Drug Detail:Trintellix (Vortioxetine [ vor-tye-ox-e-teen ])
Drug Class: Miscellaneous antidepressants
Highlights of Prescribing Information
TRINTELLIX (vortioxetine) tablets, for oral use
Initial U.S. Approval: 2013
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
See full prescribing information for complete boxed warning.
- •
- Increased risk of suicidal thinking and behavior in pediatric and young adult patients taking antidepressants.
- •
- Closely monitor for worsening and emergence of suicidal thoughts and behaviors (5.1).
- •
- TRINTELLIX is not approved for use in pediatric patients (8.4).
Recent Major Changes
|
Boxed Warning |
1/2021 |
|
Dosage and Administration, Maintenance/Continuation/Extended Treatment (2.2) |
Removed 11/2020 |
|
Dosage and Administration, Screen for Bipolar Disorder Prior to Starting TRINTELLIX (2.2) |
1/2021 |
|
Dosage and Administration, Use of TRINTELLIX with Other MAOIs Such as Linezolid or Methylene Blue (2.5) |
Removed 1/2021 |
|
Warnings and Precautions, Suicidal Thoughts and Behaviors in Adolescents and Young Adults (5.1) |
1/2021 |
|
Warnings and Precautions, Increased Risk of Bleeding (5.3) |
1/2021 |
|
Warnings and Precautions, Discontinuation Syndrome (5.5) |
1/2021 |
|
Warnings and Precautions, Sexual Dysfunction (5.8) |
9/2021 |
Indications and Usage for Trintellix
TRINTELLIX is indicated for the treatment of major depressive disorder (MDD) in adults (1, 14).
Trintellix Dosage and Administration
- •
- The recommended starting dose is 10 mg administered orally once daily without regard to meals (2.1).
- •
- The dose should then be increased to 20 mg/day, as tolerated (2.1).
- •
- Consider 5 mg/day for patients who do not tolerate higher doses (2.1).
- •
- TRINTELLIX can be discontinued abruptly. However, it is recommended that doses of 15 mg/day or 20 mg/day be reduced to 10 mg/day for one week prior to full discontinuation if possible (2.3).
- •
- The maximum recommended dose is 10 mg/day in known CYP2D6 poor metabolizers (2.5).
Dosage Forms and Strengths
Tablets: 5 mg, 10 mg and 20 mg (3).
Contraindications
- •
- Hypersensitivity to vortioxetine or any components of the TRINTELLIX formulation (4).
- •
- Monoamine Oxidase Inhibitors (MAOIs): Do not use MAOIs intended to treat psychiatric disorders with TRINTELLIX or within 21 days of stopping treatment with TRINTELLIX. Do not use TRINTELLIX within 14 days of stopping an MAOI intended to treat psychiatric disorders. In addition, do not start TRINTELLIX in a patient who is being treated with linezolid or intravenous methylene blue (4).
Warnings and Precautions
- •
- Serotonin Syndrome: Increased risk when co-administered with other serotonergic agents (SSRIs, SNRIs, and triptans), but also when taken alone. If it occurs, discontinue TRINTELLIX and initiate supportive measures (5.2).
- •
- Increased Risk of Bleeding: Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs, other antiplatelet drugs, warfarin, or other drugs that affect coagulation may increase risk (5.3).
- •
- Activation of Mania/Hypomania: Screen patients for bipolar disorder (5.4).
- •
- Angle Closure Glaucoma: Angle closure glaucoma has occurred in patients with untreated anatomically narrow angles treated with antidepressants (5.6).
- •
- Hyponatremia: Can occur in association with the syndrome of inappropriate antidiuretic hormone secretion (SIADH) (5.7).
- •
- Sexual Dysfunction: TRINTELLIX may cause symptoms of sexual dysfunction (5.8).
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were: nausea, constipation and vomiting (6).
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals America, Inc. at 1-877-TAKEDA-7 (1-877-825-3327) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- •
- Strong inhibitors of CYP2D6: Reduce TRINTELLIX dose by half when coadministered (2.5, 7.1).
- •
- Strong CYP Inducers: Consider dose increase of TRINTELLIX dose when coadministered for more than 14 days. The maximum recommended dose should not exceed 3 times the original dose (2.6, 7.1).
Use In Specific Populations
Pregnancy: Third trimester use may increase risk for persistent pulmonary hypertension and withdrawal in the newborn (8.1).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2022
Full Prescribing Information
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behavior in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors [see Warnings and Precautions (5.1)]. TRINTELLIX is not approved for use in pediatric patients [see Use in Specific Populations (8.4)].
1. Indications and Usage for Trintellix
TRINTELLIX is indicated for the treatment of major depressive disorder (MDD) in adults.
2. Trintellix Dosage and Administration
2.1 Recommended Dosage
The recommended starting dose is 10 mg administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/day, as tolerated. The efficacy and safety of doses above 20 mg/day have not been evaluated in controlled clinical trials. A dose decrease down to 5 mg/day may be considered for patients who do not tolerate higher doses [see Clinical Studies (14)].
2.2 Screen for Bipolar Disorder Prior to Starting TRINTELLIX
Prior to initiating treatment with TRINTELLIX or another antidepressant, screen patients for personal or family history of bipolar disorder, mania, or hypomania [see Warnings and Precautions (5.4)].
2.3 Discontinuing Treatment
Although TRINTELLIX can be abruptly discontinued, in placebo-controlled trials patients experienced transient adverse reactions such as headache and muscle tension following abrupt discontinuation of TRINTELLIX 15 mg/day or 20 mg/day. It is recommended that the dose be decreased to 10 mg/day for one week before full discontinuation of TRINTELLIX 15 mg/day or 20 mg/day [see Warnings and Precautions (5.5) and Adverse Reactions (6)].
2.4 Switching a Patient to or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders
At least 14 days must elapse between discontinuation of a MAOI intended to treat psychiatric disorders and initiation of therapy with TRINTELLIX to avoid the risk of Serotonin Syndrome [see Warnings and Precautions (5.2)]. Conversely, at least 21 days must elapse after stopping TRINTELLIX before starting an MAOI intended to treat psychiatric disorders [see Contraindications (4)].
2.5 Use of TRINTELLIX in Known CYP2D6 Poor Metabolizers or in Patients Taking Strong CYP2D6 Inhibitors
The maximum recommended dose of TRINTELLIX is 10 mg/day in known CYP2D6 poor metabolizers. Reduce the dose of TRINTELLIX by one-half when patients are receiving a CYP2D6 strong inhibitor (e.g., bupropion, fluoxetine, paroxetine, or quinidine) concomitantly. The dose should be increased to the original level when the CYP2D6 inhibitor is discontinued [see Drug Interactions (7.1), Use in Specific Populations (8.6)].
2.6 Use of TRINTELLIX in Patients Taking Strong CYP Inducers
Consider increasing the dose of TRINTELLIX when a strong CYP inducer (e.g., rifampin, carbamazepine, or phenytoin) is coadministered for greater than 14 days. The maximum recommended dose should not exceed three times the original dose. The dose of TRINTELLIX should be reduced to the original level within 14 days, when the inducer is discontinued [see Drug Interactions (7.1)].
3. Dosage Forms and Strengths
TRINTELLIX is available as immediate-release, film-coated tablets in the following strengths:
- •
- 5 mg: pink, almond shaped biconvex film coated tablet, debossed with "5" on one side and "TL" on the other side
- •
- 10 mg: yellow, almond shaped biconvex film coated tablet, debossed with "10" on one side and "TL" on the other side
- •
- 20 mg: red, almond shaped biconvex film coated tablet, debossed with "20" on one side and "TL" on the other side
4. Contraindications
- •
- Hypersensitivity to vortioxetine or any component of the formulation. Hypersensitivity reactions including anaphylaxis, angioedema, and urticaria have been reported in patients treated with TRINTELLIX [see Adverse Reactions (6.2)].
- •
- The use of MAOIs intended to treat psychiatric disorders with TRINTELLIX or within 21 days of stopping treatment with TRINTELLIX is contraindicated because of an increased risk of serotonin syndrome. The use of TRINTELLIX within 14 days of stopping an MAOI intended to treat psychiatric disorders is also contraindicated [see Dosage and Administration (2.4), Warnings and Precautions (5.2)].
Starting TRINTELLIX in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome [see Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Suicidal Thoughts and Behaviors in Adolescents and Young Adults
In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and 4,500 pediatric patients, the incidence of suicidal thoughts and behaviors in antidepressant-treated patients age 24 years and younger was greater than in placebo-treated patients. There was considerable variation in risk of suicidal thoughts and behaviors among drugs, but there was an increased risk identified in young patients for most drugs studied. There were differences in absolute risk of suicidal thoughts and behaviors across the different indications, with the highest incidence in patients with MDD. The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1000 patients treated are provided in Table 1.
| Age Range | Drug-Placebo Difference in Number of Patients with Suicidal Thoughts and Behaviors per 1000 Patients Treated |
|---|---|
|
Increases Compared to Placebo |
|
|
<18 years old |
14 additional patients |
|
18-24 years old |
5 additional patients |
|
Decreases Compared to Placebo |
|
|
25-64 years old |
1 fewer patient |
|
≥65 years old |
6 fewer patients |
TRINTELLIX is not approved for use in pediatric patients.
It is unknown whether the risk of suicidal thoughts and behaviors in adolescents and young adults extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with MDD that the use of antidepressants can delay the recurrence of depression and that depression itself is a risk factor for suicidal thoughts and behaviors.
Monitor all antidepressant-treated patients for all approved populations for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of drug therapy, and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behavior and to alert the healthcare provider. Consider changing the therapeutic regimen, including possibly discontinuing TRINTELLIX, in patients whose depression is persistently worse, or who are experiencing emergent suicidal thoughts and behaviors.
5.2 Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome has been reported with serotonergic antidepressants including TRINTELLIX, when used alone but more often when used concomitantly with other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, and St. John's Wort), and with drugs that impair metabolism of serotonin (in particular, MAOIs, both those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue).
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome.
The concomitant use of TRINTELLIX with MAOIs intended to treat psychiatric disorders is contraindicated. TRINTELLIX should also not be started in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue. All reports with methylene blue that provided information on the route of administration involved intravenous administration in the dose range of 1 mg/kg to 8 mg/kg. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection) or at lower doses. There may be circumstances when it is necessary to initiate treatment with a MAOI such as linezolid or intravenous methylene blue in a patient taking TRINTELLIX. TRINTELLIX should be discontinued before initiating treatment with the MAOI [see Contraindications (4), Drug Interactions (7.1)].
If concomitant use of TRINTELLIX with other serotonergic drugs, including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, buspirone, tryptophan, and St. John's Wort is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome, particularly during treatment initiation and dose increases.
Treatment with TRINTELLIX and any concomitant serotonergic agents should be discontinued immediately if the above events occur and supportive symptomatic treatment should be initiated.
5.3 Increased Risk of Bleeding
The use of drugs that interfere with serotonin reuptake inhibition, including TRINTELLIX, may increase the risk of bleeding events. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), warfarin, and other anticoagulants may add to this risk. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. Bleeding events related to drugs that inhibit serotonin reuptake have ranged from ecchymosis, hematoma, epistaxis, and petechiae to life-threatening hemorrhages.
Inform patients about the increased risk of bleeding when TRINTELLIX is coadministered with NSAIDs, aspirin, or other drugs that affect coagulation or bleeding. For patients taking warfarin, carefully monitor coagulation indices when initiating, titrating, or discontinuing TRINTELLIX [see Drug Interactions (7.1)].
5.4 Activation of Mania/Hypomania
In patients with bipolar disorder, treating a depressive episode with TRINTELLIX or another antidepressant may precipitate a mixed/manic episode. Symptoms of mania/hypomania were reported in <0.1% of patients treated with TRINTELLIX in premarketing clinical studies. Prior to initiating treatment with TRINTELLIX, screen patients for any personal or family history of bipolar disorder, mania, or hypomania.
5.5. Discontinuation Syndrome
Adverse reactions have been reported upon abrupt discontinuation of treatment with TRINTELLIX at doses of 15 mg/day and 20 mg/day [see Adverse Reactions (6.1)]. A gradual reduction in dosage rather than abrupt cessation is recommended whenever possible [see Dosage and Administration (2.3)].
Adverse reactions after discontinuation of serotonergic antidepressants, particularly after abrupt discontinuation include: nausea, sweating, dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paresthesia, such as electric shock sensations), tremor, anxiety, confusion, headache, lethargy, emotional lability, insomnia, hypomania, tinnitus, and seizures.
5.6 Angle Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs, including TRINTELLIX, may trigger an angle closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy.
5.7 Hyponatremia
Hyponatremia has occurred as a result of treatment with serotonergic drugs, including TRINTELLIX. In many cases, hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH). One case with serum sodium lower than 110 mmol/L was reported in a subject treated with TRINTELLIX in a premarketing clinical study. Elderly patients may be at greater risk of developing hyponatremia with a serotonergic antidepressant. Also, patients taking diuretics or who are otherwise volume-depleted can be at greater risk. Discontinuation of TRINTELLIX in patients with symptomatic hyponatremia and appropriate medical intervention should be instituted. Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which can lead to falls. More severe and/or acute cases have included hallucination, syncope, seizure, coma, respiratory arrest, and death.
5.8 Sexual Dysfunction
Use of serotonergic antidepressants, including TRINTELLIX, may cause symptoms of sexual dysfunction [see Adverse Reactions (6.1)]. In male patients, serotonergic antidepressant use may result in ejaculatory delay or failure, decreased libido, and erectile dysfunction. In female patients, use may result in decreased libido and delayed or absent orgasm.
It is important for prescribers to inquire about sexual function prior to initiation of TRINTELLIX and to inquire specifically about changes in sexual function during treatment, because sexual function may not be spontaneously reported. When evaluating changes in sexual function, obtaining a detailed history (including timing of symptom onset) is important because sexual symptoms may have other causes, including the underlying psychiatric disorder. Discuss potential management strategies to support patients in making informed decisions about treatment.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the label.
- •
- Hypersensitivity [see Contraindications (4)]
- •
- Clinical Worsening and Suicide Risk [see Warnings and Precautions (5.1)]
- •
- Serotonin Syndrome [see Warnings and Precautions (5.2)]
- •
- Abnormal Bleeding [see Warnings and Precautions (5.3)]
- •
- Activation of Mania/Hypomania [see Warnings and Precautions (5.4)]
- •
- Discontinuation Syndrome [see Warnings and Precautions (5.5)]
- •
- Angle Closure Glaucoma [see Warnings and Precautions (5.6)]
- •
- Hyponatremia [see Warnings and Precautions (5.7)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Patient Exposure
TRINTELLIX was evaluated for safety in 5852 patients (18 years to 88 years of age) diagnosed with MDD who participated in pre- and postmarketing clinical studies; 2616 of those patients were exposed to TRINTELLIX in 6 to 8 week, placebo-controlled studies at doses ranging from 5 mg to 20 mg once daily; 204 patients were exposed to TRINTELLIX in a 24 to 64 week placebo-controlled maintenance study at doses of 5 mg to 10 mg once daily; and 429 patients were exposed to TRINTELLIX in a 32 week placebo-controlled maintenance study in the U.S. at doses of 5 mg, 10 mg, and 20 mg, once daily. Patients from the 6 to 8 week studies continued into 12-month open-label studies. A total of 2586 patients were exposed to at least one dose of TRINTELLIX in open-label studies, 1727 were exposed to TRINTELLIX for 6 months and 885 were exposed for at least 1 year.
Adverse Reactions Reported as Reasons for Discontinuation of Treatment
In pooled 6 to 8 week placebo-controlled studies the incidence of patients who received TRINTELLIX 5 mg/day, 10 mg/day, 15 mg/day and 20 mg/day and discontinued treatment because of an adverse reaction was 5%, 6%, 8% and 8%, respectively, compared to 4% of placebo-treated patients. Nausea was the most common adverse reaction reported as a reason for discontinuation.
Common Adverse Reactions in Placebo-Controlled MDD Studies
The most commonly observed adverse reactions in MDD patients treated with TRINTELLIX in 6 to 8 week placebo-controlled studies (incidence ≥5% and at least twice the rate of placebo) were nausea, constipation and vomiting.
Table 2 shows the incidence of common adverse reactions that occurred in ≥2% of MDD patients treated with any TRINTELLIX dose and at least 2% more frequently than in placebo-treated patients in the 6 to 8 week placebo-controlled studies.
| System Organ Class
Preferred Term | TRINTELLIX
5 mg/day | TRINTELLIX
10 mg/day | TRINTELLIX
15 mg/day | TRINTELLIX
20 mg/day | Placebo |
|---|---|---|---|---|---|
| N=1013
% | N=699
% | N=449
% | N=455
% | N=1621
% |
|
|
|||||
|
Gastrointestinal disorders |
|||||
|
Nausea |
21 |
26 |
32 |
32 |
9 |
|
Diarrhea |
7 |
7 |
10 |
7 |
6 |
|
Dry mouth |
7 |
7 |
6 |
8 |
6 |
|
Constipation |
3 |
5 |
6 |
6 |
3 |
|
Vomiting |
3 |
5 |
6 |
6 |
1 |
|
Flatulence |
1 |
3 |
2 |
1 |
1 |
|
Nervous system disorders |
|||||
|
Dizziness |
6 |
6 |
8 |
9 |
6 |
|
Psychiatric disorders |
|||||
|
Abnormal dreams |
<1 |
<1 |
2 |
3 |
1 |
|
Skin and subcutaneous tissue disorders |
|||||
|
Pruritus* |
1 |
2 |
3 |
3 |
1 |
Nausea
Nausea was the most common adverse reaction and its frequency was dose-related (Table 2). It was usually considered mild or moderate in intensity and the median duration was two weeks. Nausea was more common in females than males. Nausea most commonly occurred in the first week of TRINTELLIX treatment with 15 to 20% of patients experiencing nausea after one to two days of treatment. Approximately 10% of patients taking TRINTELLIX 10 mg/day to 20 mg/day had nausea at the end of the 6 to 8 week placebo-controlled studies.
Sexual Dysfunction
Difficulties in sexual desire, sexual performance and sexual satisfaction often occur as manifestations of psychiatric disorders or comorbid conditions, but they may also be consequences of pharmacologic treatment, including TRINTELLIX. In addition to the data from the MDD studies mentioned below, TRINTELLIX has been prospectively assessed for its effects in MDD patients with existing TESD induced by prior SSRI treatment and in healthy adults with normal sexual function at baseline [see Clinical Studies (14)].
Voluntarily Reported Adverse Reactions of Sexual Dysfunction
In the MDD 6 to 8 week controlled trials of TRINTELLIX, voluntarily reported adverse reactions related to sexual dysfunction were captured as individual event terms. These event terms have been aggregated and the overall incidence was as follows. In male patients the overall incidence was 3%, 4%, 4%, 5% in TRINTELLIX 5 mg/day, 10 mg/day, 15 mg/day, 20 mg/day, respectively, compared to 2% in placebo. In female patients, the overall incidence was <1%, 1%, <1%, 2% in TRINTELLIX 5 mg/day, 10 mg/day, 15 mg/day, 20 mg/day, respectively, compared to <1% in placebo.
Adverse Reactions of Sexual Dysfunction in Patients with Normal Sexual Functioning at Baseline
Because voluntarily reported adverse sexual reactions are known to be underreported, in part because patients and physicians may be reluctant to discuss them, the Arizona Sexual Experiences Scale (ASEX), a validated measure designed to identify sexual side effects, was used prospectively in seven placebo-controlled trials. The ASEX scale includes five questions that pertain to the following aspects of sexual function: 1) sex drive, 2) ease of arousal, 3) ability to achieve erection (men) or lubrication (women), 4) ease of reaching orgasm, and 5) orgasm satisfaction.
The presence or absence of sexual dysfunction among patients entering clinical studies was based on their self-reported ASEX scores. For patients without sexual dysfunction at baseline (approximately 1/3 of the population across all treatment groups in each study), Table 3 shows the incidence of patients that developed TESD when treated with TRINTELLIX or placebo in any fixed dose group. Physicians should routinely inquire about possible sexual side effects.
| TRINTELLIX
5 mg/day N=65:67† | TRINTELLIX
10 mg/day N=94:86† | TRINTELLIX
15 mg/day N=57:67† | TRINTELLIX
20 mg/day N=67:59† | Placebo
N=135:162† |
|
|---|---|---|---|---|---|
|
|||||
|
Females |
22% |
23% |
33% |
34% |
20% |
|
Males |
16% |
20% |
19% |
29% |
14% |
Adverse Reactions Following Abrupt Discontinuation of TRINTELLIX Treatment
Discontinuation symptoms have been prospectively evaluated in patients taking TRINTELLIX 10 mg/day, 15 mg/day, and 20 mg/day using the Discontinuation-Emergent Signs and Symptoms (DESS) scale in clinical trials. Some patients experienced discontinuation symptoms such as headache, muscle tension, mood swings, sudden outbursts of anger, dizziness, and runny nose in the first week of abrupt discontinuation of TRINTELLIX 15 mg/day and 20 mg/day.
Laboratory Tests
TRINTELLIX has not been associated with any clinically important changes in laboratory test parameters in serum chemistry (except sodium), hematology and urinalysis as measured in the 6 to 8 week placebo-controlled studies. Hyponatremia has been reported with the treatment of TRINTELLIX [see Warnings and Precautions (5.7)]. In the 6-month, double-blind, placebo-controlled phase of a long-term study in patients who had responded to TRINTELLIX during the initial 12 week, open-label phase, there were no clinically important changes in lab test parameters between TRINTELLIX and placebo-treated patients.
Weight
TRINTELLIX had no significant effect on body weight as measured by the mean change from baseline in the 6 to 8 week placebo-controlled studies. In the six month, double-blind, placebo-controlled phase of a long-term study in patients who had responded to TRINTELLIX during the initial 12-week, open-label phase, there was no significant effect on body weight between TRINTELLIX and placebo-treated patients.
Vital Signs
TRINTELLIX has not been associated with any clinically significant effects on vital signs, including systolic and diastolic blood pressure and heart rate, as measured in placebo-controlled studies.
Other Adverse Reactions Observed in Clinical Studies
The following listing does not include reactions: 1) already listed in previous tables or elsewhere in labeling, 2) for which a drug cause was remote, 3) which were so general as to be uninformative, 4) which were not considered to have significant clinical implications, or 5) which occurred at a rate equal to or less than placebo.
Ear and labyrinth disorders — vertigo
Gastrointestinal disorders — dyspepsia
Nervous system disorders — dysgeusia
Vascular disorders — flushing
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of TRINTELLIX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Endocrine disorders — hyperprolactinemia
Gastrointestinal System — acute pancreatitis
Immune system disorders — hypersensitivity reactions (including anaphylaxis and urticaria)
Metabolic disorders — weight gain
Nervous system disorders — seizure, headache
Psychiatric disorders — aggression, agitation, anger, hostility, irritability
Skin and subcutaneous tissue disorders — rash, generalized rash, hyperhidrosis
7. Drug Interactions
7.1 Drugs Having Clinically Important Interactions with TRINTELLIX
|
Monoamine Oxidase Inhibitors (MAOIs) |
|
|
Clinical Impact |
The concomitant use of SSRIs and SNRIs including TRINTELLIX with MAOIs increases the risk of serotonin syndrome. |
|
Intervention |
Concomitant use of TRINTELLIX is contraindicated:
[see Dosage and Administration (2.4), Contraindications (4), Warnings and Precautions (5.2)]. |
|
Examples |
selegiline, tranylcypromine, isocarboxazid, phenelzine, linezolid, methylene blue |
|
Other Serotonergic Drugs |
|
|
Clinical Impact |
Concomitant use of TRINTELLIX with other serotonergic drugs increases the risk of serotonin syndrome. |
|
Intervention |
Monitor for symptoms of serotonin syndrome when TRINTELLIX is used concomitantly with other drugs that may affect the serotonergic neurotransmitter systems. If serotonin syndrome occurs, consider discontinuation of TRINTELLIX and/or concomitant serotonergic drugs [see Warnings and Precautions (5.2)]. |
|
Examples |
Other SNRIs, SSRIs, triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, buspirone, amphetamines, tryptophan, and St. John's Wort |
|
Strong Inhibitors of CYP2D6 |
|
|
Clinical Impact |
Concomitant use of TRINTELLIX with strong CYP2D6 inhibitors increases plasma concentrations of vortioxetine. |
|
Intervention |
Reduce TRINTELLIX dose by half when a strong CYP2D6 inhibitor is coadministered [see Dosage and Administration (2.5)]. |
|
Examples |
bupropion, fluoxetine, paroxetine, quinidine |
|
Strong CYP Inducers |
|
|
Clinical Impact |
Concomitant use of TRINTELLIX with a strong CYP inducer decreases plasma concentrations of vortioxetine. |
|
Intervention |
Consider increasing the TRINTELLIX dose when a strong CYP inducer is coadministered. The maximum dose is not recommended to exceed three times the original dose [see Dosage and Administration (2.6)]. |
|
Examples |
rifampin, carbamazepine, phenytoin |
|
Drugs that Interfere with Hemostasis (antiplatelets agents and anticoagulants) |
|
|
Clinical Impact |
Concomitant use of TRINTELLIX with an antiplatelet or anticoagulant drug may potentiate the risk of bleeding. |
|
Intervention |
Inform patients of the increased risk of bleeding associated with the concomitant use of TRINTELLIX and antiplatelet agents and anticoagulants. For patients taking warfarin, carefully monitor the international normalized ratio [see Warnings and Precautions (5.3), Drug Interactions (7.2)]. |
|
Examples |
aspirin, clopidogrel, heparin, warfarin |
|
Drugs Highly Bound to Plasma Protein |
|
|
Clinical Impact |
TRINTELLIX is highly bound to plasma protein. The concomitant use of TRINTELLIX with another drug that is highly bound to plasma protein may increase free concentrations of TRINTELLIX or other tightly-bound drugs in plasma. |
|
Intervention |
Monitor for adverse reactions and reduce dosage of TRINTELLIX or other protein bound drugs as warranted [see Drug Interactions (7.2)]. |
|
Examples |
Warfarin |
7.2 Effect of TRINTELLIX on Other Drugs
Other CNS Active Agents
No clinically relevant effect was observed on steady-state lithium exposure following coadministration with multiple daily doses of TRINTELLIX. Multiple doses of TRINTELLIX did not affect the pharmacokinetics or pharmacodynamics (composite cognitive score) of diazepam [see Clinical Pharmacology (12.3)].
A clinical study has shown that TRINTELLIX (single dose of 20 or 40 mg) did not increase the impairment of mental and motor skills caused by alcohol (single dose of 0.6 g/kg) [see Clinical Pharmacology (12.3)].
Drugs That Interfere with Hemostasis
Following coadministration of stable doses of warfarin (1 to 10 mg/day) with multiple daily doses of TRINTELLIX, no significant effects were observed in INR, prothrombin values or total warfarin (protein bound plus free drug) pharmacokinetics for both R- and S-warfarin. Coadministration of aspirin 150 mg/day with multiple daily doses of TRINTELLIX had no significant inhibitory effect on platelet aggregation or pharmacokinetics of aspirin and salicylic acid [see Clinical Pharmacology (12.3)]. Patients receiving other drugs that interfere with hemostasis should be carefully monitored when TRINTELLIX is initiated or discontinued [see Warnings and Precautions (5.3), Drug Interactions (7.1)].
7.3 Interference with Urine Enzyme Immunoassays for Methadone
False positive results in urine enzyme immunoassays for methadone have been reported in patients who have taken vortioxetine. An alternative analytical technique (e.g., chromatographic methods) should be considered to confirm positive methadone urine drug screen results.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antidepressants during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Antidepressants at 1-844-405-6185 or visiting online at https://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/antidepressants/.
Risk Summary
There are limited human data on TRINTELLIX use during pregnancy to inform any drug-associated risks. However, there are clinical considerations regarding neonates exposed to SSRIs and SNRIs, including TRINTELLIX, during the third trimester of pregnancy [see Clinical Considerations]. Vortioxetine administered to pregnant rats and rabbits during the period of organogenesis at doses ≥15 times and 10 times the maximum recommended human dose (MRHD), respectively, resulted in decreased fetal body weight and delayed ossification. No malformations were seen at doses up to 77 times and 58 times the MRHD, respectively. Vortioxetine administered to pregnant rats during gestation and lactation at oral doses ≥20 times the MRHD resulted in a decrease in the number of live-born pups and an increase in early postnatal pup mortality. Decreased pup weight at birth to weaning occurred at 58 times the MRHD and delayed physical development occurred at ≥20 times the MRHD. These effects were not seen at 5 times the MRHD [see Data]. Advise a pregnant woman of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
A prospective, longitudinal study followed 201 pregnant women with a history of major depressive disorder who were euthymic and taking antidepressants at the beginning of pregnancy. The women who discontinued antidepressants during pregnancy were more likely to experience a relapse of major depression than women who continued antidepressants. Consider the risks of untreated depression when discontinuing or changing treatment with antidepressant medication during pregnancy and postpartum.
Fetal/Neonatal adverse reactions
Exposure to serotonergic antidepressants, including TRINTELLIX, in late pregnancy may lead to an increased risk for neonatal complications requiring prolonged hospitalization, respiratory support, and tube feeding, and/or persistent pulmonary hypertension of the newborn (PPHN). Monitor neonates who were exposed to TRINTELLIX in the third trimester of pregnancy for PPHN and drug discontinuation syndrome [see Data].
Data
Human Data
Third Trimester Exposure
Neonates exposed to SSRIs or SNRIs, late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support and tube feeding. These findings are based on postmarketing reports. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremor, jitteriness, irritability and constant crying. These features are consistent with either a direct toxic effect of SSRIs and SNRIs or possibly, a drug discontinuation syndrome. In some cases, the clinical picture was consistent with serotonin syndrome [see Warnings and Precautions (5.2)].
Exposure during late pregnancy to SSRIs may have an increased risk for persistent pulmonary hypertension of the newborn (PPHN). PPHN occurs in one to two per 1,000 live births in the general population and is associated with substantial neonatal morbidity and mortality. In a retrospective case-control study of 377 women whose infants were born with PPHN and 836 women whose infants were born healthy, the risk for developing PPHN was approximately six fold higher for infants exposed to SSRIs after the 20th week of gestation compared to infants who had not been exposed to antidepressants during pregnancy. A study of 831,324 infants born in Sweden in 1997 - 2005 found a PPHN risk ratio of 2.4 (95% CI 1.2-4.3) associated with patient-reported maternal use of SSRIs "in early pregnancy" and a PPHN risk ratio of 3.6 (95% CI 1.2-8.3) associated with a combination of patient-reported maternal use of SSRIs "in early pregnancy" and an antenatal SSRI prescription "in later pregnancy."
Animal Data
In pregnant rats and rabbits, no malformations were seen when vortioxetine was given during the period of organogenesis at oral doses up to 160 and 60 mg/kg/day, respectively. These doses are 77 and 58 times the maximum recommended human dose (MRHD) of 20 mg on a mg/m2 basis, in rats and rabbits, respectively. Developmental delay, seen as decreased fetal body weight and delayed ossification, occurred in rats and rabbits at doses equal to and greater than 30 and 10 mg/kg (15 and 10 times the MRHD, respectively) in the presence of maternal toxicity (decreased food consumption and decreased body weight gain). When vortioxetine was administered to pregnant rats at oral doses of 40 and 120 mg/kg (20 and 58 times the MRHD, respectively) throughout pregnancy and lactation, the number of live-born pups was decreased and early postnatal pup mortality was increased. Additionally, pup weights were decreased at birth to weaning at 120 mg/kg and development (specifically eye opening) was slightly delayed at 40 and 120 mg/kg. These effects were not seen at 10 mg/kg (5 times the MRHD).
8.2 Lactation
Risk Summary
There is no information regarding the presence of vortioxetine in human milk, the effects on the breastfed infant, or the effects on milk production. Vortioxetine is present in rat milk [see Data]. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TRINTELLIX and any potential adverse effects on the breastfed child from TRINTELLIX or from the underlying maternal condition.
Data
Animal Data
Administration of [14C]-vortioxetine to lactating rats at an oral dose of 20 times the maximum recommended human dose (MRHD) of 20 mg on a mg/m2 basis, resulted in drug-related material in milk secretion. Milk to plasma ratio in lactating rats was 1, 1.2, 0.5, and 0.5 at 2, 6, 24, and 72 hours post dose.
8.4 Pediatric Use
The safety and effectiveness of TRINTELLIX have not been established in pediatric patients for the treatment of MDD.
Efficacy was not established in an 8-week, randomized, double-blind, placebo-controlled, active-reference study in 615 pediatric patients 12 to 17 years of age with MDD. The primary efficacy endpoint was change from double-blind baseline to Week 8 on the Children's Depression Rating Scale-Revised version. The effect of treatment with vortioxetine was not significantly different from placebo (placebo-subtracted difference of 0.21 (95% CI: -2.41, 2.82; p=0.88). In this age group, adverse reactions to TRINTELLIX were generally similar to those reported in adults.
Antidepressants, such as TRINTELLIX, increase the risk of suicidal thoughts and behaviors in pediatric patients [see the Boxed Warning and Warnings and Precautions (5.1)].
Juvenile Animal Toxicity Data
Administration of vortioxetine to juvenile rats (oral doses of 10, 20, and 40 mg/kg/day twice daily from Postnatal Day 21 to 91) resulted in a neurobehavioral effect at the highest dose of 40 mg/kg twice daily (increased peak auditory startle amplitude) during the treatment period. The effect was not seen at the end of the recovery period. When animals were mated after the 4-week recovery period, viability was decreased in the offspring of mated pairs treated with 40 mg/kg twice daily. The no-observed adverse effect dose was 20 mg/kg twice daily based on both the neurobehavioral and reproductive effects. This dose was associated with plasma vortioxetine exposure (AUC) approximately 2 times that in pediatric patients.
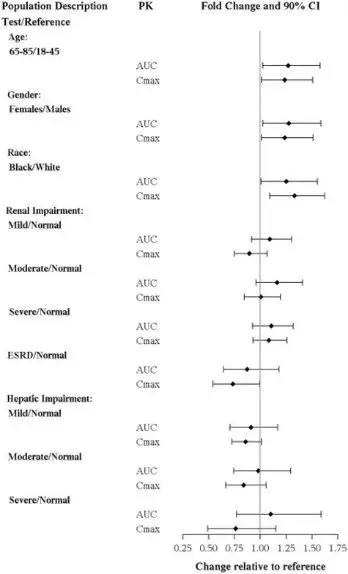
8.5 Geriatric Use
No dose adjustment is recommended on the basis of age (Figure 1). Results from a single-dose pharmacokinetic study in elderly (>65 years old) vs young (24 to 45 years old) subjects demonstrated that the pharmacokinetics were generally similar between the two age groups.
Of the 2616 subjects in clinical studies of TRINTELLIX, 11% (286) were 65 and over, which included subjects from a placebo-controlled study specifically in elderly patients [see Clinical Studies (14)]. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients.
Serotonergic antidepressants have been associated with cases of clinically significant hyponatremia in elderly patients, who may be at greater risk for this adverse event [see Warnings and Precautions (5.7)].
10. Overdosage
10.1 Human Experience
There is limited clinical trial experience regarding human overdosage with TRINTELLIX. In premarketing clinical studies, cases of overdose were limited to patients who accidentally or intentionally consumed up to a maximum dose of 40 mg of TRINTELLIX. The maximum single dose tested was 75 mg in men. Ingestion of TRINTELLIX in the dose range of 40 to 75 mg was associated with increased rates of nausea, dizziness, diarrhea, abdominal discomfort, generalized pruritus, somnolence, and flushing.
There have been postmarketing reports of overdoses of TRINTELLIX. The most frequently reported symptoms with overdoses up to 80 mg (four times the maximum recommended daily dose) were nausea and vomiting. With overdoses greater than 80 mg, a case of serotonin syndrome in combination with another serotonergic drug, and a case of seizure, have been reported.
11. Trintellix Description
TRINTELLIX is an immediate-release tablet for oral administration that contains the beta (β) polymorph of vortioxetine hydrobromide (HBr), an antidepressant. Vortioxetine HBr is known chemically as 1-[2-(2,4-Dimethyl-phenylsulfanyl)-phenyl]-piperazine, hydrobromide. The empirical formula is C18 H22 N2 S, HBr with a molecular weight of 379.36 g/mol. The structural formula is:
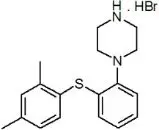
Vortioxetine HBr is a white to very slightly beige powder that is slightly soluble in water.
Each TRINTELLIX tablet contains 6.355 mg, 12.71 mg or 25.42 mg of vortioxetine HBr equivalent to 5 mg, 10 mg, or 20 mg of vortioxetine, respectively. The inactive ingredients in TRINTELLIX tablets include mannitol, microcrystalline cellulose, hydroxypropyl cellulose, sodium starch glycolate, magnesium stearate and film coating which consists of hypromellose, titanium dioxide, polyethylene glycol 400, iron oxide red (5 mg and 20 mg) and iron oxide yellow (10 mg).
12. Trintellix - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of the antidepressant effect of vortioxetine is not fully understood, but is thought to be related to its enhancement of serotonergic activity in the CNS through inhibition of the reuptake of serotonin (5-HT). It also has several other activities including 5-HT3 receptor antagonism and 5-HT1A receptor agonism. The contribution of these activities to vortioxetine's antidepressant effect has not been established.
12.2 Pharmacodynamics
Vortioxetine binds with high affinity to the human serotonin transporter (Ki=1.6 nM), but not to the norepinephrine (Ki=113 nM) or dopamine (Ki>1000 nM) transporters. Vortioxetine potently and selectively inhibits reuptake of serotonin (IC50=5.4 nM). Vortioxetine binds to 5-HT3 (Ki=3.7 nM), 5-HT1A (Ki=15 nM), 5-HT7 (Ki=19 nM), 5-HT1D (Ki=54 nM), and 5-HT1B (Ki=33 nM), receptors and is a 5-HT3, 5-HT1D, and 5-HT7 receptor antagonist, 5-HT1B receptor partial agonist, and 5-HT1A receptor agonist.
In humans, the mean 5-HT transporter occupancy, based on the results from two clinical PET studies using 5-HTT ligands ([11C]-MADAM or [11C]-DASB), was approximately 50% at 5 mg/day, 65% at 10 mg/day and approximately 80% at 20 mg/day in the regions of interest.
Effect on Cardiac Repolarization
The effect of vortioxetine 10 mg and 40 mg administered once daily on QTc interval was evaluated in a randomized, double-blind, placebo-, and active-controlled (moxifloxacin 400 mg), four-treatment-arm parallel study in 340 male subjects. In the study the upper bound of the one-sided 95% confidence interval for the QTc was below 10 ms, the threshold for regulatory concern. The oral dose of 40 mg is sufficient to assess the effect of metabolic inhibition.
12.3 Pharmacokinetics
Vortioxetine pharmacological activity is due to the parent drug. The pharmacokinetics of vortioxetine (2.5 mg to 60 mg) are linear and dose-proportional when vortioxetine is administered once daily. The mean terminal half-life is approximately 66 hours, and steady-state plasma concentrations are typically achieved within two weeks of dosing.
Absorption
The maximal plasma vortioxetine concentration (Cmax) after dosing is reached within 7 to 11 hours postdose (Tmax). Steady-state mean Cmax values were 9, 18, and 33 ng/mL following doses of 5, 10, and 20 mg/day. Absolute bioavailability is 75%.
Distribution
The apparent volume of distribution of vortioxetine is approximately 2600 L, indicating extensive extravascular distribution. The plasma protein binding of vortioxetine in humans is 98%, independent of plasma concentrations. No apparent difference in the plasma protein binding between healthy subjects and subjects with hepatic (mild, moderate or severe) or renal (mild, moderate, severe, ESRD) impairment is observed.
Elimination
Metabolism
Vortioxetine is extensively metabolized primarily through oxidation via cytochrome P450 isozymes CYP2D6, CYP3A4/5, CYP2C19, CYP2C9, CYP2A6, CYP2C8 and CYP2B6 and subsequent glucuronic acid conjugation. CYP2D6 is the primary enzyme catalyzing the metabolism of vortioxetine to its major, pharmacologically inactive, carboxylic acid metabolite, and poor metabolizers of CYP2D6 have approximately twice the vortioxetine plasma concentration of extensive metabolizers [see Dosage and Administration (2.5)].
Excretion
Following a single oral dose of [14C]-labeled vortioxetine, approximately 59% and 26% of the administered radioactivity was recovered in the urine and feces, respectively as metabolites. Negligible amounts of unchanged vortioxetine were excreted in the urine up to 48 hours. The presence of hepatic (mild, moderate or severe) or renal impairment (mild, moderate, severe and ESRD) did not affect the apparent clearance of vortioxetine.
Specific Populations
No clinically significant differences in the exposures of vortioxetine were observed based on age, gender, ethnicity, renal function, or hepatic function.
The effects of intrinsic patient factors on the pharmacokinetics of vortioxetine are presented in Figure 1.
Figure 1. Impact of Intrinsic Factors on Vortioxetine PK
Drug Interaction Studies
Clinical Studies
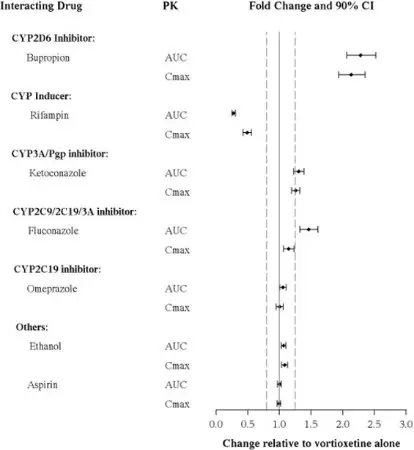
Other Drugs on TRINTELLIX
The effects of other drugs on vortioxetine exposure are summarized in Figure 2.
Figure 2. Impact of Other Drugs on Vortioxetine PK
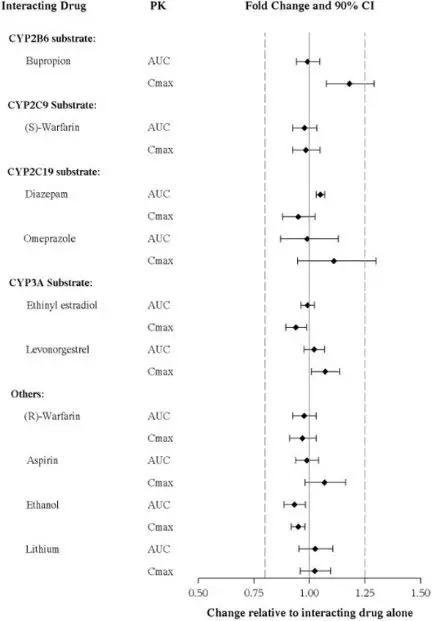
TRINTELLIX on Other Drugs
The effects of vortioxetine on the exposures of other drugs are summarized in Figure 3.
Figure 3. Impact of Vortioxetine on PK of Other Drugs
In Vitro
Vortioxetine and its metabolite(s) are unlikely to inhibit the following CYP enzymes and transporter based on in vitro data: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, P-gp, BCRP, BSEP, MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1 and OCT2. As such, no clinically relevant interactions with drugs metabolized/transported by these CYP enzymes or transporters would be expected.
In addition, vortioxetine did not induce CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4/5 in an in vitro study in cultured human hepatocytes. Chronic administration of TRINTELLIX is unlikely to induce the metabolism of drugs metabolized by these CYP isoforms. Furthermore, in a series of clinical drug interaction studies, coadministration of TRINTELLIX with substrates for CYP2B6 (e.g., bupropion), CYP2C9 (e.g., warfarin), and CYP2C19 (e.g., diazepam), had no clinical meaningful effect on the pharmacokinetics of these substrates.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies were conducted in which CD-1 mice and Wistar rats were given oral doses of vortioxetine up to 50 and 100 mg/kg/day for male and female mice, respectively, and 40 and 80 mg/kg/day for male and female rats, respectively, for two years. The doses in the two species were approximately 12, 24, 20, and 39 times, respectively, the maximum recommended human dose (MRHD) of 20 mg on a mg/m2 basis.
In rats, the incidence of benign polypoid adenomas of the rectum was statistically significantly increased in females at doses 39 times the MRHD, but not at 15 times the MRHD. These were considered related to inflammation and hyperplasia and possibly caused by an interaction with a vehicle component of the formulation used for the study. The finding did not occur in male rats at 20 times the MRHD.
In mice, vortioxetine was not carcinogenic in males or females at doses up to 12 and 24 times, respectively, the MRHD.
14. Clinical Studies
The efficacy of TRINTELLIX in treatment for MDD was established in six, 6 to 8 week randomized, double-blind, placebo-controlled, fixed-dose studies (including one study in the elderly) and one maintenance study in adult inpatients and outpatients who met the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR) criteria for MDD.
Adults (aged 18 years to 75 years)
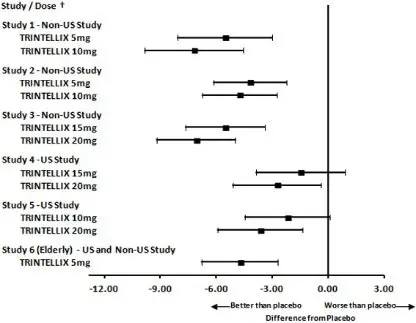
The efficacy of TRINTELLIX in patients aged 18 years to 75 years was demonstrated in five, 6 to 8 week, placebo-controlled studies (Studies 1 to 5 in Table 5). In these studies, patients were randomized to TRINTELLIX 5 mg, 10 mg, 15 mg or 20 mg or placebo once daily. For patients who were randomized to TRINTELLIX 15 mg/day or 20 mg/day, the final doses were titrated up from 10 mg/day after the first week.
The primary efficacy measures were the Hamilton Depression Scale (HAMD-24) total score in Study 2 and the Montgomery-Asberg Depression Rating Scale (MADRS) total score in all other studies. In each of these studies, at least one dose group of TRINTELLIX was superior to placebo in improvement of depressive symptoms as measured by mean change from baseline to endpoint visit on the primary efficacy measurement (see Table 5). Subgroup analysis by age, gender or race did not suggest any clear evidence of differential responsiveness. Two studies of the 5 mg dose in the U.S. (not represented in Table 5) failed to show effectiveness.
Elderly Study (aged 64 years to 88 years)
The efficacy of TRINTELLIX for the treatment of MDD was also demonstrated in a randomized, double-blind, placebo-controlled, fixed-dose study of TRINTELLIX in elderly patients (aged 64 years to 88 years) with MDD (Study 6 in Table 5). Patients meeting the diagnostic criteria for recurrent MDD with at least one previous major depressive episode before the age of 60 years and without comorbid cognitive impairment (Mini Mental State Examination score <24) received TRINTELLIX 5 mg or placebo.
| Study No.
[Primary Measure] | Treatment Group | Number of Patients | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference* (95% CI) |
|---|---|---|---|---|---|
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: unadjusted confidence interval. | |||||
|
|||||
|
Study 1 |
TRINTELLIX (5 mg/day)† |
108 |
34.1 (2.6) |
-20.4 (1.0) |
-5.9 (-8.6, -3.2) |
|
TRINTELLIX (10 mg/day)† |
100 |
34.0 (2.8) |
-20.2 (1.0) |
-5.7 (-8.5, -2.9) |
|
|
Placebo |
105 |
33.9 (2.7) |
-14.5 (1.0) |
-- |
|
|
Study 2 |
TRINTELLIX (5 mg/day) |
139 |
32.2 (5.0) |
-15.4 (0.7) |
-4.1 (-6.2, -2.1) |
|
TRINTELLIX (10 mg/day)† |
139 |
33.1 (4.8) |
-16.2 (0.8) |
-4.9 (-7.0, -2.9) |
|
|
Placebo |
139 |
32.7 (4.4) |
-11.3 (0.7) |
-- |
|
|
Study 3 |
TRINTELLIX (15 mg/day) † |
149 |
31.8 (3.4) |
-17.2 (0.8) |
-5.5 (-7.7, -3.4) |
|
TRINTELLIX (20 mg/day) † |
151 |
31.2 (3.4) |
-18.8 (0.8) |
-7.1 (-9.2, -5.0) |
|
|
Placebo |
158 |
31.5 (3.6) |
-11.7 (0.8) |
-- |
|
|
Study 4 |
TRINTELLIX (15 mg/day) |
145 |
31.9 (4.1) |
-14.3 (0.9) |
-1.5 (-3.9, 0.9) |
|
TRINTELLIX (20 mg/day) † |
147 |
32.0 (4.4) |
-15.6 (0.9) |
-2.8 (-5.1, -0.4) |
|
|
Placebo |
153 |
31.5 (4.2) |
-12.8 (0.8) |
-- |
|
|
Study 5 |
TRINTELLIX (10 mg/day) |
154 |
32.2 (4.5) |
-13.0 (0.8) |
-2.2 (-4.5, 0.1) |
|
TRINTELLIX (20 mg/day) † |
148 |
32.5 (4.3) |
-14.4 (0.9) |
-3.6 (-5.9, -1.4) |
|
|
Placebo |
155 |
32.0 (4.0) |
-10.8 (0.8) |
-- |
|
|
Study 6 (elderly) |
TRINTELLIX (5 mg/day) † |
155 |
29.2 (5.0) |
-13.7 (0.7) |
-3.3 (-5.3, -1.3) |
|
Placebo |
145 |
29.4 (5.1) |
-10.3 (0.8) |
-- |
|
TRINTELLIX was superior to placebo on the Clinical Global Impression of Improvement (CGI-I) scale, which is a clinician's impression of how much the patient's clinical condition has improved or worsened relative to baseline on a scale of 1 (very much improved) to 7 (very much worse).
Time Course of Treatment Response
In the 6 to 8 week placebo-controlled studies, an effect of TRINTELLIX based on the primary efficacy measure was generally observed starting at Week 2 and increased in subsequent weeks with the full antidepressant effect of TRINTELLIX generally not seen until Study Week 4 or later. Figure 4 depicts time course of response in U.S. based on the primary efficacy measure (MADRS) in Study 5.
Figure 4. Change from Baseline in MADRS Total Score by Study Visit (Week) in Study 5
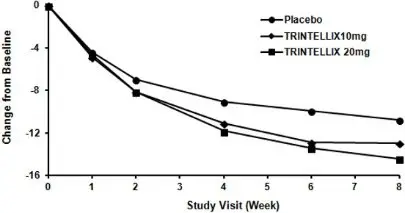
Figure 5. Difference from Placebo in Mean Change from Baseline in MADRS Total Score at Week 6 or Week 8
| † Results (point estimate and unadjusted 95% confidence interval) are from mixed model for repeated measures (MMRM) analysis. In Studies 1 and 6, the primary analysis was not based on MMRM and in Studies 2 and 6 the primary efficacy measure was not based on MADRS |
 |
Digit Symbol Substitution Test in Major Depressive Disorder
Two, eight week, randomized, double-blind, placebo-controlled studies were conducted to evaluate the effect of TRINTELLIX on the Digit Symbol Substitution Test (DSST) during the treatment of acute MDD. The DSST is a neuropsychological test that most specifically measures processing speed, an aspect of cognitive function that may be impaired in MDD. Patients are asked to match nine symbols with their corresponding number (1 to 9) according to a key; the score is the correct number of matches achieved in 90 seconds. For reference, the mean score for healthy 45 to 54 year-old subjects is 50 (SD=15).
Study 7 randomized adult patients meeting the diagnostic criteria for recurrent MDD to receive TRINTELLIX 10 mg, TRINTELLIX 20 mg, or placebo once daily. Study 8 randomized adult patients meeting the diagnostic criteria for recurrent MDD and reporting subjective difficulty concentrating or slow thinking to receive a flexible dose of TRINTELLIX (10 or 20 mg) or placebo once daily. Neither study included patients whose MDD was in remission yet who continued to experience difficulty concentrating or slow thinking. Patients' mean age was 46 (SD=12) and 45 (SD=12) in Study 7 and 8, respectively. In both studies, patients in the TRINTELLIX group had a statistically significantly greater improvement in number of correct responses on the DSST (Table 6); depressed mood as assessed by change from baseline in MADRS total score also improved in both studies.
| Study No. | Treatment Group | Number of Patients | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference
* (95% CI) |
|---|---|---|---|---|---|
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: unadjusted confidence interval. | |||||
|
|||||
|
Study 7 |
TRINTELLIX |
193 |
42.0 (12.6) |
9.0 (0.6) |
4.2 (2.5, 5.9) |
|
TRINTELLIX |
204 |
41.6 (12.7) |
9.1 (0.6) |
4.3 (2.6, 5.9) |
|
|
Placebo |
194 |
42.4 (13.8) |
4.8 (0.6) |
-- |
|
|
Study 8 |
TRINTELLIX |
175 |
42.1 (11.9) |
4.6 (0.5) |
1.8 (0.3, 3.2) |
|
Placebo |
167 |
43.0 (12.3) |
2.9 (0.5) |
-- |
|
The effects observed on DSST may reflect improvement in depression. Comparative studies have not been conducted to demonstrate a therapeutic advantage over other antidepressants on the DSST.
Maintenance Studies
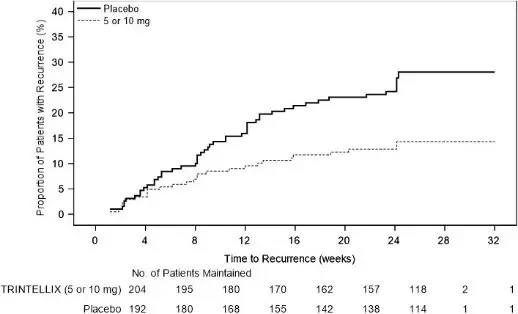
In a non-US maintenance study (Study 9 in Figure 6), 639 patients meeting DSM-IV-TR criteria for MDD received flexible doses of TRINTELLIX (5 mg or 10 mg) once daily during an initial 12 week open-label treatment phase; the dose of TRINTELLIX was fixed during Weeks 8 to 12. Three hundred ninety six (396) patients who were in remission (MADRS total score ≤10 at both Weeks 10 and 12) after open-label treatment were randomly assigned to continuation of a fixed dose of TRINTELLIX at the final dose they responded to (about 75% of patients were on 10 mg/day) during the open-label phase or to placebo for 24 to 64 weeks. Approximately 61% of randomized patients satisfied remission criterion (MADRS total score ≤10) for at least four weeks (since Week 8), and 15% for at least eight weeks (since Week 4). Patients on TRINTELLIX experienced a statistically significantly longer time to have recurrence of depressive episodes than did patients on placebo. Recurrence of depressive episode was defined as a MADRS total score ≥22 or lack of efficacy as judged by the investigator.
Figure 6. Kaplan-Meier Estimates of Proportion of Patients with Recurrence (Study 9)
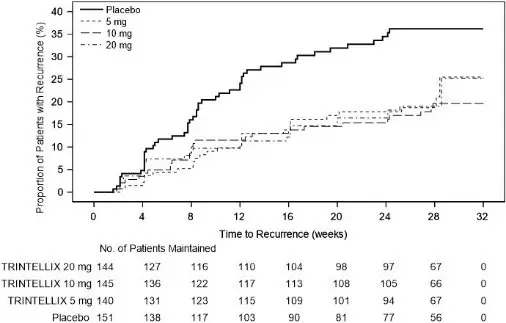
In a U.S.-based maintenance study (Study 10 in Figure 7), 1106 patients meeting DSM-IV-TR criteria for MDD were treated with a fixed dose of TRINTELLIX 10 mg once daily during an initial 16 week open-label treatment phase. Five hundred and eighty (580) patients who were in remission (MADRS total score ≤12 at both Weeks 14 and 16) after open-label treatment were randomized in a 1:1:1:1 ratio to TRINTELLIX 5 mg/day, 10 mg/day, 20 mg/day, or placebo daily for 32 weeks. The definition of recurrence of depressive episodes was the same as for Study 9. For all three doses of TRINTELLIX evaluated, patients treated with TRINTELLIX experienced a statistically significantly longer time to recurrence of depressive episodes than did patients treated with placebo.
Figure 7. Kaplan-Meier Estimates of Proportion of Patients with Recurrence (Study 10)
Prospective Evaluation of Treatment Emergent Sexual Dysfunction (TESD)
Two, randomized, double-blind, active-controlled studies were conducted to prospectively compare the incidence of TESD between TRINTELLIX and SSRIs via a validated self-rated measure of sexual function, the Changes in Sexual Functioning Questionnaire Short Form (CSFQ-14). The CSFQ-14 is designed to measure illness- and medication-related changes in sexual functioning that consists of 14 items measuring sexual functioning as a total score. The CSFQ-14 consists of subscales that assess the three phases of the sexual response cycle (desire, arousal, and orgasm). Higher scores on the CSFQ-14 indicate greater sexual function and for reference, a 2-3 point change is considered clinically meaningful.
Effect of Switching from SSRI to TRINTELLIX on TESD
The effect of TRINTELLIX on TESD induced by prior SSRI treatment in MDD patients whose depressive symptoms were adequately treated was evaluated in an eight-week, randomized, double-blind, active-controlled (escitalopram), flexible-dose study (Study 11). Patients taking citalopram, sertraline, or paroxetine for at least eight weeks duration and who were experiencing sexual dysfunction attributed to their SSRI treatment were switched to TRINTELLIX (n=217) or escitalopram (n=207). For both TRINTELLIX and escitalopram, patients were started on 10 mg, increased to 20 mg at Week 1, followed by flexible dosing. The majority of subjects received the 20 mg dose of TRINTELLIX (65.6%) or the 20 mg dose of escitalopram (71.9%) during the study.
Improvement in TESD induced by prior SSRI treatment in subjects switched to TRINTELLIX was superior to the improvement observed in those subjects who switched to escitalopram (2.2 point improvement vs escitalopram on the change from Baseline in CSFQ-14 total score, with 95% confidence interval 0.48 – 4.02), after eight weeks of treatment, while both drugs maintained the subjects' prior antidepressant response. For change from Baseline in CSFQ-14, see Figure 8.
Figure 8. Change from Baseline in CSFQ-14 Total Score by Study Visit (Week) in Study 11
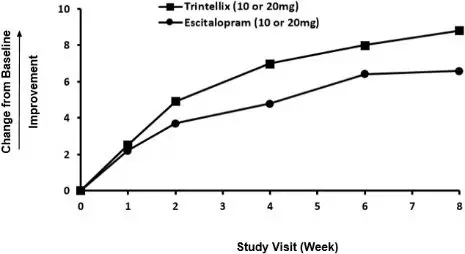
Effects in Healthy Volunteers with Normal Sexual Functioning at Baseline
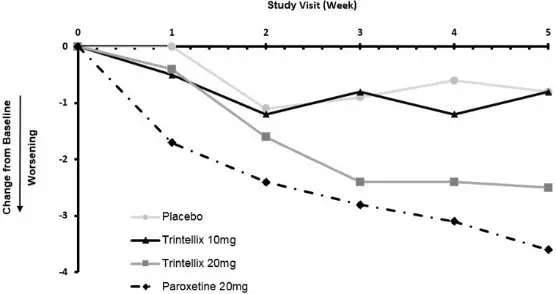
In a randomized Healthy Volunteer study (Study 12) with 348 subjects aged 18 years to 40 years with normal sexual functioning without the confounding effect of depression, TESD with TRINTELLIX 10 mg (n=85), but not with TRINTELLIX 20 mg (n=91), was statistically significantly less than with paroxetine 20 mg (n=83) [see Adverse Reactions (6.1)]. Paroxetine 20 mg was statistically significantly worse than placebo (n=89), confirming assay sensitivity in this study. For change from Baseline in CSFQ-14, see Figure 9.
Figure 9. Change from Baseline in CSFQ-14 Total Score by Study Visit (Week) in Healthy Volunteers (Study 12)
16. How is Trintellix supplied
TRINTELLIX tablets are available as follows:
Trintellix 10 mg, approximately 2520 Tablets per bottle, NDC 55154-0256-8
Trintellix 20 mg, approximately 2430 Tablets per bottle, NDC 55154-0257-8
17. Patient Counseling Information
See FDA-approved patient labeling (Medication Guide).
Suicidal Thoughts and Behaviors
Advise patients and caregivers to look for the emergence of suicidal ideation and behavior, especially early during treatment and when the dose is adjusted up or down, and instruct them to report such symptoms to the healthcare provider [see Boxed Warning, Warnings and Precautions (5.1)].
Concomitant Medication
Advise patients to inform their healthcare provider if they are taking, or plan to take, any prescription or over-the-counter medications because of a potential for interactions. Instruct patients not to take TRINTELLIX with an MAOI or within 14 days of stopping an MAOI and to allow 21 days after stopping TRINTELLIX before starting an MAOI [see Dosage and Administration (2.4), Contraindications (4), Warnings and Precautions (5.2), Drug Interactions (7.1)].
Serotonin Syndrome
Caution patients about the risk of serotonin syndrome, particularly with the concomitant use of TRINTELLIX and triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan supplements, busipirone, and St. John's Wort and with drugs that impair metabolism of serotonin (in particular, MAOIs, both those intended to treat psychiatric disorders and also others, such as linezolid) [see Warnings and Precautions (5.2), Drug Interactions (7.1, 7.2)]. Instruct patients to contact their health care provider or report to the emergency room if they experience signs or symptoms of serotonin syndrome.
Increased Risk of Bleeding
Inform patients about the concomitant use of TRINTELLIX with NSAIDs, aspirin, warfarin, or other drugs that affect coagulation because combined use has been associated with an increased risk of bleeding. Advise patients to inform their health care provider if they are taking or planning to take any prescription or over-the-counter medications that increase the risk of bleeding [see Warnings and Precautions (5.3)].
Activation of Mania/Hypomania
Advise patients and their caregivers to look for signs of activation of mania/hypomania [see Warnings and Precautions (5.4)].
Discontinuation of Treatment
Advise patients not to abruptly stop taking TRINTELLIX without first talking to their healthcare provider. Patients should be aware that discontinuation effects may occur when stopping TRINTELLIX and they should monitor for discontinuation symptoms [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
Angle Closure Glaucoma
Patients should be advised that taking TRINTELLIX can cause mild pupillary dilation, which in susceptible individuals, can lead to an episode of angle closure glaucoma. Pre-existing glaucoma is almost always open-angle glaucoma because angle closure glaucoma, when diagnosed, can be treated definitively with iridectomy. Open-angle glaucoma is not a risk factor for angle closure glaucoma. Patients may wish to be examined to determine whether they are susceptible to angle closure, and have a prophylactic procedure (e.g., iridectomy), if they are susceptible [see Warnings and Precautions (5.6)].
Hyponatremia
Advise patients that if they are treated with diuretics, or are otherwise volume depleted, or are elderly, they may be at greater risk of developing hyponatremia while taking TRINTELLIX [see Warnings and Precautions (5.7)].
Sexual Dysfunction
Advise patients that use of TRINTELLIX may cause symptoms of sexual dysfunction in both male and female patients. Inform patients that they should discuss any changes in sexual function and potential management strategies with their healthcare provider [see Warnings and Precautions (5.8)].
Nausea
Advise patients that nausea is one of the most common adverse reactions, and is dose related. Nausea commonly occurs within the first week of treatment, then decreases in frequency but can persist in some patients [see Adverse Reactions (6.1)].
Allergic Reactions
Advise patients to notify their healthcare provider if they develop an allergic reaction such as rash, hives, swelling, or difficulty breathing.
Pregnancy
Advise a pregnant woman or a woman planning to become pregnant that TRINTELLIX may cause withdrawal symptoms in the newborn or persistent pulmonary hypertension of the newborn (PPHN) [see Use in Specific Populations (8.1)]. Advise the patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to TRINTELLIX during pregnancy [see Use in Specific Populations (8.1)].
Distributed and marketed by:
Takeda Pharmaceuticals America, Inc.
Lexington, MA 02421
Marketed by:
Lundbeck
Deerfield, IL 60015
Distributed by:
Cardinal Health
Dublin, OH 43017
L72740622
L72750622
TRINTELLIX is a trademark of H. Lundbeck A/S registered with the U.S. Patent and Trademark Office and used under license by Takeda Pharmaceuticals America, Inc.
©2013-2021 Takeda Pharmaceuticals America, Inc.
LUN205 R24
| Distributed by: | |||
| Cardinal Health | |||
| Dublin, OH 43017 | |||
| L7274D Rev B | |||
| L7275D Rev. A | |||
|
MEDICATION GUIDE
|
|||
|
What is the most important information I should know about TRINTELLIX? |
|||
|
TRINTELLIX can cause serious side effects, including:
|
|||
|
Call your healthcare provider or get emergency help right away if you have any of the following symptoms, especially if they are new, worse, or worry you: |
|||
|
|
|
|
|
What is TRINTELLIX? |
|||
|
TRINTELLIX is a prescription medicine used in adults to treat a certain type of depression called Major Depressive Disorder (MDD). |
|||
|
Do not take TRINTELLIX if you:
|
|||
|
Ask your healthcare provider or pharmacist if you are not sure if you take an MAOI or one of these medicines, including the antibiotic linezolid or intravenous methylene blue. |
|||
|
Do not start TRINTELLIX if you stopped taking an MAOI in the last 14 days. |
|||
|
Do not start taking an MAOI for at least 21 days after you stop treatment with TRINTELLIX. |
|||
|
Before taking TRINTELLIX, tell your healthcare provider about all your medical conditions, including if you:
|
|||
|
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
|||
|
TRINTELLIX and some other medicines may affect each other causing possible serious side effects. |
|||
|
TRINTELLIX may affect the way other medicines work and other medicines may affect the way TRINTELLIX works. |
|||
|
Especially tell your healthcare provider if you take:
|
|||
|
Ask your healthcare provider if you are not sure if you are taking any of these medicines. Your healthcare provider can tell you if it is safe to take TRINTELLIX with your other medicines. |
|||
|
Do not start or stop any other medicines during treatment with TRINTELLIX without talking to your healthcare provider first. Stopping TRINTELLIX suddenly may cause you to have serious side effects. See, "What are the possible side effects of TRINTELLIX?" |
|||
|
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine. |
|||
|
How should I take TRINTELLIX?
|
|||
|
What are the possible side effects of TRINTELLIX? |
|||
|
TRINTELLIX may cause serious side effects, including:
|
|||
|
|
||
|
|||
|
|
||
|
|||
|
|
|
|
|
|||
|
|||
|
The most common side effects of TRINTELLIX include nausea, constipation, vomiting. |
|||
|
These are not all the possible side effects of TRINTELLIX. |
|||
|
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
|
How should I store TRINTELLIX?
|
|||
|
General information about the safe and effective use of TRINTELLIX. |
|||
|
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not take TRINTELLIX for a condition for which it was not prescribed. Do not give TRINTELLIX to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TRINTELLIX that is written for healthcare professionals. |
|||
|
What are the ingredients in TRINTELLIX? |
|||
|
Active ingredient: vortioxetine hydrobromide |
|||
|
Inactive ingredients: mannitol, microcrystalline cellulose, hydroxypropyl cellulose, sodium starch glycolate, magnesium stearate and film coating consisting of hypromellose, titanium dioxide, polyethylene glycol 400, iron oxide red (5 mg and 20 mg) and iron oxide yellow (10 mg) |
|||
|
Distributed and Marketed by: |
|||
|
Marketed by: Lundbeck, Deerfield, IL 60015 |
|||
|
TRINTELLIX is a trademark of H. Lundbeck A/S registered with the U.S. Patent and Trademark Office and used under license by Takeda Pharmaceuticals America, Inc. |
|||
|
All other trademarks are the property of their respective owners. ©2013-2021 Takeda Pharmaceuticals America, Inc. | |||
|
For more information, go to www.TRINTELLIX.com or call 1-877-TAKEDA-7 (1-877-825-3327). |
|||
| TRINTELLIX
vortioxetine tablet, film coated |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| TRINTELLIX
vortioxetine tablet, film coated |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - Cardinal Health 107, LLC (118546603) |
