Drug Detail:Synribo (Omacetaxine [ oh-ma-se-tax-een ])
Drug Class: Miscellaneous antineoplastics
Highlights of Prescribing Information
SYNRIBO® (omacetaxine mepesuccinate) for injection, for subcutaneous use
Initial U.S. Approval: 2012
Indications and Usage for Synribo
SYNRIBO for Injection is indicated for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKI) (1).
Synribo Dosage and Administration
- Induction Dose: 1.25 mg/m2 administered by subcutaneous injection twice daily for 14 consecutive days of a 28-day cycle (2.1).
- Maintenance Dose: 1.25 mg/m2 administered by subcutaneous injection twice daily for 7 consecutive days of a 28-day cycle (2.2).
- Dose modifications are needed for toxicity (2.3).
Dosage Forms and Strengths
For Injection: Single-dose vial containing 3.5 mg of omacetaxine mepesuccinate as a lyophilized powder (3).
Contraindications
None. (4)
Warnings and Precautions
-
Myelosuppression: Severe and fatal thrombocytopenia, neutropenia and anemia. Monitor hematologic parameters frequently (2.3, 5.1).
-
Bleeding: Severe thrombocytopenia and increased risk of hemorrhage. Fatal cerebral hemorrhage and severe, non-fatal gastrointestinal hemorrhage (5.1, 5.2).
-
Hyperglycemia: Glucose intolerance and hyperglycemia including hyperosmolar non-ketotic hyperglycemia (5.3).
-
Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use an effective method of contraception (5.4, 8.1, 8.3).
Adverse Reactions/Side Effects
Most common adverse reactions (frequency ≥ 20%): thrombocytopenia, anemia, neutropenia, diarrhea, nausea, fatigue, asthenia, injection site reaction, pyrexia, infection, and lymphopenia (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Teva Pharmaceuticals at 1-888-483-8279 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2022
Full Prescribing Information
1. Indications and Usage for Synribo
SYNRIBO is indicated for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKI).
2. Synribo Dosage and Administration
2.1 Induction Schedule
The recommended starting schedule for induction is 1.25 mg/m2 administered subcutaneously twice daily at approximately 12 hour intervals for 14 consecutive days every 28 days, over a 28-day cycle. Cycles should be repeated every 28 days until patients achieve a hematologic response.
2.2 Maintenance Dosing
The recommended maintenance schedule is 1.25 mg/m2 administered subcutaneously twice daily at approximately 12 hour intervals for 7 consecutive days every 28 days, over a 28-day cycle. Treatment should continue as long as patients are clinically benefiting from therapy.
2.3 Dose Adjustments and Modifications
Hematologic Toxicity:
SYNRIBO treatment cycles may be delayed and/or the number of days of dosing during the cycle reduced for hematologic toxicities (e.g. neutropenia, thrombocytopenia) [see Warnings and Precautions (5.1)].
Perform complete blood counts (CBCs) weekly during induction and initial maintenance cycles. After initial maintenance cycles, monitor CBCs every two weeks or as clinically indicated. If a patient experiences Grade 4 neutropenia (absolute neutrophil count (ANC) less than 0.5 x 109/L) or Grade 3 thrombocytopenia (platelet counts less than 50 x 109/L) during a cycle, delay starting the next cycle until ANC is greater than or equal to 1.0 x 109/L and platelet count is greater than or equal to 50 x 109/L. Also, for the next cycle, reduce the number of dosing days by 2 days (e.g. to 12 or 5 days).
Non-Hematologic Toxicity:
Manage other clinically significant non-hematologic toxicity symptomatically. Interrupt and/or delay SYNRIBO until toxicity is resolved.
2.4 Reconstitution Instructions and Handling Precautions
SYNRIBO should be prepared in a healthcare facility and must be reconstituted by a healthcare professional.
Reconstitute SYNRIBO with one mL of 0.9% Sodium Chloride Injection, USP, prior to subcutaneous injection. After addition of the diluent, gently swirl until a clear solution is obtained. The lyophilized powder should be completely dissolved in less than one minute. The resulting solution is clear and colorless and contains 3.5 mg/mL SYNRIBO. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
SYNRIBO does not contain antimicrobial preservatives. Therefore care must be taken to ensure that the solution for injection is not contaminated during preparation.
SYNRIBO is a cytotoxic drug. Follow special handling and disposal procedures1. Wear protective eyewear and gloves during handling and administration of the product. Proper aseptic technique should be used. Avoid skin and eye contact. If SYNRIBO comes into contact with skin, immediately and thoroughly wash affected area with soap and water. If contact with the eyes occurs, thoroughly flush the eyes with water.
2.5 Storage Conditions and Storage Time after Preparation of Syringes
If SYNRIBO is not used immediately after reconstitution, follow in-use storage conditions and allowable storage times prior to use as instructed in Table 1. Do not administer SYNRIBO outside of the storage conditions and timeframes listed in Table 1.
| Storage Conditions | Storage Time |
| Room temperature (20°C to 25°C [68°F to 77°F]) | Use within 12 hours of reconstitution |
| Refrigerated (2°C to 8°C [36oF to 46oF]) | Use within 6 days (144 hours) of reconstitution |
2.6 Considerations for Home Administration
Before a decision is made to allow SYNRIBO to be administered by someone other than a healthcare professional, ensure that the patient is an appropriate candidate for self-administration or for administration by a caregiver. Provide training on proper handling, storage conditions, administration, disposal, and clean-up of accidental spillage of the product. Ensure that patients receive the necessary supplies for home administration. At minimum these should include:
- Reconstituted SYNRIBO in syringe with a capped needle for subcutaneous injection. Syringe(s) should be filled to the patient-specific dose.
- Protective eyewear
- Gloves
- An appropriate biohazard container
- Absorbent pad(s) for placement of administration materials and for accidental spillage
- Alcohol swabs
- Gauze pads
- Ice packs or cooler for transportation of reconstituted SYNRIBO syringes
If a patient or caregiver cannot be trained for any reason, then in such patients, SYNRIBO should be administered by a healthcare professional.
2.7 Disposal and Accidental Spillage Procedures
After administration, any unused solution should be discarded properly1. Instruct patients planning home administration on the following: do not recap or clip the used needle, and do not place used needles, syringes, vials, and other used supplies in a household trash or recycling bin. Used needles, syringes, vials, and other used supplies should be disposed of in an appropriate biohazard container.
If accidental spillage occurs, continue to use protective eyewear and gloves, wipe the spilled liquid with the absorbent pad, and wash the area with water and soap. Then, place the pad and gloves into the biohazard container and wash hands thoroughly. Return the biohazard container to the clinic or pharmacy for final disposal.
3. Dosage Forms and Strengths
SYNRIBO for Injection contains 3.5 mg omacetaxine mepesuccinate; as a sterile, preservative-free, white to off-white lyophilized powder in a single-dose vial.
5. Warnings and Precautions
5.1 Myelosuppression
In uncontrolled trials with SYNRIBO, patients with chronic phase and accelerated phase CML experienced NCI CTC (version 3.0) Grade 3 or 4 thrombocytopenia (85%, 88%), neutropenia (81%, 71%), and anemia (62%, 80%), respectively. Fatalities related to myelosuppression occurred in 3% of patients in the safety population (N=163). Patients with neutropenia are at increased risk for infections, and should be monitored frequently and advised to contact a physician if they have symptoms of infection or fever.
Monitor complete blood counts weekly during induction and initial maintenance cycles and every two weeks during later maintenance cycles, as clinically indicated. In clinical trials myelosuppression was generally reversible and usually managed by delaying next cycle and/or reducing days of treatment with SYNRIBO [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.2 Bleeding
SYNRIBO causes severe thrombocytopenia which increases the risk of hemorrhage. In clinical trials with CP and AP CML patients, a high incidence of Grade 3 and 4 thrombocytopenia (85% and 88%, respectively) was observed. Fatalities from cerebral hemorrhage occurred in 2% of patients treated with SYNRIBO in the safety population. Severe, non-fatal, gastrointestinal hemorrhages occurred in 2% of patients in the same population. Most bleeding events were associated with severe thrombocytopenia.
Monitor platelet counts as part of the CBC monitoring as recommended [see Warnings and Precautions (5.1)]. Avoid anticoagulants, aspirin, and non-steroidal anti-inflammatory drugs (NSAIDs) when the platelet count is less than 50,000/µL as they may increase the risk of bleeding.
5.3 Hyperglycemia
SYNRIBO can induce glucose intolerance. Grade 3 or 4 hyperglycemia was reported in 11% of patients in the safety population. Hyperosmolar non-ketotic hyperglycemia occurred in 1 patient treated with SYNRIBO in the safety population. Monitor blood glucose levels frequently, especially in patients with diabetes or risk factors for diabetes. Avoid SYNRIBO in patients with poorly controlled diabetes mellitus until good glycemic control has been established.
5.4 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies and the drug’s mechanism of action, SYNRIBO can cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use an effective method of contraception during treatment with SYNRIBO and for 6 months after the final dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with SYNRIBO and for 3 months after the final dose [see Use in Specific Populations (8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions have been associated with SYNRIBO in clinical trials and are discussed in greater detail in other sections of the label.
- Myelosuppression [see Warnings and Precautions (5.1)]
- Bleeding [see Warnings and Precautions (5.2)]
- Hyperglycemia [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data for SYNRIBO are from 3 clinical trials which enrolled a total of 163 adult patients with TKI resistant and/or intolerant chronic phase (N=108) and accelerated phase (N=55) CML. All patients were treated with initial induction therapy consisting of a dose of 1.25 mg/m2 administered subcutaneously twice daily for 14 consecutive days every 28 days (induction cycle). Responding patients were then treated with the same dose and a twice daily schedule for 7 consecutive days every 28 days (maintenance cycle).
Chronic Phase CML
The median duration of exposure for the 108 patients with chronic phase CML was 7.4 months (range 0 to 43 months). The median total cycles of exposure was 6 (range 1 to 41), and the median total dose delivered during the trials was 131 mg/m2 (range 1.2 to 678). Among the patients with chronic phase CML, 87% received 14 days of treatment during cycle 1. By cycles 2 and 3, the percentage of patients receiving 14 days of treatment decreased to 42% and 16% respectively. Of the 91 patients who received at least 2 cycles of treatment, 79 (87%) had at least 1 cycle delay during the trials. The median number of days of cycle delays was greatest for cycle 2 (17 days) and cycle 3 (25 days) when more patients were receiving induction cycles.
Adverse reactions were reported for 99% of the patients with chronic phase CML. A total of 18% of patients had adverse reactions leading to withdrawal. The most frequently occurring adverse reactions leading to discontinuation were pancytopenia, thrombocytopenia, and increased alanine aminotransferase (each 2%). A total of 87% of patients reported at least 1 Grade 3 or Grade 4 treatment emergent adverse reaction (Table 2).
| Number (%) of Patients (N=108) |
||
|
Adverse reactions |
All reactions |
Grade 3 or 4 reactions |
|
Patients with at least 1 commonly occurring adverse reaction |
107 (99) |
94 (87) |
| Blood and Lymphatic System Disorders | ||
|
Thrombocytopenia |
82 (76) |
73 (68) |
|
Anemia |
66 (61) |
39 (36) |
|
Neutropenia |
57 (53) |
51 (47) |
|
Lymphopenia |
18 (17) |
17 (16) |
|
Bone Marrow Failure |
11 (10) |
11 (10) |
|
Febrile Neutropenia |
11 (10) |
11 (10) |
|
Gastrointestinal Disorders | ||
|
Diarrhea |
44 (41) |
1 (1) |
|
Nausea |
38 (35) |
1 (1) |
|
Constipation |
15 (14) |
0 |
|
Abdominal Pain/Upper Abdominal Pain |
25 (23) |
0 |
|
Vomiting |
13 (12) |
0 |
| General Disorders and Administration Site Conditions | ||
|
Fatigue |
31 (29) |
5 (5) |
|
Pyrexia |
27 (25) |
1 (1) |
|
Asthenia |
25 (23) |
1 (1) |
|
Edema Peripheral |
17 (16) |
0 |
|
Infusion and injection site related reactionsb |
38 (35) |
0 |
|
Infections and Infestationsc |
52 (48) |
12 (11) |
| Metabolism and Nutrition Disorders | ||
|
Anorexia |
11 (10) |
1 (1) |
|
Musculoskeletal and Connective Tissue Disorders | ||
|
Arthralgia |
20 (19) |
1 (1) |
|
Pain in Extremity |
14 (13) |
1 (1) |
|
Back Pain |
13 (12) |
2 (2) |
|
Myalgia |
12 (11) |
1 (1) |
| Nervous System Disorders | ||
|
Headache |
22 (20) |
1 (1) |
|
Psychiatric Disorders | ||
|
Insomnia |
13 (12) |
1 (1) |
| Respiratory, Thoracic and Mediastinal Disorders | ||
|
Cough |
17 (16) |
1 (1) |
|
Epistaxis |
18 (17) |
1 (1) |
| Skin and Subcutaneous Tissue Disorders | ||
|
Alopecia |
16 (15) |
0 |
|
Rash |
12 (11) |
0 |
a Occurred in the period between the first dose and 30 days after the last dose.
b Includes infusion related reaction, injection site erythema, injection site hematoma, injection site hemorrhage, injection site hypersensitivity, injection site induration, injection site inflammation, injection site irritation, injection site mass, injection site edema, injection site pruritus, injection site rash, and injection site reaction.
c Infection includes bacterial, viral, fungal, and non-specified.
Serious adverse reactions were reported for 51% of patients. Serious adverse reactions reported for at least 5% of patients were bone marrow failure and thrombocytopenia (each 10%), and febrile neutropenia (6%). Serious adverse reactions of infections were reported for 8% of patients.
Deaths occurred while on study in five (5%) patients with CP CML. Two patients died due to cerebral hemorrhage, one due to multi-organ failure, one due to progression of disease, and one from unknown causes.
Accelerated Phase CML
Median total cycles of exposure was 2 (range 1 to 29), and the median total dose delivered during the trials was 70 mg/m2. The median duration of exposure for the 55 patients with accelerated phase CML was 1.9 months (range 0 to 30 months). Of the patients with accelerated phase CML, 86% received 14 days of treatment during cycle 1. By cycles 2 and 3, the percentage of patients receiving 14 days of treatment decreased to 55% and 44% respectively. Of the 40 patients who received at least 2 cycles of treatment, 27 (68%) had at least 1 cycle delay during the trials. The median number of days of cycle delays was greatest for cycle 3 (31 days) and cycle 8 (36 days).
Adverse reactions regardless of investigator attribution were reported for 100% patients with accelerated phase CML. A total of 33% of patients had adverse reactions leading to withdrawal. The most frequently occurring adverse reactions leading to withdrawal were leukocytosis (6%), and thrombocytopenia (4%). A total of 84% of patients reported at least 1 Grade 3 or Grade 4 treatment emergent adverse reaction (Table 3).
| Number (%) of Patients (N=55) |
||
|
Adverse reactions |
All reactions |
Grade 3 or 4 reactions |
|
Patients with at least 1 commonly occurring adverse reaction |
55 (100) |
47 (86) |
| Blood and Lymphatic System Disorders | ||
|
Anemia |
28 (51) |
21 (38) |
|
Febrile Neutropenia |
11 (20) |
9 (16) |
|
Neutropenia |
11 (20) |
10 (18) |
|
Thrombocytopenia |
32 (58) |
27 (49) |
|
Gastrointestinal Disorders | ||
|
Diarrhea |
19 (35) |
4 (7) |
|
Nausea |
16 (29) |
2 (4) |
|
Vomiting |
9 (16) |
1 (2) |
|
Abdominal Pain/Upper Abdominal Pain |
9 (16) |
0 |
|
General Disorders and Administration Site Conditions | ||
|
Fatigue |
17 (31) |
5 (9) |
|
Pyrexia |
16 (29) |
1 (2) |
|
Asthenia |
13 (24) |
1 (2) |
|
Chills |
7 (13) |
0 |
|
Infusion and injection site related reactionsb |
12 (22) |
0 |
|
Infections and Infestationsc |
31 (56) |
11 (20) |
|
Metabolism and Nutrition Disorders | ||
|
Anorexia |
7 (13) |
1 (2) |
|
Musculoskeletal and Connective Tissue Disorders | ||
|
Pain in Extremity |
6 (11) |
1 (2) |
|
Nervous System Disorders | ||
|
Headache |
7 (13) |
0 |
| Respiratory, Thoracic and Mediastinal Disorders | ||
|
Cough |
8 (15) |
0 |
|
Dyspnea |
6 (11) |
1 (2) |
|
Epistaxis |
6 (11) |
1 (2) |
a Occurred in the period between the first dose and 30 days after the last dose.
b Includes infusion related reaction, injection site erythema, injection site hematoma, injection site hemorrhage, injection site hypersensitivity, injection site induration, injection site inflammation, injection site irritation, injection site mass, injection site edema, injection site pruritus, injection site rash, and injection site reaction.
c Infection includes bacterial, viral, fungal, and non-specified.
Serious adverse reactions were reported for 60% of patients. Serious adverse reactions reported for at least 5% of patients were febrile neutropenia (18%), thrombocytopenia (9%), anemia (7%), and diarrhea (6%). Serious adverse reactions of infections were reported for 11% of patients.
Death occurred while on study in 5 (9%) patients with AP CML. Two patients died due to cerebral hemorrhage and three due to progression of disease.
Laboratory Abnormalities in Chronic and Accelerated Phase CML
Grade 3/4 laboratory abnormalities reported in patients with chronic and accelerated phase CML are described in Table 4. Myelosuppression occurred in all patients treated with SYNRIBO [see Warnings and Precautions (5.1)]. Five patients with chronic phase CML and 4 patients with accelerated phase CML permanently discontinued SYNRIBO due to pancytopenia, thrombocytopenia, febrile neutropenia, or bone marrow necrosis. An event of hyperosmolar non-ketotic hyperglycemia was reported in one patient in the safety population and a similar case has been reported in the literature. Two patients with chronic phase CML permanently discontinued SYNRIBO due to elevated transaminases.
| Chronic Phase | Accelerated Phase | |
| % | % | |
| Hematology Parameters | ||
|
Hemoglobin Decreased |
62 |
80 |
|
Leukocytes Decreased |
72 |
61 |
|
Neutrophils Decreased |
81 |
71 |
|
Platelets Decreased |
85 |
88 |
|
Biochemistry Parameters | ||
|
Alanine aminotransferase (ALT) Increased |
6 |
2 |
|
Bilirubin Increased Creatinine Increased |
9 |
6 |
|
9 |
16 |
|
|
Glucose Increased |
10 |
15 |
|
Uric Acid Increased |
56 |
57 |
|
Glucose Decreased |
8 |
6 |
6.2 Additional Data from Safety Population
The following adverse reactions were reported in patients in the SYNRIBO clinical studies of patients with chronic phase and accelerated phase CML at a frequency of 1% to less than 10%. Within each category, adverse reactions are ranked on the basis of frequency.
Cardiac Disorders: tachycardia, palpitations, acute coronary syndrome, angina pectoris, arrhythmia, bradycardia, ventricular extrasystoles.
Ear and Labyrinth Disorders: ear pain, ear hemorrhage, tinnitus.
Eye Disorders: cataract, vision blurred, conjunctival hemorrhage, dry eye, lacrimation increased, conjunctivitis, diplopia, eye pain, eyelid edema.
Gastrointestinal Disorders: stomatitis, mouth ulceration, abdominal distension, dyspepsia, gastroesophageal reflux disease, gingival bleeding, aphthous stomatitis, dry mouth, hemorrhoids, gastritis, gastrointestinal hemorrhage, melena, mouth hemorrhage, oral pain, anal fissure, dysphagia, gingival pain, gingivitis.
General Disorders and Administration Site Conditions: mucosal inflammation, pain, chest pain, hyperthermia, influenza-like illness, catheter site pain, general edema, malaise.
Immune System Disorders: hypersensitivity.
Injury, Poisoning and Procedural Complications: contusion, transfusion reaction.
Metabolism and Nutrition Disorders: decreased appetite, diabetes mellitus, gout, dehydration.
Musculoskeletal and Connective Tissue Disorders: bone pain, myalgia, muscular weakness, muscle spasms, musculoskeletal chest pain, musculoskeletal pain, musculoskeletal stiffness, musculoskeletal discomfort.
Nervous System Disorders: dizziness, cerebral hemorrhage, paresthesia, convulsion, hypoesthesia, lethargy, sciatica, burning sensation, dysgeusia, tremor.
Psychiatric Disorders: anxiety, depression, agitation, confusional state, mental status change.
Renal and Urinary Disorders: dysuria.
Respiratory, Thoracic and Mediastinal Disorders: pharyngolaryngeal pain, nasal congestion, dysphonia, productive cough, rales, rhinorrhea, hemoptysis, sinus congestion.
Skin and Subcutaneous Tissue Disorders: erythema, pruritus, dry skin, petechiae, hyperhidrosis, night sweats, ecchymosis, purpura, skin lesion, skin ulcer, rash erythematous, rash papular, skin exfoliation, skin hyperpigmentation.
Vascular Disorders: hematoma, hypertension, hot flush, hypotension.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings from animal studies, SYNRIBO can cause fetal harm when administered to pregnant women. In animal reproduction studies, subcutaneous administration of omacetaxine mepesuccinate to pregnant mice during organogenesis at doses approximately 0.25 to 0.5 times the maximum recommended human doses (MRHD) resulted in embryo-fetal mortality, structural abnormalities, and alterations to growth (see Data). There are no available data on SYNRIBO use in pregnant women to evaluate for a drug- associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Advise pregnant women of the potential risk to a fetus [see Warnings and Precautions (5.4)].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study, pregnant mice were administered omacetaxine mepesuccinate subcutaneously during the period of organogenesis at doses of 0.63 or 1.23 mg/m2/day (approximately 0.25 to 0.5 times the MRHD on a body surface area basis). Drug-related adverse effects included embryonic death, an increase in unossified bones/reduced bone ossification, and decreased fetal body weights. Fetal toxicity occurred at doses of 1.23 mg/m2/day, which is approximately half the recommended daily human dose.
8.2 Lactation
Risk Summary
There are no data on the presence of omacetaxine mepesuccinate in either human or animal milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise patients that breastfeeding is not recommended during treatment with SYNRIBO, and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating SYNRIBO.
Contraception
Females
SYNRIBO can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use effective contraception during treatment with SYNRIBO and for 6 months after the final dose.
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with SYNRIBO and for 3 months after the final dose [see Nonclinical Toxicology (13.1)].
Infertility
Males
Based on findings from animal studies, SYNRIBO may impair male fertility. Studies in mice demonstrated adverse effects on male reproductive organs where bilateral degeneration of the seminiferous tubular epithelium in testes and hypospermia/aspermia were observed [see Nonclinical Toxicology (13.1)]. The long-term effects of SYNRIBO on male fertility, including the reversibility of adverse effects, have not been studied.
8.4 Pediatric Use
The safety and effectiveness of SYNRIBO in pediatric patients have not been established.
8.5 Geriatric Use
In the chronic and accelerated phase CML efficacy populations 23 (30%) and 16 (46%) patients were ≥65 years of age. For the age subgroups of <65 years of age and ≥65 years of age, there were differences between the subgroups, with higher rates of major cytogenetic responses (MCyRs) in younger patients with CP CML compared with older patients (23% vs. 9%, respectively) and higher rates of major hematologic responses (MaHRs) in older patients with AP CML compared with younger patients (31% vs. 0%, respectively). Patients ≥65 years of age were more likely to experience toxicity, most notably hematologic toxicity.
8.6 Effect of Gender
Of the 76 patients included in the chronic phase CML population efficacy analysis, 47 (62%) of the patients were men and 29 (38%) were women. For patients with chronic phase CML, the MCyR rate in men was higher than in women (21% vs. 14%, respectively). There were differences noted in the safety profile of omacetaxine mepesuccinate in men and women with chronic phase CML although the small number of patients in each group prevents a definitive assessment. There were inadequate patient numbers in the accelerated phase subset to draw conclusions regarding a gender effect on efficacy.
10. Overdosage
A patient in the clinical expanded access program received an overdose of 2.5 mg/m2 twice daily for 5 days in the 16th cycle. The patient presented with gastrointestinal disorders, gingival hemorrhage, alopecia, and Grade 4 thrombocytopenia and neutropenia. When SYNRIBO treatment was temporarily interrupted the gastrointestinal disorders and hemorrhagic syndrome resolved, and neutrophil values returned to within normal range. The alopecia and thrombocytopenia (Grade 1) improved, and SYNRIBO was restarted.
No specific antidote for SYNRIBO overdose is known. Management of overdosage should include general supportive measures, including monitoring of hematologic parameters.
11. Synribo Description
SYNRIBO contains the active ingredient omacetaxine mepesuccinate, a cephalotaxine ester. It is a protein synthesis inhibitor. Omacetaxine mepesuccinate is prepared by a semi-synthetic process from cephalotaxine, an extract from the leaves of Cephalotaxus sp. The chemical name of omacetaxine mepesuccinate is cephalotaxine, 4-methyl (2R)-hydroxyl-2-(4-hydroxyl-4-methylpentyl) butanedioate (ester).
Omacetaxine mepesuccinate has the following chemical structure:
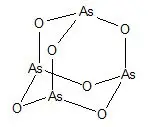
The molecular formula is C29H39NO9 with a molecular weight of 545.6 g/mol. SYNRIBO for Injection is a sterile, preservative-free, white to off-white, lyophilized powder in a single-dose vial. Each vial contains 3.5 mg omacetaxine mepesuccinate, 60 mg hydrochloric acid for drug substance solubilization, 10 mg mannitol as a bulking agent, and hydrochloric acid and sodium bicarbonate to adjust pH to 6.
SYNRIBO is intended for subcutaneous administration after reconstitution with 1.0 mL of 0.9% Sodium Chloride Injection, USP. The pH of the reconstituted solution is between 5.5 and 7.0.
12. Synribo - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of action of omacetaxine mepesuccinate has not been fully elucidated but includes inhibition of protein synthesis and is independent of direct Bcr-Abl binding. Omacetaxine mepesuccinate binds to the A-site cleft in the peptidyl-transferase center of the large ribosomal subunit from a strain of archaeabacteria. In vitro, omacetaxine mepesuccinate reduced protein levels of the Bcr-Abl oncoprotein and Mcl-1, an anti-apoptotic Bcl-2 family member. Omacetaxine mepesuccinate showed activity in mouse models of wild-type and T315I mutated Bcr-Abl CML.
12.2 Pharmacodynamics
Cardiac Electrophysiology
In an uncontrolled pharmacokinetic study there were no reports of QTcF > 480 ms or ΔQTcF > 60 ms in 21 treated patients who received omacetaxine mepesuccinate 1.25 mg/m2 BID for 14 consecutive days. There was no evidence for concentration-dependent increases in QTc for omacetaxine mepesuccinate or 4’-DMHHT. Although the mean effect on QTc was 4.2 ms (upper 95% CI: 9.5 ms), QTc effects less than 10 ms cannot be verified due to the absence of a placebo and positive controls.
12.3 Pharmacokinetics
The dose proportionality of omacetaxine mepesuccinate is unknown. A 90% increase in systemic exposure to omacetaxine mepesuccinate was observed between the first dose and steady state.
Absorption
The absolute bioavailability of omacetaxine mepesuccinate has not been determined. Omacetaxine mepesuccinate is absorbed following subcutaneous administration, and maximum concentrations are achieved after approximately 30 minutes.
Distribution
The steady-state (mean ± SD) volume of distribution of omacetaxine mepesuccinate is approximately 141 ± 93.4 L following subcutaneous administration of 1.25 mg/m2 twice daily for 11 days . The plasma protein binding of omacetaxine mepesuccinate is less than or equal to 50%.
Elimination
The terminal elimination half-life of omacetaxine mepesuccinate in plasma is 14.6 hours.
Metabolism
Omacetaxine mepesuccinate is primarily hydrolyzed to 4′-DMHHT via plasma esterases with little hepatic microsomal oxidative and/or esterase-mediated metabolism in vitro.
Excretion
Following a single subcutaneous dose of radiolabeled omacetaxine mepesuccinate, the mean total recovery of radioactivity in excreta was approximately 81% of the radioactive dose. Approximately 37% of the radioactivity was recovered in urine and approximately 44% in feces.
Drug Interaction Studies
Cytochrome P450 (CYP) Enzymes: Omacetaxine mepesuccinate and 4′-DMHHT do not inhibit major CYP enzymes in vitro at concentrations that can be expected clinically. The potential for omacetaxine mepesuccinate or 4′-DMHHT to induce CYP enzymes has not been determined.
Transporter Systems: Omacetaxine mepesuccinate is a P-glycoprotein (P-gp) substrate in vitro. Omacetaxine mepesuccinate and 4′-DMHHT do not inhibit P-gp mediated efflux of loperamide in vitro at concentrations that can be expected clinically.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity studies have been conducted with omacetaxine mepesuccinate.
Omacetaxine mepesuccinate was genotoxic in an in vitro chromosomal aberration test system in Chinese hamster ovary (CHO) cells, but was not mutagenic when tested in an in vitro bacterial cell assay (Ames test), and it did not induce genetic damage using an in vivo mouse micronucleus assay.
SYNRIBO may impair male fertility. Studies in mice demonstrated adverse effects on male reproductive organs. Bilateral degeneration of the seminiferous tubular epithelium in testes and hypospermia/aspermia in the epididymides were reported in the highest dose group (2.33 mg/kg/day reduced to 1.67 mg/kg/day; 7 to 5 mg/m2/day) following subcutaneous injection of omacetaxine mepesuccinate for six cycles over six months. The doses used in the mice were approximately two to three times the recommended daily human dose based on body surface area.
14. Clinical Studies
The efficacy of SYNRIBO was evaluated using a combined cohort of adult patients with CML from two trials. The combined cohort consisted of patients who had received 2 or more approved TKIs and had, at a minimum, documented evidence of resistance or intolerance to dasatinib and/or nilotinib. Resistance was defined as one of the following: no complete hematologic response (CHR) by 12 weeks (whether lost or never achieved); or no cytogenetic response by 24 weeks (i.e., 100% Ph positive [Ph+]) (whether lost or never achieved); or no major cytogenetic response (MCyR) by 52 weeks (i.e., ≥35% Ph+) (whether lost or never achieved); or progressive leukocytosis. Intolerance was defined as one of the following: 1) Grade 3-4 non-hematologic toxicity that does not resolve with adequate intervention; or 2) Grade 4 hematologic toxicity lasting more than 7 days; or 3) any Grade ≥ 2 toxicity that is unacceptable to the patient. Patients with NYHA class III or IV heart disease, active ischemia or other uncontrolled cardiac conditions were excluded.
Patients were treated with omacetaxine mepesuccinate at a dose of 1.25 mg/m2 administered subcutaneously twice daily for 14 consecutive days every 28 days (induction cycle). Responding patients were then treated with the same dose and twice daily schedule for 7 consecutive days every 28 days (maintenance cycle). Patients were allowed to continue to receive maintenance treatment for up to 24 months. Responses were adjudicated by an independent Data Monitoring Committee (DMC).
Chronic Phase CML (CP CML)
A total of 76 patients with chronic phase CML were included in the efficacy analysis. The demographics were: median age 59 years, 62% were male, 30% were 65 years of age or older, 80% were Caucasian, 5% were African-American, 4% were Asian and 4% were Hispanic. Thirty-six (47%) patients had failed treatment with imatinib, dasatinib, and nilotinib. Most patients had also received prior non-TKI treatments, most commonly hydroxyurea (54%), interferon (30%), and/or cytarabine (29%). The efficacy endpoint was based on MCyR (adjudicated by a DMC).
Table 5: Efficacy Results Evaluated by DMC for Patients with CP CML
| Patients (N=76) |
|
| Primary Response – MCyR | |
| Total with MCyR, n (%) | 14 (18.4) |
| 95% confidence interval | (10.5% – 29.0%) |
| Cytogenetic Response, n (%) | |
| Confirmed complete | 6 (7.9) |
| Confirmed partial | 3 (3.9) |
Cytogenetic response evaluation is based on standard cytogenetic analysis (at least 20 metaphases).
Complete: 0% Ph+ cells, Partial > 0% to 35% Ph+ cells
The mean time to MCyR onset in the 14 patients was 3.5 months. The median duration of MCyR for the 14 patients was 12.5 months (Kaplan-Meier estimate).
Accelerated Phase CML (AP CML)
A total of 35 patients with accelerated phase CML were included in the efficacy analysis. The demographics were: median age was 63 years, 57% were male, 46% were 65 years of age or older, 68% were Caucasian, 23% were African-American, 3% were Asian and 3% were Hispanic. Twenty-two (63%) of 35 patients with accelerated phase had failed treatment with imatinib, dasatinib, and nilotinib. Most patients had also received prior non-TKI treatments, most commonly hydroxyurea (43%), interferon (31%), and/or cytarabine (29%). The efficacy endpoint was assessed based on MCyR and MaHR (complete hematologic response [CHR] or no evidence of leukemia [NEL]). The efficacy results for the patients with accelerated phase as adjudicated by the DMC are shown in Table 6.
Table 6: Efficacy Results Evaluated by DMC for Patients with AP CML
| Patients (N=35) |
|
| Primary Response – MaHR | |
| Total with MaHR, n (%) | 5 (14.3) |
| 95% confidence interval | (4.5% - 30.3%) |
| CHR | 4 (11.4) |
| NEL | 1 (2.9) |
| Primary Response – MCyR | |
| Total with MCyR, n (%) | 0 |
MaHR is defined as complete hematologic response (CHR) or no evidence of leukemia (NEL): CHR - absolute neutrophil count ≥ 1.5 × 109/liter, platelets ≥ 100 × 109/liter, no blood blasts, bone marrow blasts < 5%, no extramedullary disease; NEL - Morphologic leukemia-free state, defined as <5% bone marrow blasts.
The mean time to response onset in the 5 patients was 2.3 months. The median duration of MaHR for the 5 patients was 4.7 months (Kaplan-Meier estimate).
16. How is Synribo supplied
16.1 How Supplied
SYNRIBO (omacetaxine mepesuccinate) for Injection is supplied in 8 mL clear glass single-dose vial in individual cartons. Each vial contains 3.5 mg of SYNRIBO (omacetaxine mepesuccinate) for Injection (NDC 63459-177-14).
16.2 Storage and Handling
Store unopened vials at 20oC to 25ºC (68o F to 77ºF); excursions permitted from 15ºC to 30ºC (59ºF to 86ºF) [see USP Controlled Room Temperature]. Prior to re-constitution, keep product in carton to protect from light.
Omacetaxine mepesuccinate is a cytotoxic drug. Follow special handling and disposal procedures1.
17. Patient Counseling Information
Availability of Medication Guide and Instructions for Use
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use). Assist patients and caregivers in understanding their contents and give them the opportunity to discuss the contents of the Medication Guide and Instructions for Use and to obtain answers to any questions they may have prior to initiating therapy. The complete text of the Medication Guide and Instructions for Use are attached to the prescribing information.
Patient Training
Once it is determined that a patient is an appropriate candidate for self-administration or administration by a caregiver, ensure that patients receive the necessary supplies for home administration of SYNRIBO and train them on the following [see Dosage and Administration (2.4, 2.5, 2.6, 2.7)]:
- How to transport reconstituted SYNRIBO in a secure container or packaging and under recommended temperature conditions.
- Acceptable storage conditions and use times for reconstituted SYNRIBO:
- When stored in a refrigerator (2°C to 8°C [36°F to 46°F]), use within 6 days (144 hours).
- When stored at room temperature (not to exceed 25°C [77°F]), use within 12 hours.
- If stored in a refrigerator, keep SYNRIBO from coming into contact with food or drink.
- To wear disposable gloves and protective eyewear when handling SYNRIBO.
- To wash hands before putting on gloves and after removing gloves.
- Not to eat or drink while handling SYNRIBO. To administer SYNRIBO in an area away from food or food preparation areas.
- To administer SYNRIBO in a location away from children and pregnant women.
- Proper subcutaneous injection technique including acceptable sites.
- The importance of body site selection for administering the injection, as well as the importance of alternating the injection sites. Advise patients to not inject SYNRIBO into areas of the skin that are tender, red, bruised, hard, or that have scars or stretch marks.
- In the case of a missed dose: If a patient misses an injection, skip the missed dose and the patient should give the next scheduled injection at the next scheduled time. Inform patients NOT to give two injections to make up for a missed injection.
- In the case that SYNRIBO comes into contact with a patient’s skin or eyes: Advise patients to wash exposed skin with soap and water and in the case of eye exposure, thoroughly flush the eye with water. After washing or flushing, advise patients to call their healthcare provider immediately.
- In the case that too much SYNRIBO is injected or that SYNRIBO is accidentally swallowed: Instruct patients to contact their healthcare provider immediately if they have injected too much SYNRIBO, or if someone has swallowed SYNRIBO.
- Disposal procedures, including use of an appropriate biohazard container and return of the container to the clinic or pharmacy for final disposal. Inform patients NOT to recap or clip the used needle and not to place used needles, syringes, vials, and other used supplies in a household trash or recycle container.
- Accidental spillage procedures, including wiping the spilled liquid with the absorbent pad (using protective eyewear and gloves), washing the area with water and soap, and proper disposal of materials.
Myelosuppression
Advise patients of the likelihood that SYNRIBO will cause a decrease in white blood cells, platelets, and red blood cells and that monitoring of these parameters will be needed. Instruct patients to contact a health care professional if they develop a fever, or other signs/symptoms of infection; shortness of breath, significant fatigue, or bleeding [see Warnings and Precautions (5.1)].
Bleeding
Advise patients of the possibility of serious bleeding due to low platelet counts. Instruct patients to report immediately any signs or symptoms suggestive of hemorrhage (unusual bleeding, easy bruising or blood in urine or stool; confusion, slurred speech, or altered vision). Instruct patients to report in advance if they plan to have any dental or surgical procedures [see Warnings and Precautions (5.2)].
Hyperglycemia
Advise patients with diabetes of the possibility of hyperglycemia and the need for careful monitoring of blood glucose levels. Patients with poorly controlled diabetes mellitus should not be treated with omacetaxine mepesuccinate until good glycemic control has been established [see Warnings and Precautions (5.3)].
Gastrointestinal Distress
Advise patients that they may experience nausea, diarrhea, abdominal pain, constipation, and vomiting. If these symptoms persist, they should seek medical attention.
Fatigue
Advise patients that SYNRIBO may cause tiredness and to avoid driving any vehicle or operating any dangerous tools or machinery if they experience this side effect.
Rash
Advise patients that they may experience skin rash. Advise patients to immediately report severe or worsening rash or itching.
Alopecia
Advise patients that they may experience hair loss.
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1, 8.3)]. Advise female patients of reproductive potential to use effective contraception during treatment with SYNRIBO and for 6 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with SYNRIBO and for 3 months after the final dose [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Lactation
Advise females not to breastfeed during treatment with SYNRIBO and for 2 weeks after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise males of reproductive potential that SYNRIBO may impair fertility [see Use in Specific Populations (8.3)].
SYN-007
Distributed by:
Teva Pharmaceuticals USA, Inc.
Parsippany, NJ 07054
© Cephalon, Inc., a wholly-owned subsidiary of Teva Pharmaceutical Industries Ltd. All rights reserved.
Medication Guide
|
SYNRIBO® (sin-RYE-bo) (omacetaxine mepesuccinate) for injection, for subcutaneous use |
|
What is the most important information I should know about SYNRIBO? SYNRIBO can cause serious side effects including:
See “What are the possible side effects of SYNRIBO?” for more information about side effects. |
|
What is SYNRIBO? SYNRIBO is a prescription medicine used to treat adults with a type of blood cancer called chronic myeloid leukemia (CML):
It is not known if SYNRIBO is safe and effective in children. |
|
Before using SYNRIBO, tell your healthcare provider about all of your medical conditions, including if you:
Females who are able to become pregnant:
Males with female partners who are able to become pregnant should use effective birth control during treatment with SYNRIBO and for 3 months after the final dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|
How should I use SYNRIBO?
|
|
What should I avoid while using SYNRIBO? SYNRIBO may cause tiredness. Avoid driving or operating dangerous tools or machinery if you develop tiredness when using SYNRIBO. |
|
What are the possible side effects of SYNRIBO? SYNRIBO may cause serious side effects, including:
The most common side effects of SYNRIBO include:
Tell your healthcare provider or get medical help right away if you get nausea, diarrhea, stomach (abdominal) pain, severe or worsening skin rash, or itching that does not go away. You may have hair loss during treatment with SYNRIBO. SYNRIBO may cause fertility problems in males, which may affect your ability to have children. Talk to your healthcare provider if this is a concern for you. These are not all of the side effects of SYNRIBO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store SYNRIBO?
Keep SYNRIBO and all medicines out of the reach of children. |
|
General information about the safe and effective use of SYNRIBO. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use SYNRIBO for a condition for which it was not prescribed. Do not give SYNRIBO to other people even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about SYNRIBO that is written for health professionals. |
|
What are the ingredients in SYNRIBO? Active ingredient: omacetaxine mepesuccinate Inactive ingredients: hydrochloric acid, mannitol, sodium bicarbonate
Distributed by: Teva Pharmaceuticals USA, Inc.  SYNMG-003 For more information, go to www.synribo.com or call Teva Pharmaceuticals at 1-888-483-8279. |
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: May 2021
© Cephalon, Inc., a wholly-owned subsidiary of Teva Pharmaceutical Industries Ltd.
All rights reserved.
Instructions for Use
SYNRIBO® (sin-RYE-bo)
(omacetaxine mepesuccinate)
for injection, for subcutaneous use
Read this Instructions for Use before you inject SYNRIBO for the first time and each time you get a refill. Also read the Medication Guide for SYNRIBO.
Before you or your caregiver injects SYNRIBO, your healthcare provider will show you how to properly:
- handle syringes and inject SYNRIBO
- dispose of used supplies for injecting SYNRIBO
- clean up any spilled SYNRIBO
Important:
- Be sure that you store SYNRIBO exactly as your healthcare provider tells you to. See the section “How should I store SYNRIBO?” in the Medication Guide.
- Never try to re-cap the needle. This could cause a needle-stick injury.
- If SYNRIBO comes into contact with your skin, wash the area well with soap and water.
Your healthcare provider will arrange for you to receive all of the supplies that you will need for each injection of SYNRIBO:
- syringe with attached needle, containing SYNRIBO for injection
- protective eyewear, such as protective eyeglasses (not regular eyeglasses) or face shield
- gloves
- disposal biohazard container
- absorbent pads for use to clean up an accidental spill of SYNRIBO
- alcohol swabs
- gauze pads
You may also need an adhesive bandage.
Never mix SYNRIBO yourself. If you don’t receive syringes already filled with SYNRIBO, contact your doctor or pharmacy.
Step 1. Preparing to give an injection of SYNRIBO.
- Find a clean flat work surface.
- Wash your hands well with soap and water.
- Put on a pair of gloves and your protective eyewear before you handle the syringe containing SYNRIBO. Wearing gloves and protective eyewear (even if you wear glasses) protects you from splashes or spills. See Figure A.
- Look at the date printed on the syringe label to make sure that the expiration date has not passed. Do not use if the expiration date has passed and contact your doctor or pharmacy immediately.
- Gather the rest of your supplies and place them on your work surface.
Figure A

Step 2. Choose an injection site.
- You may inject SYNRIBO into your thigh or stomach-area (abdomen). See Figure B. The injection can be given in the back of your arm if a caregiver is giving the injection. See Figure C.
- Use a different site for each injection to help decrease tenderness at the injection site. Each injection site should be at least 1 inch away from any recently used injection site.
- Do not inject SYNRIBO into areas of your skin that are tender, red, bruised, hard, or that have scars or stretch marks.
Figure B

Figure C
Step 3. Prepare the injection site.
- Clean the injection site well with an alcohol wipe and allow it to air dry. See Figure D.
Figure D
Step 4. Inject SYNRIBO.
- Carefully remove the needle cap by pulling, taking care not to stick yourself. See Figure E.
- Do not press down on the plunger.
Figure E
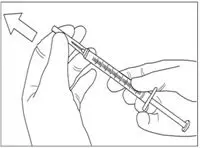
- With one hand, pinch skin of injection site between your thumb and forefinger. See Figure F.
Figure F
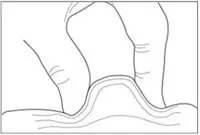
- With your other hand, hold the syringe at a 45 degree or 90 degree angle to your skin. Use a quick dart-like motion to insert the needle through the skin at the injection site. See Figures G and H. The needle should go through the skin but not into your muscle.
Figure G
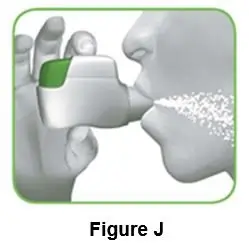
Figure H

- Slowly push down on the plunger with your thumb until syringe is empty. See Figure I.
Figure I

- Stop pinching your skin. Quickly remove needle and then apply pressure on injection site with a dry gauze pad. You can put a small adhesive bandage over the injection site if there is bleeding. See Figure J.
Figure J
- Follow the instructions below for how to dispose of the syringe, needle, and other supplies used to give your injection. Never try to re-cap the needle. This could cause a needle-stick injury.
- Remove your gloves. Wash your hands right away with soap and water, and then remove your protective eyewear.
How should I throw away (dispose of) used SYNRIBO syringes, needles, and other supplies?
- Throw away (dispose of) used SYNRIBO syringes, needles, gloves, and other used supplies in an appropriate biohazard container.
- Return the biohazard container to your healthcare provider for disposal.
- Do not place used syringes, needles, or other supplies in a household trash or recycle container. Do not re-cap or clip the used needle. This could cause a needle-stick injury.
- Do not throw away the protective eyewear. You will need them for each dose of SYNRIBO.
What should I do in case of an accidental SYNRIBO spill?
- Your healthcare provider will arrange for you to receive supplies to use in case you spill SYNRIBO.
- Follow your healthcare provider’s instructions about how to clean up a SYNRIBO spill.
- Do not touch a spill unless you are wearing gloves and protective eyewear.
- Use an absorbent pad to wipe up the spill. Wash the area with soap and water. Use an extra absorbent pad or paper towel to dry the area.
- Place the pad, gloves, and other supplies that were used to clean the spill in the biohazard container.
- Call your healthcare provider right away to report the spill.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Distributed by:

Teva Pharmaceuticals USA, Inc.
Parsippany, NJ 07054
SYNIFU-004
Revised: May 2021
© Cephalon, Inc., a wholly-owned subsidiary of Teva Pharmaceutical Industries Ltd.
All rights reserved.
Package/Label Display Panel

NDC 63459-177-14 Rx only
Single-Dose Vial.
Discard unused portion.
Synribo® (omacetaxine mepesuccinate) for Injection
3.5 mg/vial
For Subcutaneous Use Only
Must be Reconstituted Before Use
Medication Guide Required: Each time SYNRIBO is dispensed, give the patient a Medication Guide.
Distributed by:
Teva Pharmaceuticals USA, Inc.
Parsippany, NJ 07054
TEVA
| SYNRIBO
omacetaxine mepesuccinate injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Cephalon, LLC (183236314) |
