Drug Detail:Somatuline depot (Lanreotide [ lan-ree-oh-tide ])
Drug Class: Somatostatin and somatostatin analogs
Highlights of Prescribing Information
SOMATULINE® DEPOT (lanreotide) injection, for subcutaneous use
Initial U.S. Approval: 2007
Indications and Usage for Somatuline Depot
SOMATULINE DEPOT is a somatostatin analog indicated for:
- the long-term treatment of acromegalic patients who have had an inadequate response to or cannot be treated with surgery and/or radiotherapy. (1.1)
- the treatment of adult patients with unresectable, well- or moderately-differentiated, locally advanced or metastatic gastroenteropancreatic neuroendocrine tumors (GEP-NETs) to improve progression-free survival. (1.2)
- the treatment of adults with carcinoid syndrome; when used, it reduces the frequency of short-acting somatostatin analog rescue therapy. (1.3)
Somatuline Depot Dosage and Administration
Administration (2.1):
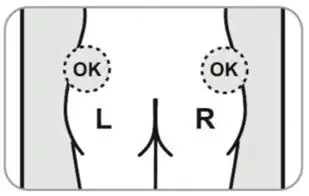
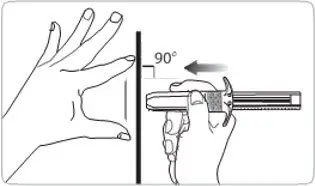
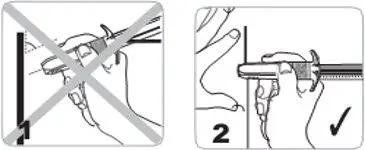
- For deep subcutaneous injection only.
- Intended for administration by a healthcare provider.
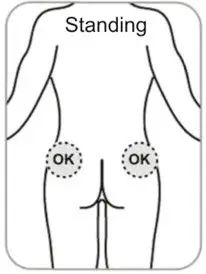
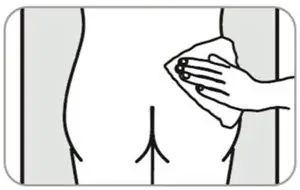
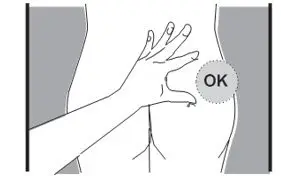
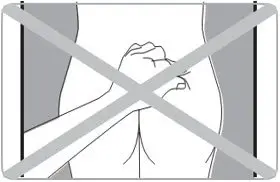
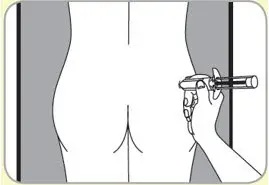
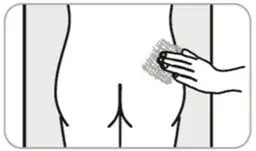
- Administer in the superior external quadrant of the buttock.
- Alternate injection sites.
Recommended Dosage (2.1)
- Acromegaly: 90 mg every 4 weeks for 3 months. Adjust thereafter based on GH and/or IGF-1 levels. See full prescribing information for titration regimen.
- GEP-NETs: 120 mg every 4 weeks.
- Carcinoid Syndrome: 120 mg every 4 weeks. If patients are already being treated with SOMATULINE DEPOT for GEP-NET, do not administer an additional dose for carcinoid syndrome.
Dosage Adjustment:
- See full prescribing information for dosage adjustment in patients with acromegaly and renal or hepatic impairment. (2.3, 2.4)
Dosage Forms and Strengths
Injection: 60 mg/0.2 mL, 90 mg/0.3 mL, and 120 mg/0.5 mL single-dose prefilled syringes (3)
Contraindications
Hypersensitivity to lanreotide. (4)
Warnings and Precautions
- Cholelithiasis and Complications of Cholelithiasis: Monitor periodically. Discontinue if complications of cholelithiasis are suspected. Gallstones may occur; consider periodic monitoring. (5.1)
- Hyperglycemia and Hypoglycemia: Glucose monitoring is recommended and antidiabetic treatment adjusted accordingly. (5.2, 7.1)
- Cardiovascular Abnormalities: Decrease in heart rate may occur. Use with caution in at-risk patients. (5.3)
- Thyroid Function Abnormalities: Decreases in thyroid function may occur; perform tests where clinically indicated. (5.4)
Adverse Reactions/Side Effects
Most common adverse reactions are:
- Acromegaly (>5%): diarrhea, cholelithiasis, abdominal pain, nausea and injection site reactions. (6.1)
- GEP-NET (>10%): abdominal pain, musculoskeletal pain, vomiting, headache, injection site reaction, hyperglycemia, hypertension, and cholelithiasis. (6.1)
- Carcinoid Syndrome: (≥5% and at least 5% greater than placebo): headache, dizziness and muscle spasm. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Ipsen Biopharmaceuticals, Inc. at 1-855-463-5127 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Drug Interactions
- Cyclosporine: SOMATULINE DEPOT may decrease the absorption of cyclosporine. Dosage adjustment of cyclosporine may be needed. (7.2)
- Bromocriptine: SOMATULINE DEPOT may increase the absorption of bromocriptine. (7.3)
- Bradycardia-Inducing Drugs (e.g., beta-blockers): SOMATULINE DEPOT may decrease heart rate. Dosage adjustment of the coadministered drug may be necessary. (7.3)
Use In Specific Populations
Lactation: Advise women not to breastfeed during treatment and for 6 months after the last dose. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2023
Full Prescribing Information
1. Indications and Usage for Somatuline Depot
1.1 Acromegaly
SOMATULINE DEPOT is indicated for the long-term treatment of acromegalic patients who have had an inadequate response to surgery and/or radiotherapy, or for whom surgery and/or radiotherapy is not an option.
The goal of treatment in acromegaly is to reduce growth hormone (GH) and insulin growth factor-1 (IGF-1) levels to normal.
2. Somatuline Depot Dosage and Administration
2.1 Important Administration Instructions
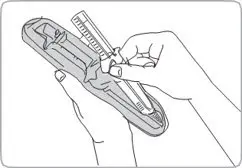
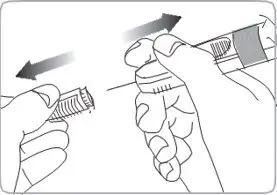
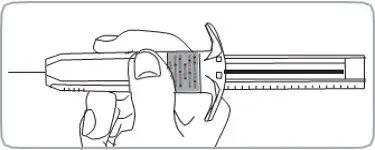
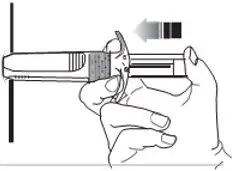
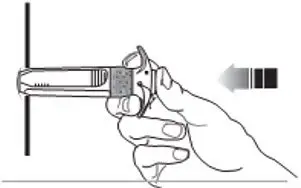
- For deep subcutaneous injection only.
- SOMATULINE DEPOT is intended for administration by a healthcare provider.
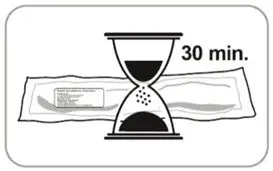
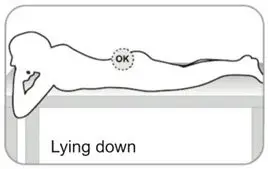
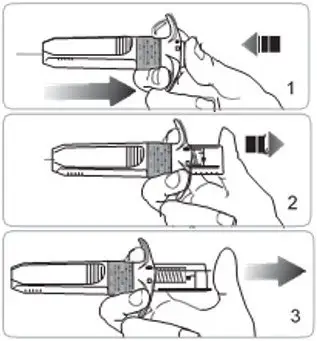
- Refer to the Instructions For Use (IFU) for complete administration instructions with illustrations.
3. Dosage Forms and Strengths
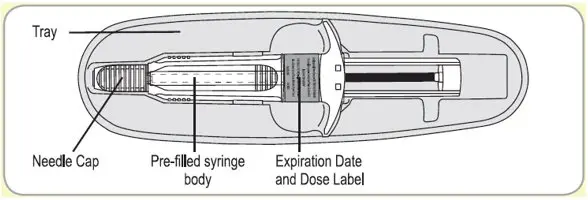
Injection: 60 mg/0.2 mL, 90 mg/0.3 mL, and 120 mg/0.5 mL sterile, single-dose, prefilled syringes fitted with an automatic safety system (attached retractable needle and needle guard). The prefilled syringes contain a white to pale yellow, semi-solid formulation.
4. Contraindications
SOMATULINE DEPOT is contraindicated in patients with history of a hypersensitivity to lanreotide. Allergic reactions (including angioedema and anaphylaxis) have been reported following administration of lanreotide [see Adverse Reactions (6.3)].
5. Warnings and Precautions
5.1 Cholelithiasis and Complications of Cholelithiasis
SOMATULINE DEPOT may reduce gallbladder motility and lead to gallstone formation; therefore, patients may need to be monitored periodically [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)]. There have been postmarketing reports of cholelithiasis (gallstones) resulting in complications, including cholecystitis, cholangitis, and pancreatitis, and requiring cholecystectomy in patients taking SOMATULINE DEPOT. If complications of cholelithiasis are suspected, discontinue SOMATULINE DEPOT and treat appropriately.
5.2 Hyperglycemia and Hypoglycemia
Pharmacological studies in animals and humans show that lanreotide, like somatostatin and other somatostatin analogs, inhibits the secretion of insulin and glucagon. Hence, patients treated with SOMATULINE DEPOT may experience hypoglycemia or hyperglycemia. Blood glucose levels should be monitored when lanreotide treatment is initiated, or when the dose is altered, and antidiabetic treatment should be adjusted accordingly [see Adverse Reactions (6.1)].
5.3 Cardiovascular Abnormalities
The most common overall cardiac adverse reactions observed in three pooled SOMATULINE DEPOT cardiac studies in patients with acromegaly were sinus bradycardia (12/217, 5.5%), bradycardia (6/217, 2.8%), and hypertension (12/217, 5.5%) [see Adverse Reactions (6.1)].
In 81 patients with baseline heart rates of 60 beats per minute (bpm) or greater treated with SOMATULINE DEPOT in Study 3, the incidence of heart rate less than 60 bpm was 23% (19/81) as compared to 16% (15/94) of placebo treated patients; 10 patients (12%) had documented heart rates less than 60 bpm on more than one visit. The incidence of documented episodes of heart rate less than 50 bpm as well as the incidence of bradycardia reported as an adverse event was 1% in each treatment group. Initiate appropriate medical management in patients who develop symptomatic bradycardia.
In patients without underlying cardiac disease, SOMATULINE DEPOT may lead to a decrease in heart rate without necessarily reaching the threshold of bradycardia. In patients suffering from cardiac disorders prior to SOMATULINE DEPOT treatment, sinus bradycardia may occur. Care should be taken when initiating treatment with SOMATULINE DEPOT in patients with bradycardia.
6. Adverse Reactions/Side Effects
The following adverse reactions to SOMATULINE DEPOT are discussed in greater detail in other sections of the labeling:
- Cholelithiasis and Complications of Cholelithiasis [see Warnings and Precautions (5.1)]
- Hyperglycemia and Hypoglycemia [see Warnings and Precautions (5.2)]
- Cardiovascular Abnormalities [see Warnings and Precautions (5.3)]
- Thyroid Function Abnormalities [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Acromegaly
The data described below reflect exposure to SOMATULINE DEPOT in 416 acromegalic patients in seven studies. One study was a fixed-dose pharmacokinetic study. The other six studies were open-label or extension studies, one had a placebo-controlled, run-in period, and another had an active control. The population was mainly Caucasian (329/353, 93%) with a median age of 53 years of age (range 19 to 84 years). Fifty-four subjects (13%) were age 66 to 74 and 18 subjects (4.3%) were 75 years of age and older.
Patients were evenly matched for sex (205 males and 211 females). The median average monthly dose was 91.2 mg (e.g., 90 mg injected via the deep subcutaneous route every 4 weeks) over 385 days with a median cumulative dose of 1290 mg. Of the patients reporting acromegaly, severity at baseline (N=265), serum GH levels were less than 10 ng/mL for 69% (183/265) of the patients and 10 ng/mL or greater for 31% (82/265) of the patients.
The most commonly reported adverse reactions reported by greater than 5% of patients who received SOMATULINE DEPOT (N=416) in the overall pooled safety studies in acromegaly patients were gastrointestinal disorders (diarrhea, abdominal pain, nausea, constipation, flatulence, vomiting, loose stools), cholelithiasis, and injection site reactions.
Tables 1 and 2 present adverse reaction data from clinical studies with SOMATULINE DEPOT in acromegalic patients. The tables include data from a single clinical study and pooled data from seven clinical studies.
Adverse Reactions in Parallel Fixed-Dose Phase of Study 1
The incidence of treatment-emergent adverse reactions for SOMATULINE DEPOT 60, 90, and 120 mg by dose as reported during the first 4 months (fixed-dose phase) of Study 1 [see Clinical Studies (14.1)] are provided in Table 1.
| Placebo-Controlled Double-Blind Phase Weeks 0 to 4 | Fixed-Dose Phase Double-Blind + Single-Blind Weeks 0 to 20 |
|||||
|---|---|---|---|---|---|---|
| Body System Preferred Term | Placebo (N=25) | SOMATULINE DEPOT Overall (N=83) | SOMATULINE DEPOT 60 mg (N=34) | SOMATULINE DEPOT 90 mg (N=36) | SOMATULINE DEPOT 120 mg (N=37) | SOMATULINE DEPOT Overall (N=107) |
| N (%) | N (%) | N (%) | N (%) | N (%) | N (%) | |
| A patient is counted only once for each body system and preferred term. | ||||||
| Dictionary = WHOART. | ||||||
| Gastrointestinal System Disorders | 1 (4%) | 30 (36%) | 12 (35%) | 21 (58%) | 27 (73%) | 60 (56%) |
| Diarrhea | 0 | 26 (31%) | 9 (26%) | 15 (42%) | 24 (65%) | 48 (45%) |
| Abdominal pain | 1 (4%) | 6 (7%) | 3 (9%) | 6 (17%) | 7 (19%) | 16 (15%) |
| Flatulence | 0 | 5 (6%) | 0 (0%) | 3 (8%) | 5 (14%) | 8 (7%) |
| Application Site Disorders | 0 (0%) | 5 (6%) | 3 (9%) | 4 (11%) | 8 (22%) | 15 (14%) |
| (Injection site mass/ pain/ reaction/ inflammation) | ||||||
| Liver and Biliary System Disorders | 1 (4%) | 3 (4%) | 9 (26%) | 7 (19%) | 4 (11%) | 20 (19%) |
| Cholelithiasis | 0 | 2 (2%) | 5 (15%) | 6 (17%) | 3 (8%) | 14 (13%) |
| Heart Rate & Rhythm Disorders | 0 | 8 (10%) | 7 (21%) | 2 (6%) | 5 (14%) | 14 (13%) |
| Bradycardia | 0 | 7 (8%) | 6 (18%) | 2 (6%) | 2 (5%) | 10 (9%) |
| Red Blood Cell Disorders | 0 | 6 (7%) | 2 (6%) | 5 (14%) | 2 (5%) | 9 (8%) |
| Anemia | 0 | 6 (7%) | 2 (6%) | 5 (14%) | 2 (5%) | 9 (8%) |
| Metabolic & Nutritional Disorders | 3 (12%) | 13 (16%) | 8 (24%) | 9 (25%) | 4 (11%) | 21 (20%) |
| Weight decrease | 0 | 7 (8%) | 3 (9%) | 4 (11%) | 2 (5%) | 9 (8%) |
In Study 1, the adverse reactions of diarrhea, abdominal pain, and flatulence increased in incidence with increasing dose of SOMATULINE DEPOT.
Adverse Reactions in Long-Term Clinical Trials
Table 2 provides the most common adverse reactions (greater than 5%) that occurred in 416 acromegalic patients treated with SOMATULINE DEPOT pooled from 7 studies compared to those patients from the 2 efficacy studies (Studies 1 and 2). Patients with elevated GH and IGF-1 levels were either naive to somatostatin analog therapy or had undergone a 3-month washout [see Clinical Studies (14.1)].
| System Organ Class | Number and Percentage of Patients | |||
|---|---|---|---|---|
| Studies 1 & 2 | Overall Pooled Data | |||
| (N=170) | (N=416) | |||
| N | % | N | % | |
| Dictionary = MedDRA 7.1 | ||||
| Patients with any Adverse Reactions | 157 | 92 | 356 | 86 |
| Gastrointestinal disorders | 121 | 71 | 235 | 57 |
| Diarrhea | 81 | 48 | 155 | 37 |
| Abdominal pain | 34 | 20 | 79 | 19 |
| Nausea | 15 | 9 | 46 | 11 |
| Constipation | 9 | 5 | 33 | 8 |
| Flatulence | 12 | 7 | 30 | 7 |
| Vomiting | 8 | 5 | 28 | 7 |
| Loose stools | 16 | 9 | 23 | 6 |
| Hepatobiliary disorders | 53 | 31 | 99 | 24 |
| Cholelithiasis | 45 | 27 | 85 | 20 |
| General disorders and administration site conditions | 51 | 30 | 91 | 22 |
| (Injection site pain /mass /induration/ nodule/pruritus) | 28 | 17 | 37 | 9 |
| Musculoskeletal and connective tissue disorders | 44 | 26 | 70 | 17 |
| Arthralgia | 17 | 10 | 30 | 7 |
| Nervous system disorders | 34 | 20 | 80 | 19 |
| Headache | 9 | 5 | 30 | 7 |
In addition to the adverse reactions listed in Table 2, the following reactions were also seen:
- Sinus bradycardia occurred in 7% (12) of patients in the pooled Study 1 and 2 and in 3% (13) of patients in the overall pooled studies.
- Hypertension occurred in 7% (11) of patients in the pooled Study 1 and 2 and in 5% (20) of patients in the overall pooled studies.
- Anemia occurred in 7% (12) of patients in the pooled Study 1 and 2 and in 3% (14) of patients in the overall pooled studies.
Gastroenteropancreatic Neuroendocrine Tumors
The safety of SOMATULINE DEPOT 120 mg for the treatment of patients with gastroenteropancreatic neuroendocrine tumors (GEP-NETs) was evaluated in Study 3, a double-blind, placebo-controlled trial. Patients in Study 3 were randomized to receive SOMATULINE DEPOT (N=101) or placebo (N=103) administered by deep subcutaneous injection once every 4 weeks. The data below reflect exposure to SOMATULINE DEPOT in 101 patients with GEP-NETs, including 87 patients exposed for at least 6 months and 72 patients exposed for at least 1 year (median duration of exposure 22 months). Patients treated with SOMATULINE DEPOT had a median age of 64 years (range 30 to 83 years), 53% were men and 96% were Caucasian. Eighty-one percent of patients (83/101) in the SOMATULINE DEPOT arm and 82% of patients (82/103) in the placebo arm did not have disease progression within 6 months of enrollment and had not received prior therapy for GEP-NETs. The rates of discontinuation due to treatment-emergent adverse reactions were 5% (5/101 patients) in the SOMATULINE DEPOT arm and 3% (3/103 patients) in the placebo arm.
Table 3 compares the adverse reactions reported with an incidence of 5% and greater in patients receiving SOMATULINE DEPOT 120 mg administered every 4 weeks and reported more commonly than placebo.
| Adverse Reaction | SOMATULINE DEPOT 120 mg N=101 | Placebo N=103 |
||
|---|---|---|---|---|
| Any (%) | Severe* (%) | Any (%) | Severe* (%) | |
|
||||
| Any Adverse Reactions | 88 | 26 | 90 | 31 |
| Abdominal pain† | 34‡ | 6‡ | 24‡ | 4 |
| Musculoskeletal pain§ | 19‡ | 2‡ | 13 | 2 |
| Vomiting | 19‡ | 2‡ | 9‡ | 2‡ |
| Headache | 16 | 0 | 11 | 1 |
| Injection site reaction¶ | 15 | 0 | 7 | 0 |
| Hyperglycemia# | 14‡ | 0 | 5 | 0 |
| HypertensionÞ | 14‡ | 1‡ | 5 | 0 |
| Cholelithiasis | 14‡ | 1‡ | 7 | 0 |
| Dizziness | 9 | 0 | 2‡ | 0 |
| Depressionß | 7 | 0 | 1 | 0 |
| Dyspnea | 6 | 0 | 1 | 0 |
6.2 Immunogenicity
As with all peptides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to lanreotide in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Laboratory investigations of acromegalic patients treated with SOMATULINE DEPOT in clinical studies show that the percentage of patients with putative antibodies at any time point after treatment is low (less than 1% to 4% of patients in specific studies whose antibodies were tested). The antibodies did not appear to affect the efficacy or safety of SOMATULINE DEPOT.
In Study 3, development of anti-lanreotide antibodies was assessed using a radioimmunoprecipitation assay. In patients with GEP NETs receiving SOMATULINE DEPOT, the incidence of anti-lanreotide antibodies was 4% (3 of 82) at 24 weeks, 10% (7 of 67) at 48 weeks, 11% (6 of 57) at 72 weeks, and 10% (8 of 84) at 96 weeks. Assessment for neutralizing antibodies was not conducted. In Study 4, less than 2% (2 of 108) of the patients treated with SOMATULINE DEPOT developed anti-lanreotide antibodies.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of SOMATULINE DEPOT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hepatobiliary: steatorrhea; cholecystitis, cholangitis, pancreatitis, which have sometimes required cholecystectomy
Hypersensitivity: angioedema and anaphylaxis
Injection site reactions: injection site abscess
7. Drug Interactions
7.1 Insulin and Oral Hypoglycemic Drugs
Lanreotide, like somatostatin and other somatostatin analogs, inhibits the secretion of insulin and glucagon. Therefore, blood glucose levels should be monitored when SOMATULINE DEPOT treatment is initiated or when the dose is altered, and antidiabetic treatment should be adjusted accordingly [see Warnings and Precautions (5.2)].
7.2 Cyclosporine
Concomitant administration of cyclosporine with SOMATULINE DEPOT may decrease the absorption of cyclosporine, and therefore, may necessitate adjustment of cyclosporine dose to maintain therapeutic drug concentrations. [see Clinical Pharmacology (12.3)]
7.3 Bromocriptine
Limited published data indicate that concomitant administration of a somatostatin analog and bromocriptine may increase the absorption of bromocriptine [see Clinical Pharmacology (12.3)].
7.4 Bradycardia-Inducing Drugs
Concomitant administration of bradycardia-inducing drugs (e.g., beta-blockers) may have an additive effect on the reduction of heart rate associated with lanreotide. Dosage adjustments of concomitant drugs may be necessary.
7.5 Drug Metabolism Interactions
The limited published data available indicate that somatostatin analogs may decrease the metabolic clearance of compounds known to be metabolized by cytochrome P450 enzymes, which may be due to the suppression of growth hormone. Since it cannot be excluded that SOMATULINE DEPOT may have this effect, avoid other drugs mainly metabolized by CYP3A4 and which have a low therapeutic index (e.g., quinidine, terfenadine). Drugs metabolized by the liver may be metabolized more slowly during SOMATULINE DEPOT treatment and dose reductions of the concomitantly administered medications should be considered [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.4 Pediatric Use
The safety and effectiveness of SOMATULINE DEPOT in pediatric patients have not been established.
8.5 Geriatric Use
No overall differences in safety or effectiveness were observed between elderly patients with acromegaly compared with younger patients and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Studies 3 and 4, conducted in patients with neuroendocrine tumors, did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.
Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
11. Somatuline Depot Description
SOMATULINE DEPOT (lanreotide) Injection 60 mg/0.2 mL, 90 mg/0.3 mL, and 120 mg/0.5 mL is a prolonged-release formulation for deep subcutaneous injection. It contains the drug substance lanreotide acetate, a synthetic octapeptide with a biological activity similar to naturally occurring somatostatin, water for injection and acetic acid (for pH adjustment).
SOMATULINE DEPOT is available as sterile, ready-to-use, single-dose prefilled syringes containing lanreotide acetate supersaturated bulk solution of 24.6% w/w lanreotide base.
| Each syringe contains: | SOMATULINE DEPOT 60 mg/0.2 mL | SOMATULINE DEPOT 90 mg/0.3 mL | SOMATULINE DEPOT 120 mg/0.5 mL |
|---|---|---|---|
| Lanreotide acetate | 77.9 mg | 113.6 mg | 149.4 mg |
| Acetic Acid | q.s. | q.s. | q.s. |
| Water for injection | 186.6 mg | 272.3 mg | 357.8 mg |
| Total Weight | 266 mg | 388 mg | 510 mg |
Lanreotide acetate is a synthetic cyclical octapeptide analog of the natural hormone, somatostatin. Lanreotide acetate is chemically known as [cyclo S-S]-3-(2-naphthyl)-D-alanyl-L-cysteinyl-L-tyrosyl-D-tryptophyl-L-lysyl-L-valyl-L-cysteinyl-L-threoninamide, acetate salt. Its molecular weight is 1096.34 (base) and its amino acid sequence is:

The SOMATULINE DEPOT in the prefilled syringe is a white to pale yellow, semi-solid formulation.
12. Somatuline Depot - Clinical Pharmacology
12.1 Mechanism of Action
Lanreotide, the active component of SOMATULINE DEPOT is an octapeptide analog of natural somatostatin. The mechanism of action of lanreotide is believed to be similar to that of natural somatostatin.
12.2 Pharmacodynamics
Lanreotide has a high affinity for human somatostatin receptors (SSTR) 2 and 5 and a reduced binding affinity for human SSTR1, 3, and 4. Activity at human SSTR2 and 5 is the primary mechanism believed responsible for GH inhibition. Like somatostatin, lanreotide is an inhibitor of various endocrine, neuroendocrine, exocrine, and paracrine functions.
The primary pharmacodynamic effect of lanreotide is a reduction of GH and/or IGF-1 levels enabling normalization of levels in acromegalic patients [see Clinical Studies (14.1)]. In acromegalic patients, lanreotide reduces GH levels in a dose-dependent way. After a single injection of SOMATULINE DEPOT, plasma GH levels fall rapidly and are maintained for at least 28 days.
In Study 4, patients with carcinoid syndrome treated with SOMATULINE DEPOT 120 mg every 4 weeks had reduced levels of urinary 5-hydroxyindoleacetic acid (5-HIAA) compared with placebo [see Clinical Studies (14.3)].
Lanreotide inhibits the basal secretion of motilin, gastric inhibitory peptide, and pancreatic polypeptide, but has no significant effect on the secretion of secretin. Lanreotide inhibits postprandial secretion of pancreatic polypeptide, gastrin, and cholecystokinin (CCK). In healthy subjects, lanreotide produces a reduction and a delay in postprandial insulin secretion, resulting in transient, mild glucose intolerance.
Lanreotide inhibits meal-stimulated pancreatic secretions, and reduces duodenal bicarbonate and amylase concentrations, and produces a transient reduction in gastric acidity.
Lanreotide has been shown to inhibit gallbladder contractility and bile secretion in healthy subjects [see Warnings and Precautions (5.1)].
In healthy subjects, lanreotide inhibits meal-induced increases in superior mesenteric artery and portal venous blood flow, but has no effect on basal or meal-stimulated renal blood flow. Lanreotide has no effect on renal plasma flow or renal vascular resistance. However, a transient decrease in glomerular filtration rate (GFR) and filtration fraction has been observed after a single injection of lanreotide.
In healthy subjects, non-significant reductions in glucagon levels were seen after lanreotide administration. In diabetic non-acromegalic subjects receiving a continuous infusion (21-day) of lanreotide, serum glucose concentrations were temporarily decreased by 20% to 30% after the start and end of the infusion. Serum glucose concentrations returned to normal levels within 24 hours. A significant decrease in insulin concentrations was recorded between baseline and Day 1 only [see Warnings and Precautions (5.2)].
Lanreotide inhibits the nocturnal increase in thyroid-stimulating hormone (TSH) seen in healthy subjects. Lanreotide reduces prolactin levels in acromegalic patients treated on a long-term basis [see Warnings and Precautions (5.4)].
12.3 Pharmacokinetics
SOMATULINE DEPOT is thought to form a drug depot at the injection site due to the interaction of the formulation with physiological fluids. The most likely mechanism of drug release is a passive diffusion of the precipitated drug from the depot towards the surrounding tissues, followed by the absorption to the bloodstream.
After a single, deep subcutaneous administration, the mean absolute bioavailability of SOMATULINE DEPOT in healthy subjects was 73.4, 69.0, and 78.4% for the 60 mg, 90 mg, and 120 mg doses, respectively. Mean Cmax values ranged from 4.3 to 8.4 ng/mL during the first day. Single-dose linearity was demonstrated with respect to AUC and Cmax, and showed high inter-subject variability. SOMATULINE DEPOT showed sustained release of lanreotide with a half-life of 23 to 30 days. Mean serum concentrations were > 1 ng/mL throughout 28 days at 90 mg and 120 mg and > 0.9 ng/mL at 60 mg.
In studies evaluating excretion, <5% of lanreotide was excreted in urine and less than 0.5% was recovered unchanged in feces, indicative of some biliary excretion.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Standard lifetime carcinogenicity bioassays were conducted in mice and rats. Mice were given daily subcutaneous doses of lanreotide at 0.5, 1.5, 5, 10, and 30 mg/kg for 104 weeks. Cutaneous and subcutaneous tumors of fibrous connective tissues at the injection sites were observed at the high dose of 30 mg/kg/day. Fibrosarcomas in both genders and malignant fibrous histiocytomas were observed in males at 30 mg/kg/day resulting in exposures 3 times higher than the clinical therapeutic exposure at the maximum therapeutic dose of 120 mg given by monthly subcutaneous injection based on the AUC values. Rats were given daily subcutaneous doses of lanreotide at 0.1, 0.2, and 0.5 mg/kg for 104 weeks. Increased cutaneous and subcutaneous tumors of fibrous connective tissues at the injection sites were observed at the dose of 0.5 mg/kg/day resulting in exposures less than the clinical therapeutic exposure at 120 mg given by monthly subcutaneous injection. The increased incidence of injection site tumors in rodents is likely related to the increased dosing frequency (daily) in animals compared to monthly dosing in humans and therefore may not be clinically relevant.
Lanreotide was not genotoxic in tests for gene mutations in a bacterial mutagenicity (Ames) assay, or mouse lymphoma cell assay with or without metabolic activation. Lanreotide was not genotoxic in tests for the detection of chromosomal aberrations in a human lymphocyte and in vivo mouse micronucleus assay.
In a fertility study conducted with lanreotide in rats, reduced female fecundity was observed at estimated exposure corresponding to approximately 10-fold the plasma exposure at the MRHD of 120 mg. The fertility of male rats was unaffected by the treatment up to an estimated exposure corresponding to approximately 11-fold the plasma exposure at the MRHD of 120 mg.
14. Clinical Studies
14.1 Acromegaly
The effect of SOMATULINE DEPOT on reducing GH and IGF-levels and control of symptoms in patients with acromegaly was studied in 2 long-term, multiple-dose, randomized, multicenter studies.
Study 1
This 1-year study included a 4-week, double-blind, placebo-controlled phase; a 16-week single-blind, fixed-dose phase; and a 32-week, open-label, dose-titration phase. Patients with active acromegaly, based on biochemical tests and medical history, entered a 12-week washout period if there was previous treatment with a somatostatin analog or a dopaminergic agonist.
Upon entry, patients were randomly allocated to receive a single, deep subcutaneous injection of SOMATULINE DEPOT 60, 90, or 120 mg or placebo. Four weeks later, patients entered a fixed-dose phase where they received 4 injections of SOMATULINE DEPOT followed by a dose-titration phase of 8 injections for a total of 13 injections over 52 weeks (including the placebo phase). Injections were given at 4-week intervals. During the dose-titration phase of the study, the dose was titrated twice (every fourth injection), as needed, according to individual GH and IGF-1 levels.
A total of 108 patients (51 males, 57 females) were enrolled in the initial placebo-controlled phase of the study. Half (54/108) of the patients had never been treated with a somatostatin analog or dopamine agonist, or had stopped treatment for at least 3 months prior to their participation in the study and were required to have a mean GH level greater than 5 ng/mL at their first visit. The other half of the patients had received prior treatment with a somatostatin analog or a dopamine agonist before study entry and at study entry were required to have a mean GH concentration greater than 3 ng/mL and at least a 100% increase in mean GH concentration after washout of medication.
One hundred and seven (107) patients completed the placebo-controlled phase, 105 patients completed the fixed-dose phase, and 99 patients completed the dose-titration phase. Patients not completing withdrew due to adverse events (5) or lack of efficacy (4).
In the double-blind phase of Study 1, a total of 52 (63%) of the 83 lanreotide-treated patients had a greater than 50% decrease in mean GH from baseline to Week 4, including 52%, 44%, and 90% of patients in the 60, 90, and 120 mg groups, respectively, compared to placebo (0%, 0/25). In the fixed-dose phase at Week 16, 72% of all 107 lanreotide-treated patients had a decrease from baseline in mean GH of greater than 50%, including 68% (23/34), 64% (23/36), and 84% (31/37) of patients in the 60, 90, and 120 mg lanreotide treatment groups, respectively. Efficacy achieved in the first 16 weeks was maintained for the duration of the study (see Table 4).
| Baseline | Before Titration 1 | Before Titration 2 | Last Value Available* | ||
|---|---|---|---|---|---|
| N=107 | (16 weeks) N=107 | (32 weeks) N=105 | N=107 | ||
|
|||||
| GH | |||||
| ≤5.0 ng/mL | Number of Responders (%) | 20 (19%) | 72 (67%) | 76 (72%) | 74 (69%) |
| ≤2.5 ng/mL | Number of Responders (%) | 0 (0%) | 52 (49%) | 59 (56%) | 55 (51%) |
| ≤1.0 ng/mL | Number of Responders (%) | 0 (0%) | 15 (14%) | 18 (17%) | 17 (16%) |
| Median GH | ng/mL | 10.27 | 2.53 | 2.20 | 2.43 |
| GH Reduction | Median % Reduction | -- | 75.5 | 78.2 | 75.5 |
| IGF-1 | |||||
| Normal† | Number of Responders (%) | 9 (8%) | 58 (54%) | 57 (54%) | 62 (58%) |
| Median IGF-1 | ng/mL | 775.0 | 332.0‡ | 316.5§ | 326.0 |
| IGF-1 Reduction | Median % Reduction | -- | 52.3‡ | 54.5§ | 55.4 |
| IGF-1 Normal† + GH ≤2.5 ng/mL | Number of Responders (%) | 0 (0%) | 41 (38%) | 46 (44%) | 44 (41%) |
Study 2
This was a 48-week, open-label, uncontrolled, multicenter study that enrolled patients who had an IGF-1 concentration 1.3 times or greater than the upper limit of the normal age-adjusted range. Patients receiving treatment with a somatostatin analog (other than SOMATULINE DEPOT) or a dopaminergic agonist had to attain this IGF-1 concentration after a washout period of up to 3 months.
Patients were initially enrolled in a 4-month, fixed-dose phase where they received 4 deep subcutaneous injections of SOMATULINE DEPOT 90 mg, at 4-week intervals. Patients then entered a dose-titration phase where the dose of SOMATULINE DEPOT was adjusted based on GH and IGF-1 levels at the beginning of the dose-titration phase and, if necessary, again after another 4 injections. Patients titrated up to the maximum dose (120 mg) were not allowed to titrate down again.
A total of 63 patients (38 males, 25 females) entered the fixed-dose phase of the trial and 57 patients completed 48 weeks of treatment. Six patients withdrew due to adverse reactions (3), other reasons (2), or lack of efficacy (1).
After 48 weeks of treatment with SOMATULINE DEPOT at 4-week intervals, 43% (27/63) of the acromegalic patients in this study achieved normal age-adjusted IGF-1 concentrations. Mean IGF-1 concentrations after treatment completion were 1.3 ± 0.7 times the upper limit of normal compared to 2.5 ± 1.1 times the upper limit of normal at baseline.
The reduction in IGF-1 concentrations over time correlated with a corresponding marked decrease in mean GH concentrations. The proportion of patients with mean GH concentrations less than 2.5 ng/mL increased significantly from 35% to 77% after the fixed-dose phase and 85% at the end of the study. At the end of treatment, 24/63 (38%) of patients had both normal IGF-1 concentrations and a GH concentration of less than or equal to 2.5 ng/mL (see Table 5) and 17/63 patients (27%) had both normal IGF-1 concentrations and a GH concentration of less than 1 ng/mL.
| Baseline | Before Titration 1 (12 wks) | Before Titration 2 (28 wks) | Last Value Available* | ||
|---|---|---|---|---|---|
| N=63 | N=63 | N=59 | N=63 | ||
|
|||||
| IGF-1 | |||||
| Normal† | Number of Responders (%) | 0 (0%) | 17 (27%) | 22 (37%) | 27 (43%) |
| Median IGF-1 | ng/mL | 689.0 | 382.0 | 334.0 | 317.0 |
| IGF-1 Reduction | Median % Reduction | -- | 41.0 | 51.0 | 50.3 |
| GH | |||||
| ≤5.0 ng/mL | Number of Responders (%) | 40 (64%) | 59 (94%) | 57 (97%) | 62 (98%) |
| ≤2.5 ng/mL | Number of Responders (%) | 21 (33%) | 47 (75%) | 47 (80%) | 54 (86 %) |
| ≤1.0 ng/mL | Number of Responders (%) | 8 (13%) | 19 (30%) | 18 (31%) | 28 (44%) |
| Median GH | ng/Ml | 3.71 | 1.65 | 1.48 | 1.13 |
| GH Reduction | Median % Reduction | -- | 63.2 | 66.7 | 78.6‡ |
| IGF-1 normal† + GH ≤2.5 ng/mL | Number of Responders (%) | 0 (0%) | 14 (22%) | 20 (34%) | 24 (38%) |
Examination of age and gender subgroups did not identify differences in response to SOMATULINE DEPOT among these subgroups. The limited number of patients in the different racial subgroups did not raise any concerns regarding efficacy of SOMATULINE DEPOT in these subgroups.
14.2 Gastroenteropancreatic Neuroendocrine Tumors
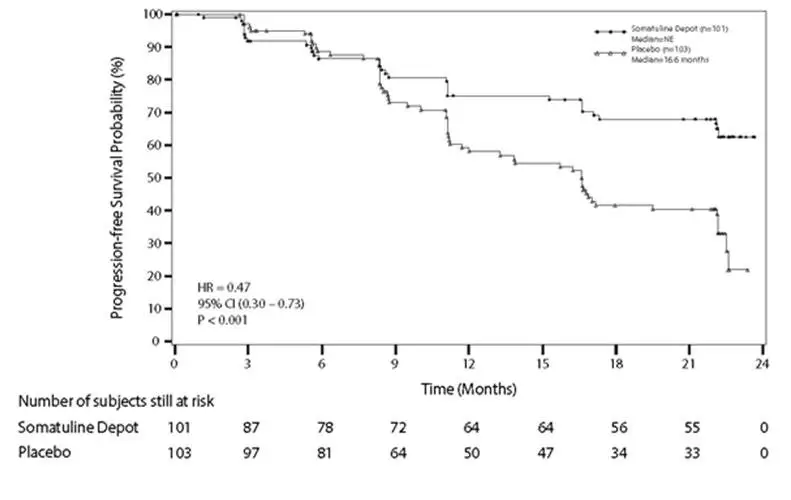
The efficacy of SOMATULINE DEPOT was established in a multicenter, randomized, double-blind, placebo-controlled trial of 204 patients with unresectable, well or moderately differentiated, metastatic or locally advanced, gastroenteropancreatic neuroendocrine tumors. Patients were required to have non-functioning tumors without hormone-related symptoms. Patients were randomized 1:1 to receive SOMATULINE DEPOT 120 mg (n=101) or placebo (n=103) every 4 weeks until disease progression, unacceptable toxicity, or a maximum of 96 weeks of treatment. Randomization was stratified by the presence or absence of prior therapy and by the presence or absence of disease progression within 6 months of enrollment. The major efficacy outcome measure was progression-free survival (PFS), defined as time to disease progression as assessed by central independent radiological review using the Response Evaluation Criteria in Solid Tumors (RECIST 1.0) or death.
The median patient age was 63 years (range 30 to 92 years) and 95% were Caucasian. Disease progression was present in nine of 204 patients (4.4%) in the 6 months prior to enrollment and 29 patients (14%) received prior chemotherapy. Ninety-one patients (45%) had primary sites of disease in the pancreas, with the remainder originating in the midgut (35%), hindgut (7%), or unknown primary location (13%). The majority (69%) of the study population had grade 1 tumors. Baseline prognostic characteristics were similar between arms with one exception; there were 39% of patients in the SOMATULINE DEPOT arm and 27% of patients in the placebo arm who had hepatic involvement by tumor of greater than 25%.
Patients on the SOMATULINE DEPOT arm had a statistically significant improvement in PFS compared to patients receiving placebo (see Table 6 and Figure 1).
| SOMATULINE DEPOT | Placebo | |
|---|---|---|
| n=101 | n=103 | |
|
||
| Number of Events (%) | 32 (31.7%) | 60 (58.3%) |
| Median PFS (months)(95% CI) | NE* (NE, NE) | 16.6 (11.2, 22.1) |
| HR (95% CI) | 0.47 (0.30, 0.73)† | |
| Log-rank p-value | <0.001 | |
Figure 1: Kaplan-Meier Curves of Progression-Free Survival
14.3 Carcinoid Syndrome
Study 4 was a multicenter, randomized, 16-week, double-blind, placebo-controlled trial in 115 patients with histopathologically-confirmed neuroendocrine tumors and a history of carcinoid syndrome (flushing and/or diarrhea) who were treatment naïve or stable on another somatostatin analog and who were randomized 1:1 to receive SOMATULINE DEPOT 120 mg (n=59) or placebo (n=56) by deep subcutaneous injection every 4 weeks. Patients were instructed to self-administer a short-acting somatostatin analog (octreotide) as rescue medication as needed for symptom control. The use of rescue therapy and the severity and frequency of diarrhea and flushing symptoms were reported daily in electronic patient diaries. During the 16 week double-blind phase, the primary efficacy outcome measure was the percentage of days in which patients administered at least one injection of rescue medication for symptom control. Average daily frequencies of diarrhea and flushing events were assessed secondarily.
The patient population had a mean age of 59 years (range 27 to 85 years), 58% were female and 77% were Caucasian. Patients in the SOMATULINE DEPOT arm experienced 15% fewer days on rescue medication compared to patients in the placebo arm (34% vs. 49% of days, respectively; p=0.02). The average daily frequencies of diarrhea and flushing events in patients treated with SOMATULINE DEPOT (and rescue medication) were numerically lower relative to patients treated with placebo (and rescue medication), but were not statistically significantly different via hierarchical testing.
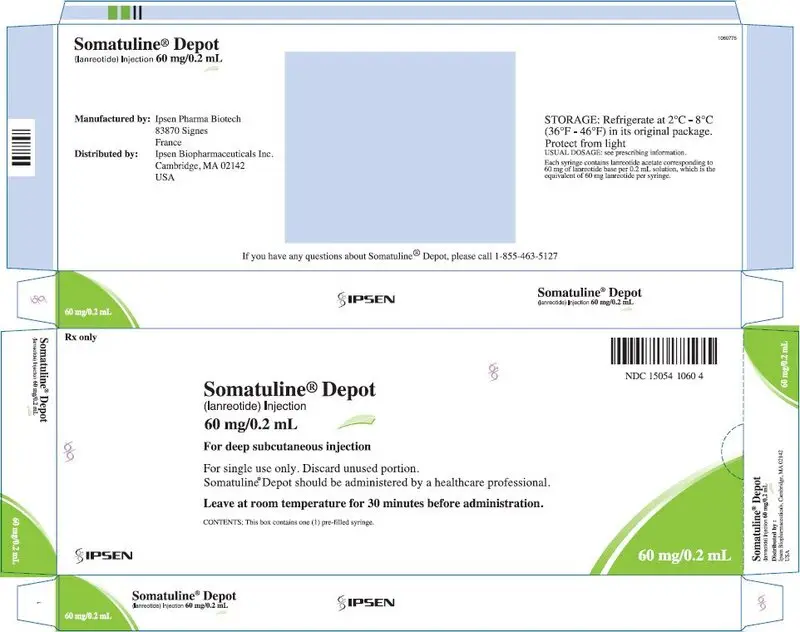
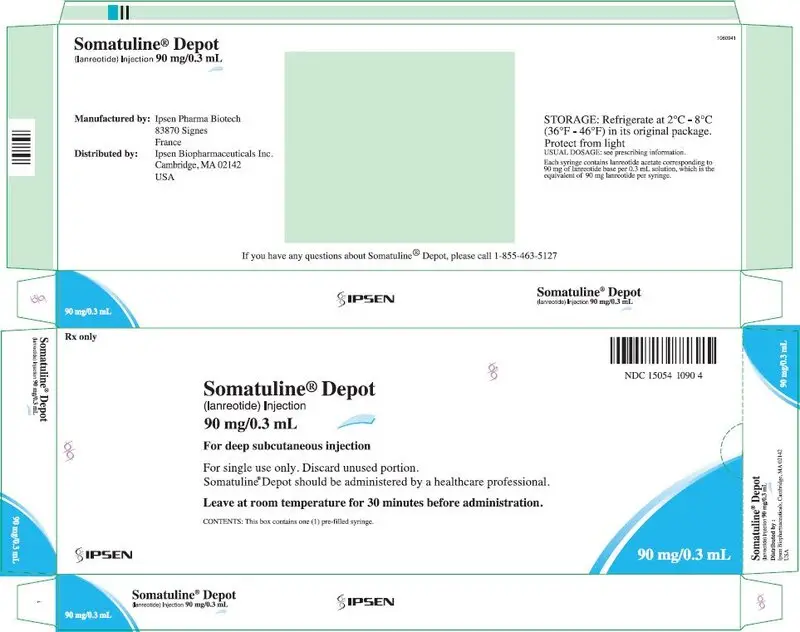
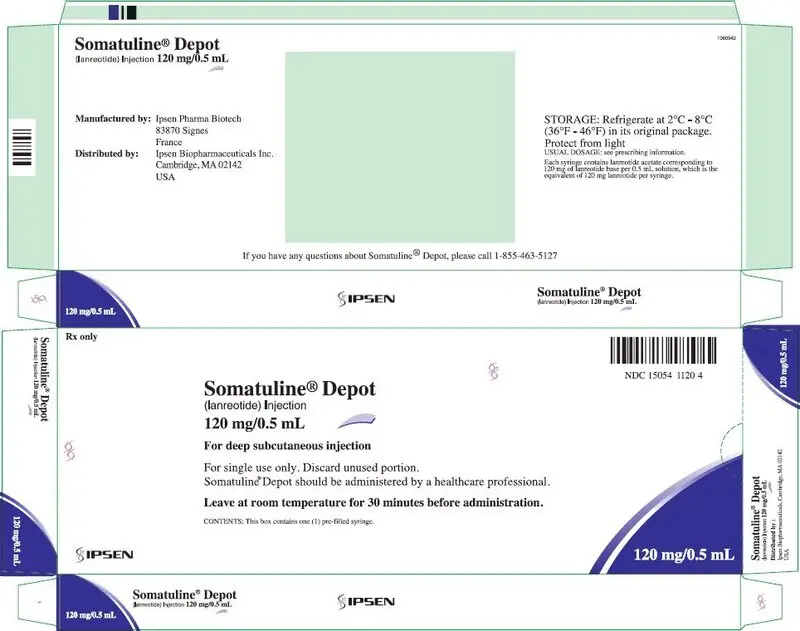
16. How is Somatuline Depot supplied
SOMATULINE DEPOT is supplied in strengths of 60 mg/0.2 mL, 90 mg/0.3 mL, and 120 mg/0.5 mL as a white to pale yellow, semi-solid formulation in a single, sterile, prefilled, ready-to-use, polypropylene syringe fitted with an automatic safety system, a bromobutyl rubber plunger stopper and a 20 mm attached needle covered by a plastic cap.
Each prefilled syringe is placed in a plastic tray, sealed in a laminated pouch and packed in a carton.
| NDC 15054-1060-4 | 60 mg/0.2 mL, sterile, prefilled syringe |
| NDC 15054-1090-4 | 90 mg/0.3 mL, sterile, prefilled syringe |
| NDC 15054-1120-4 | 120 mg/0.5 mL, sterile, prefilled syringe |


| SOMATULINE DEPOT
lanreotide acetate injection |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| SOMATULINE DEPOT
lanreotide acetate injection |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| SOMATULINE DEPOT
lanreotide acetate injection |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Ipsen Biopharmaceuticals, Inc. (118461578) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| IPSEN PHARMA BIOTECH | 502570286 | MANUFACTURE(15054-1060, 15054-1090, 15054-1120) | |
