies varied from 1.2% to 1.6% of samples. After the first exposure, the frequency of anti-C1INH antibodies varied from 0.6% to 1.0% of samples tested. After repeated exposures, the frequency of anti-C1INH antibodies varied from 0.5 to 2.2% of samples tested. The frequency of anti-HRI antibodies was 1.0% in pre-exposure samples, and after the first exposure varied from 3.5% to 4.6%. After repeated exposure, the frequency of anti-HRI antibodies varied from 5.7% to 17% of samples. At least 10% of subjects formed a specific antibody response to RUCONEST after five treated HAE attacks. No anti-C1INH neutralizing antibodies were detected. Observed anti-C1INH and anti-HRI antibodies were not associated with adverse clinical findings.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to RUCONEST with the incidence of antibodies to other products may be misleading.
Postmarketing Experience
Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Category B. Studies performed in rats and rabbits at doses up to 12.5 times the human dose of 50 U/kg could not exclude an effect on embryofetal development. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, RUCONEST should only be used during pregnancy if clearly needed.
8.2 Labor and Delivery
The safety and efficacy of RUCONEST administration prior to or during labor and delivery have not been established. Use only if clearly needed.
8.3 Nursing Mothers
It is not known if RUCONEST is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when RUCONEST is administered to a nursing woman.
8.4 Pediatric Use
The safety and efficacy of RUCONEST were evaluated in 17 adolescent patients (13-17 years of age) treated for 52 HAE attacks. Eight out of 17 (47%) adolescent patients experienced adverse reactions. No serious adverse reactions were reported in these patients. The most common reactions (occurring in at least 2 patients) were: abdominal pain, headache, and oropharyngeal pain.
11. Ruconest Description
RUCONEST is a recombinant analogue of human complement component 1 esterase inhibitor for intravenous injection. RUCONEST is purified from the milk of transgenic rabbits, and supplied as a sterile, preservative-free, white/off-white lyophilized powder for reconstitution for injection. One U of rhC1INH activity is defined as the equivalent of C1 esterase inhibiting activity present in 1 mL of pooled normal plasma.
RUCONEST is a soluble, single-chain glycoprotein containing 478 amino acids, with a molecular mass of 68 kDa, of which approximately 22% comprises oligosaccharide structures. The primary and secondary structures of the molecule and target protease selectivity are consistent with those of plasma-derived C1 esterase inhibitor.
Each vial of RUCONEST contains 2100 U of rhC1INH, 937 mg of sucrose, 83.3 mg of sodium citrate dihydrate and 1.0 mg of citric acid monohydrate. After reconstitution with 14 mL of sterile Water for Injection, each vial of RUCONEST contains 150 U of rhC1INH per 1 mL in a 20 mM sodium citrate buffer with a pH of 6.8. RUCONEST does not contain preservatives and each vial is for single use only.
RUCONEST is purified from the milk of transgenic rabbits. The rabbits are maintained in a closed colony that is controlled and routinely monitored for specific pathogens. The skimmed milk is screened for adventitious contaminants prior to further manufacture. The manufacturing process has been validated to demonstrate adequate capacity for removal and/or inactivation of viruses ( Table 4). RUCONEST contains less than 0.002% of host-related impurities.
| a MLV: Murine leukemia virus; REO-3: Reovirus type 3; ORF: Scab-mouth ORF virus; FCV: Feline calicivirus; PPV: Porcine parvovirus;
b SP BB: SP Sepharose BB; SD: Solvent/detergent; Q HP: Q Sepharose HP; Zn FF: Zinc Chelating Sepharose FF; c Not added to the total reduction factor since independence of clearance mechanism has not been experimentally verified; d Not applicable since indicated model virus is not enveloped; e Not tested because SD chemicals are present in the starting material. |
|||||
|
Virus Reduction Factor (Log 10) |
|||||
|
Step |
MLV a |
REO 3 |
ORF |
FCV |
PPV |
|
SP BB chromatography b |
1.8 |
2.2 c |
1.5 c |
2.3 |
1.5 b |
|
SD incubation b |
≥ 5.8 |
NA d |
3.7 |
NA |
NA |
|
Q HP chromatography b |
NT e |
4.8 |
NT |
0.8 c |
2.2 |
|
Zn FF chromatography b |
1.1 c |
3.2 c |
3.3 |
1.9 c |
0.4 c |
|
Nanofiltration |
≥ 5.5 |
≥ 6.5 |
≥ 5.8 |
≥ 6.9 |
5.8 |
|
Total reduction factor |
≥ 13.1 |
≥ 11.3 |
≥ 12.8 |
≥ 9.2 |
8.0 |
12. Ruconest - Clinical Pharmacology
12.1 Mechanism of Action
C1 esterase inhibitor (C1INH) is a normal constituent of human blood and is one of the serine protease inhibitors (serpins). The primary function of C1INH is to regulate the activation of the complement and contact system pathways. Regulation of these systems is performed through the formation of complexes between the protease and the inhibitor, resulting in inactivation of both and consumption of the C1INH.
C1INH exerts its inhibitory effect by irreversibly binding several proteases (target proteases) of the contact and complement systems. The effect of RUCONEST on the following target proteases was assessed in vitro: activated C1s, kallikrein, factor XIIa and factor XIa. Inhibition kinetics were found to be comparable with those observed for plasma-derived human C1INH.
HAE patients have low levels of endogenous or functional C1INH. Although the events that induce attacks of angioedema in HAE patients are not well defined, it is thought that contact system activation, and resulting increased vascular permeability lead to the clinical manifestation of HAE attacks. Suppression of contact system activation by C1INH through the inactivation of plasma kallikrein and factor XIIa is thought to modulate vascular permeability by preventing the generation of bradykinin. 5
Administration of RUCONEST increases plasma levels of functional C1INH activity.
12.2 Pharmacodynamics
The complement component (protein) C4 is a substrate for activated C1. Patients with HAE have low levels of C4 in the circulation; RUCONEST shows a dose-dependent restoration of complement homeostasis of C4 in HAE patients. A dose of 50 U/kg of RUCONEST increases plasma C1INH activity levels to greater than 0.7 U/mL (the lower limit of normal) in HAE patients.
12.3 Pharmacokinetics
The pharmacokinetics of RUCONEST was evaluated in a study of 12 asymptomatic HAE patients (dose ranged from 6.25 U/kg to 100 U/kg). Pharmacokinetics evaluation was performed by non-compartmental analysis, using functional C1INH levels. Following administration of RUCONEST (50 U/kg) in asymptomatic HAE patients ( Table 5), the mean Cmax was 1.2 U/mL, and the elimination half-life was approximately 2.5 hours. The clearance of RUCONEST was nonlinear (clearance decreased with increasing dose) over the dose range of 25-100 U/kg.
|
Parameters |
50 U/kg |
100 U/kg |
|
C baseline (U/mL) |
0.18 ± 0.12 |
0.14 ± 0.08 |
|
C max (U/mL) |
1.2 ± 0.2 |
2.3 ± 0.2 |
|
T max (hours) |
0.31 ± 0.10 |
0.31 ± 0.10 |
|
AUC (U x hr/mL) |
3.3 ± 1.0 |
10.6 ± 2.5 |
|
CL (mL/hr) |
1207 ± 414 |
781 ± 147 |
|
Half-life (hours) |
2.4 ± 0.6 |
2.7 ± 0.3 |
|
V ss (L) |
3.0 ± 0.9 |
2.4 ± 0.5 |
Studies have not been conducted to evaluate the PK of RUCONEST in special patient populations, identified by race, age (pediatric or geriatric), or the presence of renal or hepatic impairment.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been conducted to evaluate the effects of RUCONEST on carcinogenesis and mutagenesis. Fertility was not evaluated in animal studies.
13.2 Animal Toxicology and/or Pharmacology
Single- and repeat-dose studies of up to 14 days in rats, dogs, and cynomolgus monkeys with daily doses of RUCONEST up to 80 times the human dose (50 U/kg) were conducted. In a 14-day monkey toxicology study of intravenous doses up to 2000 U/kg twice daily, increases in AST and ALP were observed at doses of 500 U/kg and higher. No histopathological correlates were observed in the liver and the increases in liver enzymes were reversible. Histologic findings included changes in the size of thymic cortex and medulla, and microvacuoles in the epithelial cells lining the renal tubules. The renal tubular vacuolation was dose dependent, but was not accompanied by other histological changes in the kidney. This finding was only partially reversible at the highest dose level of 2000 U/kg twice daily. It was concluded that the NOAEL of RUCONEST was 1000 U/kg twice daily in this species. There were no adverse findings in a safety pharmacology study in dogs and a local tolerance study in rabbits. It is concluded that preclinical toxicology data for daily doses up to 40 times the proposed human dose of RUCONEST (50 U/kg) do not indicate a safety concern for the use of RUCONEST in humans.
Embryo-fetal studies have been conducted in rabbits and rats at a dose of RUCONEST 12.5 times the human dose of 50 U/kg. In rats, no malformed fetuses were observed. In rabbits, an increase in the incidence of fetal cardiac vessel defects was observed (1.12% [2 cases] in the treatment group compared to 0.03% in historical controls). These defects are considered to be a chance finding, but a RUCONEST-related effect cannot be excluded.
14. Clinical Studies
The safety and efficacy of RUCONEST for treatment of acute angioedema attacks in patients with HAE was established in Study 1, a double-blind, randomized, placebo-controlled trial (RCT) which included an open-label extension (OLE) phase; and supported by the results of 2 additional RCTs and 2 additional OLE studies.
Randomized, Controlled Trials
The safety and efficacy of RUCONEST in the treatment of acute attacks in patients with hereditary angioedema were demonstrated in a placebo-controlled, double-blind, randomized study (Study 1). Supportive evidence of effectiveness is provided by two double-blind, randomized, placebo-controlled studies (Studies 2 and 3). Evidence for the efficacy of repeat treatment of HAE attacks is provided from the open-label extensions (OLE) of each of the three randomized studies.
Study 1 was a randomized, double-blind, placebo-controlled trial that included an open-label extension (OLE) phase to assess the efficacy and safety of RUCONEST 50 U/kg in the treatment of acute attacks in patients with HAE. Seventy-five (75) adults and adolescent patients were randomized (3:2) to receive RUCONEST 50 U/kg (N = 44) or placebo (N = 31). Patients ranged in age from 17 to 69 years of age; 63% were female and 37% were male; 96% were Caucasian.
The primary efficacy endpoint was the time to beginning of relief of symptoms, assessed using patient-reported responses to two questions from a Treatment Effect Questionnaire (TEQ). The TEQ required patients to assess the severity of their attack symptoms at each affected anatomic location, using a seven-point scale (“much worse” to “much better” [TEQ Question 1]), and whether their symptoms had begun to decrease notably since receiving the study medication (“yes” or “no” [TEQ Question 2]). To achieve the primary endpoint, a patient had to have a positive response to both questions along with persistence of improvement at the next assessment time (i.e., the same or better response).
Rescue treatment with RUCONEST was available for patients who did not experience the beginning of relief at 4 hours after study drug administration, or earlier to patients who experienced life-threatening oropharyngeal-laryngeal angioedema symptoms. If a patient received a medication which could have impacted the efficacy evaluation or open-label RUCONEST as rescue medication, prior to achieving beginning of relief of symptoms, the time to beginning of relief of symptoms was censored at the last assessed time prior to medication use.
In the RCT phase, the median time to beginning of relief of symptoms was statistically significantly shorter in patients treated with RUCONEST 50 U/kg compared with patients treated with placebo as assessed by the TEQ; ( Table 6, Figure 1).
| Time to Beginning of Relief of Symptoms, minutes | RUCONEST 50 U/kg
N=44 | Placebo
N=31 |
|---|---|---|
| Values that are not estimable are displayed as ‘- ’. | ||
|
Median |
90 |
152 |
|
95% CI |
(61, 150) |
(93, -) |
|
Log-rank p-value |
0.031 |
|
Figure 1. Kaplan-Meier Plot of Time to Beginning of Relief of Symptoms (Study 1, RCT Phase)
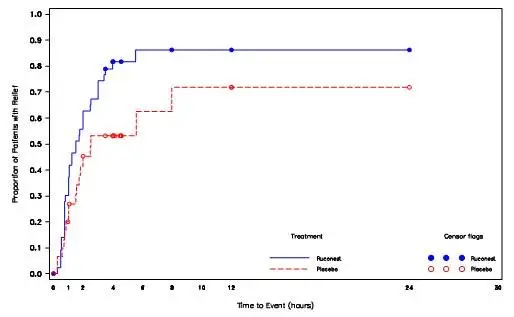
Among several planned subgroup analyses, descriptive statistics showed that in US patients a median time to beginning of relief of symptoms with persistence at the primary attack location (based on TEQ) was 98 minutes [95% CI:(45, 240); n=22] for those receiving RUCONEST and 90 minutes [95% CI: (50, -); n=16] for those receiving placebo. The hazard ratio for time to the beginning of relief of symptoms in this subpopulation was 1.20 [95% CI: 0.48 to 3.01] for patient receiving RUCONEST as compared with patient receiving placebo. Non-US patients receiving RUCONEST had a median time to beginning of relief of 90 minutes [95% CI: (63, 120); n=22] and non-US patients receiving placebo had a median time to beginning of relief of 334 minutes [95% CI: (150, -); n=15]. The hazard ratio for the non-US subgroup was 4.82 [95% CI: 1.58 to 14.72] for patients receiving RUCONEST compared to placebo.
Examination of gender subgroups suggested a larger treatment effect in men than women. For women receiving RUCONEST, the median time to beginning of relief was 113 minutes [95% CI: (63, 151); n=28], and for women receiving placebo, the median time to beginning of relief was 105 minutes [95% CI: (60, 334); n=19]. The hazard ratio for women receiving RUCONEST versus placebo was 1.22 [95% CI: 0.60 to 2.48]. For men receiving RUCONEST, the median time to beginning of relief was 75 minutes [95% CI: (45, 210); n=16], and for men receiving placebo, the median time to beginning of relief was 480 minutes [95% CI: (150, -) n=12]. The hazard ratio for men receiving RUCONEST versus placebo was 3.94 [95% CI: 1.23 to 12.68].
No plausible biological explanations for the regional or gender subgroup effects were found. One possible explanation is a larger-than-expected placebo response among US women. None of the subgroup confidence intervals were adjusted for multiplicity.
Because almost all of the patients were Caucasian and were between 18 and 65 years of age, race and age subgroup analyses were not considered meaningful.
Among patients who achieved relief within 4 hours, there were 4 (27%) patients in the placebo group who had a relapse of their symptoms within 24 hours as compared with 1 (3%) in the RUCONEST group. The proportion of patients who received RUCONEST as rescue medication was greater in patients randomized to placebo (13 of 31 patients; 42%) than in patients randomized to RUCONEST (5 of 44 patients; 11%).
The efficacy of RUCONEST 50 U/kg for different anatomical locations of HAE attacks is summarized in Table 7.
| *Life-threatening laryngeal attacks were excluded from the randomized-controlled phase of Study 1 | |||||
|
Attack Type * |
RUCONEST 50 U/kg n/N (%) |
Placebo n/N (%) |
|||
|
Abdominal |
14/16 (88%) |
7/12 (58%) |
|||
|
Facial |
3/6 (50%) |
0/2 (0%) |
|||
|
Peripheral (extremities) |
17/20 (85%) |
7/14 (50%) |
|||
In the OLE phase of Study 1, patients were treated with open-label RUCONEST 50 U/kg for repeated attacks of HAE. Forty-four patients who completed the RCT phase were enrolled into the OLE phase where they were treated for a total of 170 attacks. In this phase, the median time to beginning of relief of symptoms was 75 minutes (95% CI: 64, 90), consistent with the results of the RCT phase of the study ( Table 6). Results were also comparable across attacks, suggesting that the efficacy of RUCONEST 50 U/kg was maintained over repeated attacks of HAE. In the OLE phase of Study 1, 5/170 (3%) attacks received a second dose of RUCONEST 50 U/kg.
In Study 2 (North American RCT), patients were randomized to receive a single administration of either RUCONEST 50 U/kg (N=12), RUCONEST 100 U/kg (N=13) or placebo (N=13). Patients ranged in age from 17 to 66 years of age; 74% were female and 26% were male; and 92% were Caucasian.
In Study 3 (European RCT), patients were randomized to receive a single administration of either RUCONEST 100 U/kg (N=16) or placebo (N=16). Patients ranged in age from 17 to 71 years of age: 53% were female and 47% were male; and 100% were Caucasian.
Patients scored their symptoms using a visual analog scale (VAS) ranging from 0-100 mm. A VAS decrease of > 20 mm compared with baseline with persistence of the improvement at two consecutive time points was considered the onset of relief in Studies 2 and 3.
In both Study 2 and 3, the efficacy of RUCONEST in the treatment of acute angioedema attacks was demonstrated by significantly shorter times to beginning of relief of symptoms based on the VAS (Figure 2).
Figure 2. Mean VAS scores over time with 95% Confidence Intervals (Study 2 and 3, RCT Phase)
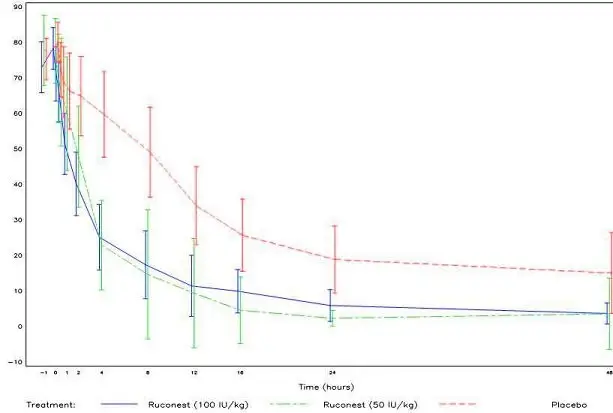
In open-label extension studies of Study 2 and 3, 119 patients were treated with RUCONEST for a total of 362 acute angioedema attacks. As observed in Study 1, the efficacy of RUCONEST was maintained for repeat attacks.
15. References
- Sulikowski T, Patston PA. The inhibition of TNK-t-PA by C1-inhibitor. Blood Coagul Fibrinolysis. 2001 Jan;12(1):75-7.
- Chandler WL, Alessi MC, Aillaud MF, Henderson P, Vague P, Juhan-Vague I. Clearance of tissue plasminogen activator (TPA) and TPA/plasminogen activator inhibitor type 1 (PAI-1) complex: relationship to elevated TPA antigen in patients with high PAI-1 activity levels. Circulation. 1997 Aug 5;96(3):761-8.
- Huisman LG, van Griensven JM, Kluft C. On the role of C1-inhibitor as inhibitor of tissue-type plasminogen activator in human plasma. Thromb Haemost. 1995 Mar;73(3):466-71.
- Gurewich V, Pannell R. Recombinant human C1-inhibitor prevents non-specific proteolysis by mutant pro-urokinase during optimal fibrinolysis. Thromb Haemost. 2009 Aug;102(2):279-86.
- Caliezi C, Wuillemin WA, Zeerleder S, Redondo M, Eisele B, Hack CE. C1-esterase inhibitor: an anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharmacol Rev. 2000 Mar;52(1):91-112.
- German Medical Profession’s Drugs Committee [Arzneimittelkommission der deutschen Arzteschaft]. Severe thrombus formation of Berinert ® HS [Schwerwiegende Thrombenbildung nach Berinert ® HS]. Deutsches Ärzteblatt. 2000 April 2000; 97(Heft 15): A-1016.
- Horstick, G, Berg O, Heimann A, Gotze O, Loos M, Hafner G, et al. Application of C1-esterase inhibitor during reperfusion of ischemic myocardium: dose-related beneficial versus detrimental effects. Circulation. 2001 Dec 18;104(25):3125-31.
16. How is Ruconest supplied
16.1 How supplied
- RUCONEST is supplied in single-use 25 mL glass vials with a stopper (siliconized chlorobutyl rubber) and a flip-off seal (aluminum and colored plastic).
- Each carton contains one single-use vial.
- Each vial contains 2100 U rhC1INH lyophilized powder for reconstitution for injection.
16.2 Storage and handling
- Store in the original package in order to protect from light.
- Shelf life: 48 months when stored at 36ºF - 77ºF (2ºC - 25ºC).
- Do not freeze.
- Each vial of RUCONEST should be reconstituted with 14 mL Water for Injection (not supplied). The reconstituted solution contains 150 U/mL rhC1INH and is clear and colorless.
- Each vial of RUCONEST is for single use only. RUCONEST contains no preservative. Any product that has been reconstituted should be used immediately, or within 8 hours stored at 36ºF - 46ºF (2ºC - 8ºC). Discard partially used vials after treatment.
17. Patient Counseling Information
Advise the patient to read the FDA-Approved patient labeling (Product Information and Instructions for Use).
Patients being treated with RUCONEST should receive the following information and instructions. This information is intended to aid the patient in the safe and effective use of RUCONEST.
- Advise female patients to notify their physician if they are pregnant or intend to become pregnant during the treatment of acute attacks of HAE with RUCONEST.
- Advise patients to notify their physician if they are breastfeeding or plan to breastfeed.
- Inform patients of the risks and benefits of RUCONEST before prescribing or administering it to the patient.
Advise patients to immediately report :
- Signs and symptoms of allergic hypersensitivity reactions, such as hives, urticaria, tightness of the chest, wheezing, hypotension and/or anaphylaxis experienced during or after injection of RUCONEST ( see WARNINGS AND PRECAUTIONS/Hypersensitivity [ 5] ).
Manufactured by:
Pharming Americas B.V.
Darwinweg 24
2333 CR Leiden
The Netherlands
RUCONEST
® is a registered trademark of Pharming Intellectual Property B.V.
Distributed by:
Pharming Healthcare Inc.
Bridgewater
NJ 08807
Product protected by U.S patent Nos. 7,067,713 and RE43691.
Please see www.ruconest.com for patent information.
US License No. 2079

REV NOV 2017
FDA-Approved Patient Labeling - Patient Product Information (PPI)
RUCONEST ® (ROO-Ko-nest)
( C1 esterase inhibitor [recombinant])
Lyophilized powder for reconstitution for injection in a single-use vial
This leaflet summarizes important information about RUCONEST. Please read it carefully each time before using RUCONEST. There may be new information provided. This information does not take the place of talking with your healthcare provider, and it does not include all of the important information about RUCONEST. If you have any questions after reading this, ask your healthcare provider.
What is RUCONEST?
RUCONEST is an injectable medicine that is used to treat acute angioedema attacks in adult and adolescent patients with Hereditary Angioedema (HAE). HAE is caused by a shortage of a protein called C1 esterase inhibitor, that is present in your blood and helps control inflammation (swelling) and parts of the immune system. A shortage of C1 esterase inhibitor can lead to repeated attacks of swelling, pain in the abdomen, difficulty breathing and other symptoms. RUCONEST contains C1 esterase inhibitor.
Who should not use RUCONEST?
You should not use RUCONEST if you have a known or suspected allergy (hypersensitivity) to rabbits or rabbit-derived products.
You should not use RUCONEST if you have experienced life-threatening immediate hypersensitivity reactions, including anaphylaxis, to RUCONEST or to any other C1 esterase inhibitor product.
RUCONEST is not indicated for use in children under the age of 13 years.
What should I tell my healthcare provider before using RUCONEST?
Tell your healthcare provider about all of your medical conditions, including if you:
- have an allergy to rabbits since this can put you at high risk of a serious allergic reaction with RUCONEST. These allergy symptoms could include runny nose, itchy nose, sneezing, coughing, wheezing, difficulty breathing or watery eyes when you are near rabbits.
- are pregnant or planning to become pregnant. It is not known if RUCONEST can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if RUCONEST passes to your milk and if it can harm your baby.
Tell your healthcare provider and pharmacist about all of the medicines you take, including all prescription and non-prescription medicines such as over-the-counter medicines, supplements, or herbal remedies.
How is RUCONEST given?
RUCONEST will be slowly injected into your vein (intravenous injection). Before injection, the RUCONEST powder must be dissolved using sterile Water for Injection. The dose will be determined based on your weight.
Most of the time a single dose of RUCONEST is enough to treat an attack, but a second dose may be needed.
What are the possible side effects of RUCONEST?
Like all medicines, RUCONEST can cause side effects, although not everybody gets them.
Allergic reactions may occur with RUCONEST. Call your healthcare provider or the emergency department immediately if you have any of the following symptoms after receiving RUCONEST:
- wheezing
- difficulty breathing
- chest tightness
- turning blue (look at lips and gums)
- fast heartbeat
- swelling
- faintness
- rash
- hives
In clinical studies, the most severe side effect reported in a person who received RUCONEST was a severe allergic reaction in a subject who was allergic to rabbits.
Other side effects patients experienced during clinical research studies include:
- headache
- nausea
- diarrhea
These are not all the possible side effects of RUCONEST.
If any of the side effects get serious, or if you notice any side effects not listed in the leaflet, please inform your healthcare provider or pharmacist. You can also report side effects to the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch or contact Pharming Healthcare Inc. at 1-800-930-5221.
You can ask your healthcare provider for information about RUCONEST that is written for healthcare providers.
General Information about RUCONEST
Do not use RUCONEST for a condition for which it is not prescribed.
If you would like more information, talk to your healthcare provider. You can ask your healthcare provider or pharmacist for information about RUCONEST that was written for healthcare professionals.
For more information go to www.ruconest.com or call 1-800-930-5221.
|
RECOMMENDED PREPARATION OF MEDICATION AND MATERIALS |
|
|
Use aseptic (sterile) technique when preparing and administering RUCONEST. | |
|
Step 1: Assemble supplies from medication box on a clean flat surface.
|  |
|
Step 2: Inspect the RUCONEST vial. Do not use if:
|  |
|
Step 3: Wash hands.
|  |
DOSAGE VERIFICATION
Discuss this section with your healthcare provider to ensure that the correct dose of RUCONEST is administered.
- The dose of RUCONEST and volume (mL) of reconstituted solution to be administered is based on body weight show in the chart below. No more than 2 vials may be combined for a single dose.
|
Body weight |
RUCONEST dose for intravenous injection |
Volume (mL) of reconstituted solution (150 U/mL) to be administered |
|
<84 kg (185.2 lb) |
50 U per kg of body weight |
Body weight in kg divided by 3 |
|
≥84 kg (185.2 lb) |
4200 U (2 vials) |
28 mL |
|
RECONSTITUTION The procedures below are provided as general guidelines for the reconstitution of RUCONEST. |
|
|
Step 1: Ensure that the RUCONEST vials and the diluent vial are at room temperature (range of 68°F - 77°F or 20°C - 25°C). The RUCONEST vial is for single | |
|
Step 2: Place the 2 RUCONEST vials and the diluent vial on a flat surface and remove the flip caps on each vial
|  |
|
Step 3: Peel back the covers from the clear plastic vial adapter packages.
| |
|
Step 4: While holding the adapter in its package, place the first adapter over the diluent vial and press down until the device snaps into place.
|
|
|
Step 5: Now, place a 2nd adapter over a RUCONEST vial and press down until it snaps into place. Repeat if your dose requires a 2nd RUCONEST vial. |  |
|
Step 6: Lift the package away from the diluent adapter. | |
|
Step 7: Remove syringe from package and withdraw sterile water for injection from the diluent vial.
|  |
|
Step 8: Transfer the diluent to the RUCONEST vial.
|  |
|
Step 9: Swirl the vial to mix. Do this slowly to avoid foaming. Repeat Steps 8 and 9 if you are using a second RUCONEST vial. Remove syringe from the first RUCONEST vial prior to attaching to the second vial. |  |
|
Step 10: Inspect RUCONEST vial(s) visually.
|  |
|
Step 11: With the syringe still connected to the RUCONEST vial, turn the vial upside down.
|
|
|
Step 12: Repeat Step 11 and withdraw additional reconstituted RUCONEST from the second RUCONEST vial (if needed).
| |
|
SELF-ADMINISTRATION (intravenous injection) Your healthcare provider will teach you how to safely administer RUCONEST. It is important that you inject RUCONEST directly into a visible vein. Do not inject into surrounding tissues or an artery. Once you learn how to self-administer, follow the instructions provided below. The reconstituted solution should be used within 8 hours and should be cooled but not be frozen. |
|
|
Step 1: Assemble supplies.
|  |
|
Step 2: Clean surface.
| |
|
Step 3: Prime the infusion set. As instructed by your healthcare provider:
|
|
|
Step 4: Prepare the approved IV injection site.
|  |
|
Step 5: Start infusion. Follow the instructions provided by your healthcare provider.
|
 |
|
Step 6: Cover the injection site and clean up. After all of the RUCONEST has been infused:
|  |
|
POST-ADMINISTRATION The procedures below are provided as general guidelines for after the infusion of RUCONEST. |
|
|
Record each injection into treatment journal.
| |


This Patient Package Insert has been approved by the US Food and Drug Administration.
Manufactured by:
Pharming Americas B.V.
Darwinweg 24
2333 CR Leiden
The Netherlands
RUCONEST ® is a registered trademark of Pharming Intellectual Property B.V.
Distributed by:
Pharming Healthcare Inc.
Bridgewater
NJ 08807
Product protected by U.S. patent Nos. 7,067,713 and RE43691.
Please see www.ruconest.com for patent information.
US License No. 2079

REV NOV 2017


| RUCONEST
c1 esterase inhibitor recombinant injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Bioconnection B.V. (414325873) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bioconnection B.V. | 414325873 | manufacture(70383-350) | |
