Drug Detail:Tikosyn (Dofetilide)
Drug Class: Group III antiarrhythmics
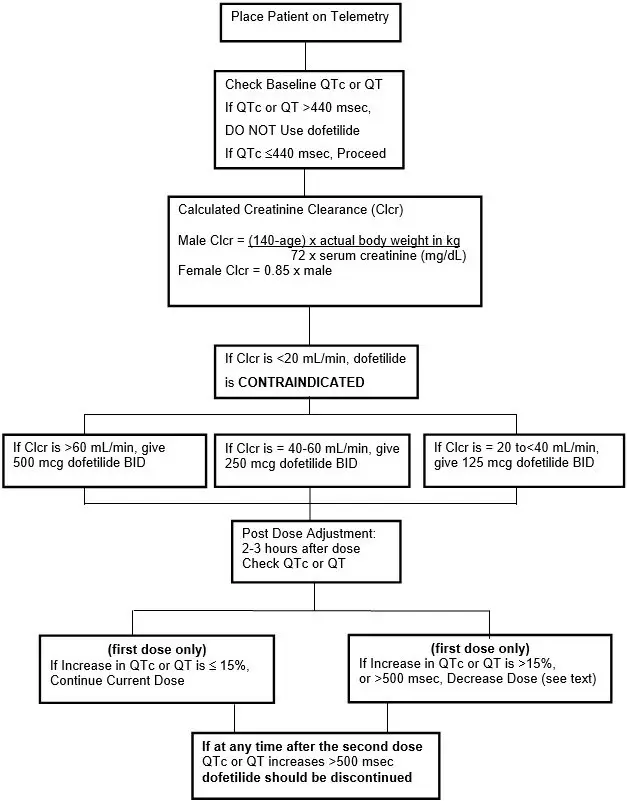
To minimize the risk of induced arrhythmia, patients initiated or re-initiated on TIKOSYN should be placed for a minimum of 3 days in a facility that can provide calculations of creatinine clearance, continuous electrocardiographic monitoring, and cardiac resuscitation. For detailed instructions regarding dose selection, see DOSAGE AND ADMINISTRATION.
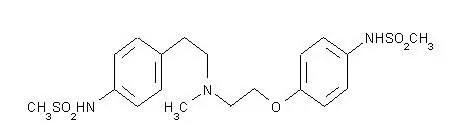
Tikosyn - Clinical Pharmacology
Pharmacokinetics in Special Populations
Renal Impairment
In volunteers with varying degrees of renal impairment and patients with arrhythmias, the clearance of dofetilide decreases with decreasing creatinine clearance. As a result, and as seen in clinical studies, the half-life of dofetilide is longer in patients with lower creatinine clearances. Because increase in QT interval and the risk of ventricular arrhythmias are directly related to plasma concentrations of dofetilide, dosage adjustment based on calculated creatinine clearance is critically important (see DOSAGE AND ADMINISTRATION). Patients with severe renal impairment (creatinine clearance <20 mL/min) were not included in clinical or pharmacokinetic studies (see CONTRAINDICATIONS).
Dose-Response and Concentration Response for Increase in QT Interval
Increase in QT interval is directly related to dofetilide dose and plasma concentration. Figure 1 shows that the relationship in normal volunteers between dofetilide plasma concentrations and change in QTc is linear, with a positive slope of approximately 15–25 msec/(ng/mL) after the first dose and approximately 10–15 msec/(ng/mL) at Day 23 (reflecting a steady state of dosing). A linear relationship between mean QTc increase and dofetilide dose was also seen in patients with renal impairment, in patients with ischemic heart disease, and in patients with supraventricular and ventricular arrhythmias.
Figure 1: Mean QTc-Concentration Relationship in Young Volunteers Over 24 Days
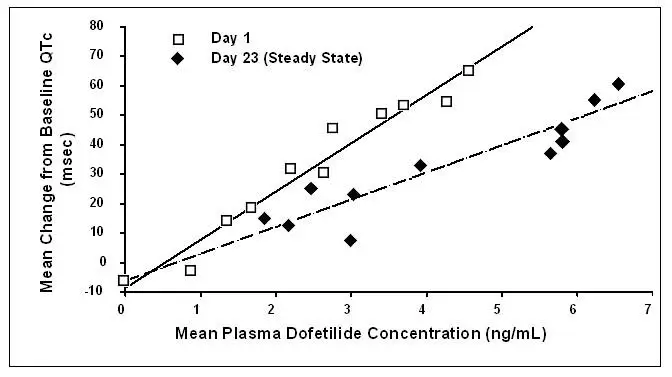
Note: The range of dofetilide plasma concentrations achieved with the 500 mcg BID dose adjusted for creatinine clearance is 1–3.5 ng/mL.
The relationship between dose, efficacy, and the increase in QTc from baseline at steady state for the two randomized, placebo-controlled studies (described further below) is shown in Figure 2. The studies examined the effectiveness of TIKOSYN in conversion to sinus rhythm and maintenance of normal sinus rhythm after conversion in patients with atrial fibrillation/flutter of >1 week duration. As shown, both the probability of a patient's remaining in sinus rhythm at six months and the change in QTc from baseline at steady state of dosing increased in an approximately linear fashion with increasing dose of TIKOSYN. Note that in these studies, doses were modified by results of creatinine clearance measurement and in-hospital QTc prolongation.
Figure 2: Relationship Between TIKOSYN Dose, QTc Increase and Maintenance of NSR
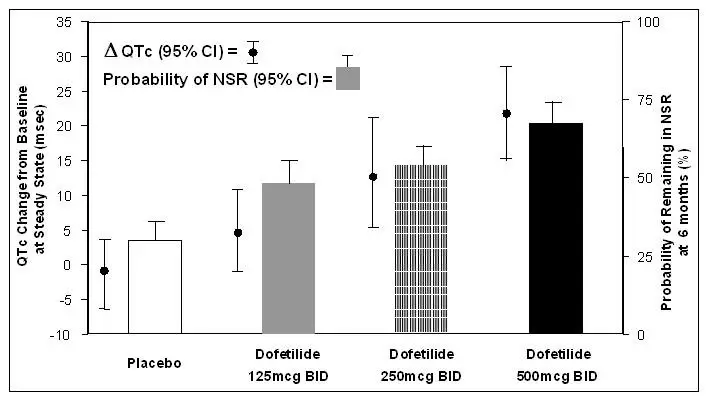
Number of patients evaluated for maintenance of NSR: 503 TIKOSYN, 174 placebo.
Number of patients evaluated for QTc change: 478 TIKOSYN, 167 placebo.
Clinical Studies


Safety in Patients with Structural Heart Disease: DIAMOND Studies (The Danish Investigations of Arrhythmia and Mortality on Dofetilide)
The two DIAMOND studies were 3-year trials comparing the effects of TIKOSYN and placebo on mortality and morbidity in patients with impaired left ventricular function (ejection fraction ≤35%). Patients were treated for at least one year. One study was in patients with moderate to severe (60% NYHA Class III or IV) congestive heart failure (DIAMOND CHF) and the other was in patients with recent myocardial infarction (DIAMOND MI) (of whom 40% had NYHA Class III or IV heart failure). Both groups were at relatively high risk of sudden death. The DIAMOND trials were intended to determine whether TIKOSYN could reduce that risk. The trials did not demonstrate a reduction in mortality; however, they provide reassurance that, when initiated carefully, in a hospital or equivalent setting, TIKOSYN did not increase mortality in patients with structural heart disease, an important finding because other antiarrhythmics [notably the Class IC antiarrhythmics studied in the Cardiac Arrhythmia Suppression Trial (CAST) and a pure Class III antiarrhythmic, d-sotalol (SWORD)] have increased mortality in post-infarction populations. The DIAMOND trials therefore provide evidence of a method of safe use of TIKOSYN in a population susceptible to ventricular arrhythmias. In addition, the subset of patients with AF in the DIAMOND trials provide further evidence of safety in a population of patients with structural heart disease accompanying the AF. Note, however, that this AF population was given a lower (250 mcg BID) dose (see CLINICAL STUDIES, DIAMOND Patients with Atrial Fibrillation).
In both DIAMOND studies, patients were randomized to 500 mcg BID of TIKOSYN, but this was reduced to 250 mcg BID if calculated creatinine clearance was 40–60 mL/min, if patients had AF, or if QT interval prolongation (>550 msec or >20% increase from baseline) occurred after dosing. Dose reductions for reduced calculated creatinine clearance occurred in 47% and 45% of DIAMOND CHF and MI patients, respectively. Dose reductions for increased QT interval or QTc occurred in 5% and 7% of DIAMOND CHF and MI patients, respectively. Increased QT interval or QTc (>550 msec or >20% increase from baseline) resulted in discontinuation of 1.8% of patients in DIAMOND CHF and 2.5% of patients in DIAMOND MI.
In the DIAMOND studies, all patients were hospitalized for at least 3 days after treatment was initiated and monitored by telemetry. Patients with QTc greater than 460 msec, second or third degree AV block (unless with pacemaker), resting heart rate <50 bpm, or prior history of polymorphic ventricular tachycardia were excluded.
DIAMOND CHF studied 1518 patients hospitalized with severe CHF who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 311 deaths from all causes in patients randomized to TIKOSYN (n=762) and 317 deaths in patients randomized to placebo (n=756). The probability of survival at one year was 73% (95% CI: 70% – 76%) in the TIKOSYN group and 72% (95% CI: 69% – 75%) in the placebo group. Similar results were seen for cardiac deaths and arrhythmic deaths. Torsade de Pointes occurred in 25/762 patients (3.3%) receiving TIKOSYN. The majority of cases (76%) occurred within the first 3 days of dosing. In all, 437/762 (57%) of patients on TIKOSYN and 459/756 (61%) on placebo required hospitalization. Of these, 229/762 (30%) of patients on TIKOSYN and 290/756 (38%) on placebo required hospitalization because of worsening heart failure.
DIAMOND MI studied 1510 patients hospitalized with recent myocardial infarction (2–7 days) who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 230 deaths in patients randomized to TIKOSYN (n=749) and 243 deaths in patients randomized to placebo (n=761). The probability of survival at one year was 79% (95% CI: 76% – 82%) in the TIKOSYN group and 77% (95% CI: 74% – 80%) in the placebo group. Cardiac and arrhythmic mortality showed a similar result. Torsade de Pointes occurred in 7/749 patients (0.9%) receiving TIKOSYN. Of these, 4 cases occurred within the first 3 days of dosing and 3 cases occurred between Day 4 and the conclusion of the study. In all, 371/749 (50%) of patients on TIKOSYN and 419/761 (55%) on placebo required hospitalization. Of these, 200/749 (27%) of patients on TIKOSYN and 205/761 (27%) on placebo required hospitalization because of worsening heart failure.
Of the 506 patients in the DIAMOND studies who had atrial fibrillation or flutter at baseline, 12% of patients in the TIKOSYN group and 2% of patients in the placebo group had converted to normal sinus rhythm after one month. In those patients converted to normal sinus rhythm, 79% of the TIKOSYN group and 42% of the placebo group remained in normal sinus rhythm for one year.
In the DIAMOND studies, although Torsade de Pointes occurred more frequently in the TIKOSYN-treated patients (see ADVERSE REACTIONS), TIKOSYN, given with an initial 3-day hospitalization and with dose modified for reduced creatinine clearance and increased QT interval, was not associated with an excess risk of mortality in these populations with structural heart disease in the individual studies or in an analysis of the combined studies. The presence of atrial fibrillation did not affect outcome.
Contraindications
TIKOSYN is contraindicated in patients with congenital or acquired long QT syndromes. TIKOSYN should not be used in patients with a baseline QT interval or QTc >440 msec (500 msec in patients with ventricular conduction abnormalities). TIKOSYN is also contraindicated in patients with severe renal impairment (calculated creatinine clearance <20 mL/min).
The concomitant use of verapamil or the cation transport system inhibitors cimetidine, trimethoprim (alone or in combination with sulfamethoxazole), or ketoconazole with TIKOSYN is contraindicated (see WARNINGS and PRECAUTIONS, Drug-Drug Interactions), as each of these drugs cause a substantial increase in dofetilide plasma concentrations. In addition, other known inhibitors of the renal cation transport system such as prochlorperazine, dolutegravir and megestrol should not be used in patients on TIKOSYN.
The concomitant use of hydrochlorothiazide (alone or in combinations such as with triamterene) with TIKOSYN is contraindicated (see PRECAUTIONS, Drug-Drug Interactions) because this has been shown to significantly increase dofetilide plasma concentrations and QT interval prolongation.
TIKOSYN is also contraindicated in patients with a known hypersensitivity to the drug.
Warnings
Ventricular Arrhythmia
TIKOSYN (dofetilide) can cause serious ventricular arrhythmias, primarily Torsade de Pointes (TdP) type ventricular tachycardia, a polymorphic ventricular tachycardia associated with QT interval prolongation. QT interval prolongation is directly related to dofetilide plasma concentration. Factors such as reduced creatinine clearance or certain dofetilide drug interactions will increase dofetilide plasma concentration. The risk of TdP can be reduced by controlling the plasma concentration through adjustment of the initial dofetilide dose according to creatinine clearance and by monitoring the ECG for excessive increases in the QT interval.
Treatment with dofetilide must therefore be started only in patients placed for a minimum of three days in a facility that can provide electrocardiographic monitoring and in the presence of personnel trained in the management of serious ventricular arrhythmias. Calculation of the creatinine clearance for all patients must precede administration of the first dose of dofetilide. For detailed instructions regarding dose selection, see DOSAGE AND ADMINISTRATION.
The risk of dofetilide induced ventricular arrhythmia was assessed in three ways in clinical studies: 1) by description of the QT interval and its relation to the dose and plasma concentration of dofetilide; 2) by observing the frequency of TdP in TIKOSYN-treated patients according to dose; 3) by observing the overall mortality rate in patients with atrial fibrillation and in patients with structural heart disease.
Drug-Drug Interactions
(see CONTRAINDICATIONS)
Because there is a linear relationship between dofetilide plasma concentration and QTc, concomitant drugs that interfere with the metabolism or renal elimination of dofetilide may increase the risk of arrhythmia (Torsade de Pointes). Dofetilide is metabolized to a small degree by the CYP3A4 isoenzyme of the cytochrome P450 system and an inhibitor of this system could increase systemic dofetilide exposure. More important, dofetilide is eliminated by cationic renal secretion, and three inhibitors of this process have been shown to increase systemic dofetilide exposure. The magnitude of the effect on renal elimination by cimetidine, trimethoprim, and ketoconazole (all contraindicated concomitant uses with dofetilide) suggests that all renal cation transport inhibitors should be contraindicated.
Hypokalemia and Potassium-Depleting Diuretics
Hypokalemia or hypomagnesemia may occur with administration of potassium-depleting diuretics, increasing the potential for Torsade de Pointes. Potassium levels should be within the normal range prior to administration of TIKOSYN and maintained in the normal range during administration of TIKOSYN (see DOSAGE AND ADMINISTRATION).
Precautions
Adverse Reactions/Side Effects
The TIKOSYN clinical program involved approximately 8,600 patients in 130 clinical studies of normal volunteers and patients with supraventricular and ventricular arrhythmias. TIKOSYN was administered to 5,194 patients, including two large, placebo-controlled mortality trials (DIAMOND CHF and DIAMOND MI) in which 1,511 patients received TIKOSYN for up to three years.
In the following section, adverse reaction data for cardiac arrhythmias and non-cardiac adverse reactions are presented separately for patients included in the supraventricular arrhythmia development program and for patients included in the DIAMOND CHF and MI mortality trials (see CLINICAL STUDIES, Safety in Patients with Structural Heart Disease, DIAMOND Studies, for a description of these trials).
In studies of patients with supraventricular arrhythmias, a total of 1,346 and 677 patients were exposed to TIKOSYN and placebo for 551 and 207 patient years, respectively. A total of 8.7% of patients in the dofetilide groups were discontinued from clinical trials due to adverse events compared to 8.0% in the placebo groups. The most frequent reason for discontinuation (>1%) was ventricular tachycardia (2.0% on dofetilide vs. 1.3% on placebo). The most frequent adverse events were headache, chest pain, and dizziness.
Tikosyn Dosage and Administration
- Therapy with TIKOSYN must be initiated (and, if necessary, re-initiated) in a setting that provides continuous electrocardiographic (ECG) monitoring and in the presence of personnel trained in the management of serious ventricular arrhythmias. Patients should continue to be monitored in this way for a minimum of three days. Additionally, patients should not be discharged within 12 hours of electrical or pharmacological conversion to normal sinus rhythm.
- The dose of TIKOSYN must be individualized according to calculated creatinine clearance and QTc. (QT interval should be used if the heart rate is <60 beats per minute. There are no data on use of TIKOSYN when the heart rate is <50 beats per minute.) The usual recommended dose of TIKOSYN is 500 mcg BID, as modified by the dosing algorithm described below. For consideration of a lower dose, see Special Considerations below.
- Serum potassium should be maintained within the normal range before TIKOSYN treatment is initiated and should be maintained within the normal range while the patient remains on TIKOSYN therapy. (See WARNINGS, Hypokalemia and Potassium-Depleting Diuretics). In clinical trials, potassium levels were generally maintained above 3.6–4.0 mEq/L.
- Patients with atrial fibrillation should be anticoagulated according to usual medical practice prior to electrical or pharmacological cardioversion. Anticoagulant therapy may be continued after cardioversion according to usual medical practice for the treatment of people with AF. Hypokalemia should be corrected before initiation of TIKOSYN therapy (see WARNINGS, Ventricular Arrhythmia).
- Patients to be discharged on TIKOSYN therapy from an inpatient setting as described above must have an adequate supply of TIKOSYN, at the patient's individualized dose, to allow uninterrupted dosing until the patient can fill a TIKOSYN prescription.










| TIKOSYN
dofetilide capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| TIKOSYN
dofetilide capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| TIKOSYN
dofetilide capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Pharmaceuticals LLC | 829084545 | ANALYSIS(0069-5800, 0069-5810, 0069-5820) , MANUFACTURE(0069-5800, 0069-5810, 0069-5820) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Ireland Pharmaceuticals | 985052076 | ANALYSIS(0069-5800, 0069-5810, 0069-5820) , API MANUFACTURE(0069-5800, 0069-5810, 0069-5820) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Pharmaceuticals LLC | 829084552 | PACK(0069-5800, 0069-5810, 0069-5820) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Italia S.r.l. | 458521908 | ANALYSIS(0069-5800, 0069-5810, 0069-5820) , MANUFACTURE(0069-5800, 0069-5810, 0069-5820) , PACK(0069-5800, 0069-5810, 0069-5820) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Laboratories Div Pfizer Inc | 001147495 | ANALYSIS(0069-5800, 0069-5810, 0069-5820) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Inc | 943955690 | ANALYSIS(0069-5800, 0069-5810, 0069-5820) | |
