Drug Detail:Trodelvy (Sacituzumab govitecan-hziy)
Drug Class: Miscellaneous antineoplastics
Highlights of Prescribing Information
TRODELVY® (sacituzumab govitecan-hziy) for injection, for intravenous use
Initial U.S. Approval: 2020
WARNING: NEUTROPENIA AND DIARRHEA
See full prescribing information for complete boxed warning.
Severe or life threatening neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patients with febrile neutropenia without delay. (2.3, 5.1)
Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses. (2.3, 5.2)
Recent Major Changes
| Boxed Warning | 02/2023 |
| Indications and Usage (1.1) | 02/2023 |
| Dosage and Administration (2.3) | 02/2023 |
| Dosage and Administration (2.4) | 12/2022 |
| Warnings and Precautions (5.1, 5.2, 5.3, 5.4, 5.5) | 02/2023 |
Indications and Usage for Trodelvy
TRODELVY is a Trop-2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with:
Locally Advanced or Metastatic Breast Cancer
- Unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. (1.1, 14.1)
- Unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting. (1.1, 14.2)
Locally Advanced or Metastatic Urothelial Cancer
- Locally advanced or metastatic urothelial cancer (mUC) who have previously received a platinum-containing chemotherapy and either programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor.a (1.2)
a This indication is approved under accelerated approval based on tumor response rate and duration of response [see Clinical Studies (14.3)].
Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
Trodelvy Dosage and Administration
- Do NOT substitute TRODELVY for or use with other drugs containing irinotecan or its active metabolite SN-38. (2.1)
- For intravenous infusion only. Do not administer as an intravenous push or bolus.
- The recommended dose is 10 mg/kg once weekly on Days 1 and 8 of continuous 21-day treatment cycles until disease progression or unacceptable toxicity. (2.2)
- Premedication for prevention of infusion reactions and prevention of chemotherapy-induced nausea and vomiting is recommended. (2.2)
- Monitor patients during the infusion and for at least 30 minutes after completion of infusion. Treatment interruption and/or dose reduction may be needed to manage adverse reactions. (2.2)
- See Full Prescribing Information for preparation and administration instructions. (2.4)
Dosage Forms and Strengths
For injection: 180 mg lyophilized powder in single-dose vials for reconstitution. (3)
Contraindications
Severe hypersensitivity reaction to TRODELVY. (4, 5.3)
Warnings and Precautions
- Hypersensitivity and Infusion-Related Reactions: Hypersensitivity reactions including severe anaphylactic reactions have been observed. Monitor patients for infusion-related reactions. Permanently discontinue TRODELVY if severe or life-threatening reactions occur. (5.3)
- Nausea/Vomiting: Use antiemetic preventive treatment and withhold TRODELVY for patients with Grade 3 nausea or Grade 3–4 vomiting at the time of scheduled treatment. (5.4)
- Patients with Reduced UGT1A1 Activity: Individuals who are homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia following initiation of TRODELVY treatment. (5.5)
- Embryo-Fetal Toxicity: TRODELVY can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.6, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥25%) are (including laboratory abnormalities) were decreased leukocyte count, decreased neutrophil count, decreased hemoglobin, diarrhea, nausea, decreased lymphocyte count, fatigue, alopecia, constipation, increased glucose, decreased albumin, vomiting, decreased appetite, decreased creatinine clearance, increased alkaline phosphatase, decreased magnesium, decreased potassium, and decreased sodium. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-888-983-4668 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- UGT1A1 inhibitors or inducers: Avoid concomitant use. (7)
Use In Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2023
Full Prescribing Information
WARNING: NEUTROPENIA AND DIARRHEA
- Severe or life threatening neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis [see Dosage and Administration (2.3)]. Initiate anti-infective treatment in patient with febrile neutropenia without delay [see Warnings and Precautions (5.1)].
- Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide [see Warnings and Precautions (5.2)]. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses [see Dosage and Administration (2.3)].
1. Indications and Usage for Trodelvy
1.1 Locally Advanced or Metastatic Breast Cancer
- TRODELVY is indicated for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease.
- TRODELVY is indicated for the treatment of adult patients with unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
1.2 Locally Advanced or Metastatic Urothelial Cancer
- TRODELVY is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC) who have previously received a platinum-containing chemotherapy and either programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor.
This indication is approved under accelerated approval based on tumor response rate and duration of response [see Clinical Studies (14.3)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
2. Trodelvy Dosage and Administration
2.1 Important Use Information
Do NOT substitute TRODELVY for or use with other drugs containing irinotecan or its active metabolite SN-38.
2.2 Recommended Dosage
The recommended dosage of TRODELVY is 10 mg/kg administered as an intravenous infusion once weekly on Days 1 and 8 of 21-day treatment cycles. Continue treatment until disease progression or unacceptable toxicity. Do not administer TRODELVY at doses greater than 10 mg/kg.
Administer TRODELVY as an intravenous infusion only. Do not administer as an intravenous push or bolus.
First infusion: Administer infusion over 3 hours. Observe patients during the infusion and for at least 30 minutes following the initial dose, for signs or symptoms of infusion-related reactions [see Warning and Precautions (5.3)].
Subsequent infusions: Administer infusion over 1 to 2 hours if prior infusions were tolerated. Observe patients during the infusion and for at least 30 minutes after infusion.
3. Dosage Forms and Strengths
For injection: 180 mg off-white to yellowish lyophilized powder in a single-dose vial.
4. Contraindications
TRODELVY is contraindicated in patients who have experienced a severe hypersensitivity reaction to TRODELVY [see Warnings and Precautions (5.3)].
5. Warnings and Precautions
5.1 Neutropenia
Severe, life-threatening, or fatal neutropenia can occur in patients treated with TRODELVY. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3–4 neutropenia occurred in 49% of patients. Febrile neutropenia occurred in 6% of patients. The median time to first onset of neutropenia (including febrile neutropenia) was 16 days and has occurred earlier in some patient populations [see Warnings and Precautions (5.5)]. Neutropenic colitis occurred in 1.4% of patients.
Withhold TRODELVY for absolute neutrophil count below 1500/mm3 on Day 1 of any cycle or neutrophil count below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Dose modifications may be required due to neutropenia. Administer G-CSF as clinically indicated or indicated in Table 1 [see Dosage and Administration (2.3)].
5.2 Diarrhea
TRODELVY can cause severe diarrhea. Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3–4 diarrhea occurred in 11% of all patients treated with TRODELVY. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.7% of all patients.
Withhold TRODELVY for Grade 3–4 diarrhea at the time of scheduled treatment administration and resume when resolved to ≤Grade 1 [see Dosage and Administration (2.3)].
At the onset of diarrhea, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated.
Patients who exhibit an excessive cholinergic response to treatment with TRODELVY (e.g., abdominal cramping, diarrhea, salivation, etc.) can receive appropriate premedication (e.g., atropine) for subsequent treatments.
5.3 Hypersensitivity and Infusion-Related Reactions
Serious hypersensitivity reactions including life-threatening anaphylactic reactions have occurred with TRODELVY treatment. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, pneumonitis, and skin reactions [see Contraindications (4)].
Hypersensitivity reactions within 24 hours of dosing occurred in 35% of patients treated with TRODELVY. Grade 3–4 hypersensitivity occurred in 2% of patients treated with TRODELVY. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.2%. The incidence of anaphylactic reactions was 0.2%.
Premedication for infusion reactions in patients receiving TRODELVY is recommended. Have medications and emergency equipment to treat infusion-related reactions, including anaphylaxis, available for immediate use when administering TRODELVY [see Dosage and Administration (2.2)].
Closely monitor patients for hypersensitivity and infusion-related reactions during each TRODELVY infusion and for at least 30 minutes after completion of each infusion [see Dosage and Administration (2.3)].
Permanently discontinue TRODELVY for Grade 4 infusion-related reactions [see Dosage and Administration (2.3)].
5.4 Nausea and Vomiting
TRODELVY is emetogenic. Nausea occurred in 64% of all patients treated with TRODELVY. Grade 3–4 nausea occurred in 3% of patients.
Vomiting occurred in 35% of all patients treated with TRODELVY. Grade 3–4 vomiting occurred in 2% of these patients.
Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV) [see Dosage and Administration (2.2)].
Withhold TRODELVY doses for Grade 3 nausea or Grade 3–4 vomiting at the time of scheduled treatment administration and resume with additional supportive measures when resolved to ≤Grade 1 [see Dosage and Administration (2.3)].
Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
5.5 Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity
Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia; and may be at increased risk for other adverse reactions when treated with TRODELVY.
The incidence of neutropenia and anemia was analyzed in 948 patients who received TRODELVY and had UGT1A1 genotype results. In patients homozygous for the UGT1A1 *28 allele (n=112), the incidence of Grade 3–4 neutropenia was 58%. In patients heterozygous for the UGT1A1*28 allele (n=420), the incidence of Grade 3–4 neutropenia was 49%. In patients homozygous for the wild-type allele (n=416), the incidence of Grade 3–4 neutropenia was 43% [see Clinical Pharmacology (12.5)]. In patients homozygous for the UGT1A1 *28 allele, the incidence of Grade 3–4 anemia was 21%. In patients heterozygous for the UGT1A1*28 allele, the incidence of Grade 3–4 anemia was 10%. In patients homozygous for the wild-type allele, the incidence of Grade 3–4 anemia was 9%.
The median time to first neutropenia including febrile neutropenia was 9 days in patients homozygous for the UGT1A1*28 allele, 15 days in patients heterozygous for the UGT1A1*28 allele, and 20 days in patients homozygous for the wild-type allele. The median time to first anemia was 21 days in patients homozygous for the UGT1A1*28 allele, 25 days in patients heterozygous for the UGT1A1*28 allele, and 28 days in patients homozygous for the wild-type allele.
Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 enzyme activity [see Dosage and Administration (2.3)].
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)]. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the label:
- Neutropenia [see Warnings and Precautions (5.1)]
- Diarrhea [see Warnings and Precautions (5.2)]
- Hypersensitivity and Infusion-Related Reactions [see Warnings and Precautions (5.3)]
- Nausea and Vomiting [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The pooled safety population described in the Warnings and Precautions section reflect exposure to TRODELVY in 1063 patients from four studies, IMMU-132-01, ASCENT, TROPiCS-02, and TROPHY which included 366 patients with mTNBC, 322 patients with hormone receptor-positive (HR+)/human epidermal growth factor receptor 2-negative (HER2-) breast cancer, and 180 patients with mUC. TRODELVY was administered as an intravenous infusion once weekly on Days 1 and 8 of 21-day treatment cycles at doses of 10 mg/kg until disease progression or unacceptable toxicity. Among the 1063 patients treated with TRODELVY, the median duration of treatment was 4.1 months (range: 0 to 63 months). In this pooled safety population, the most common (≥ 25%) adverse reactions including laboratory abnormalities were decreased leukocyte count (84%), decreased neutrophil count (75%), decreased hemoglobin (69%), diarrhea (64%), nausea (64%), decreased lymphocyte count (63%), fatigue (51%), alopecia (45%), constipation (37%), increased glucose (37%), decreased albumin (35%), vomiting (35%), decreased appetite (30%), decreased creatinine clearance (28%), increased alkaline phosphatase (28%), decreased magnesium (27%), decreased potassium (26%), and decreased sodium (26%).
8. Use In Specific Populations
8.4 Pediatric Use
Safety and effectiveness of TRODELVY have not been established in pediatric patients.
8.5 Geriatric Use
Of the 366 patients with TNBC who were treated with TRODELVY, 19% of patients were 65 years and 3% were 75 years and older. No overall differences in safety and effectiveness were observed between patients ≥ 65 years of age and younger patients.
Of the 322 patients with HR+/HER2- breast cancer who were treated with TRODELVY, 26% of patients were 65 years and older and 6% were 75 years and older. No overall differences in effectiveness were observed between patients ≥ 65 years of age and younger patients. There was a higher discontinuation rate due to adverse reactions in patients aged 65 years or older (14%) compared with younger patients (3%).
Of the 180 patients with UC who were treated with TRODELVY, 59% of patients were 65 years and older and 27% were 75 years and older. No overall differences in effectiveness were observed between patients ≥ 65 years of age and younger patients. There was a higher discontinuation rate due to adverse reactions in patients aged 65 years or older (14%) compared with younger patients (8%).
8.6 Hepatic Impairment
No adjustment to the starting dosage is required when administering TRODELVY to patients with mild hepatic impairment [see Clinical Pharmacology (12.3)].
The safety of TRODELVY in patients with moderate (total bilirubin > 1.5 to 3.0 × ULN) or severe (total bilirubin > 3.0 × upper limit of normal [ULN]) hepatic impairment has not been established. TRODELVY has not been tested in patients with AST or ALT > 3 ULN without liver metastases, or AST or ALT > 5 ULN with liver metastases. No recommendations can be made for the starting dosage in these patients.
10. Overdosage
In a clinical trial, planned doses of up to 18 mg/kg (approximately 1.8 times the maximum recommended dose of 10 mg/kg) of TRODELVY were administered. In these patients, a higher incidence of severe neutropenia was observed.
11. Trodelvy Description
Sacituzumab govitecan-hziy is a Trop-2 directed antibody and topoisomerase inhibitor conjugate, composed of the following three components:
- the humanized monoclonal antibody, hRS7 IgG1κ (also called sacituzumab), which binds to Trop-2 (the trophoblast cell-surface antigen-2);
- the drug SN-38, a topoisomerase inhibitor;
- a hydrolysable linker (called CL2A), which links the humanized monoclonal antibody to SN-38.
The recombinant monoclonal antibody is produced by mammalian (murine myeloma) cells, while the small molecule components SN-38 and CL2A are produced by chemical synthesis. Sacituzumab govitecan-hziy contains on average 7 to 8 molecules of SN-38 per antibody molecule. Sacituzumab govitecan-hziy has a molecular weight of approximately 160 kilodaltons. Sacituzumab govitecan-hziy has the following chemical structure.
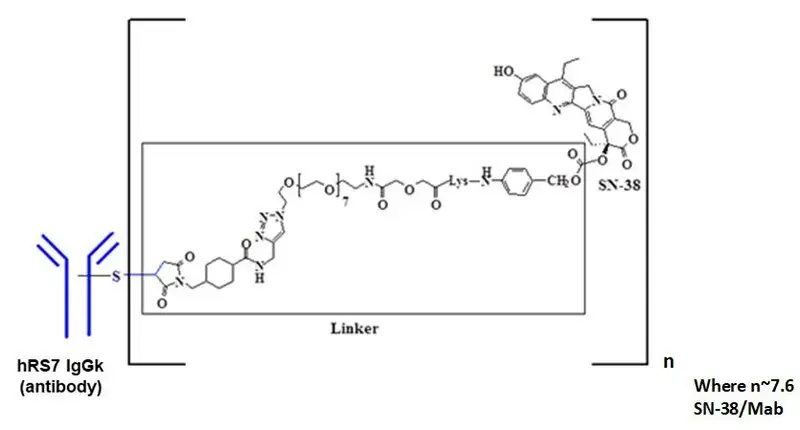
TRODELVY (sacituzumab govitecan-hziy) for injection is a sterile, preservative-free, off-white to yellowish lyophilized powder for intravenous use in a 50 mL clear glass single-dose vial, with a rubber stopper and crimp-sealed with an aluminum flip-off cap.
Each single-dose vial of TRODELVY delivers 180 mg sacituzumab govitecan-hziy, 77.3 mg 2-(N-morpholino) ethane sulfonic acid (MES), 1.8 mg polysorbate 80 and 154 mg trehalose dihydrate. Reconstitution with 20 mL of 0.9% Sodium Chloride Injection, USP, results in a concentration of 10 mg/mL with a pH of 6.5.
12. Trodelvy - Clinical Pharmacology
12.1 Mechanism of Action
Sacituzumab govitecan-hziy is a Trop-2-directed antibody-drug conjugate. Sacituzumab is a humanized antibody that recognizes Trop-2. The small molecule, SN-38, is a topoisomerase I inhibitor, which is covalently attached to the antibody by a linker. Pharmacology data suggest that sacituzumab govitecan-hziy binds to Trop-2-expressing cancer cells and is internalized with the subsequent release of SN-38 via hydrolysis of the linker. SN-38 interacts with topoisomerase I and prevents re-ligation of topoisomerase I-induced single strand breaks. The resulting DNA damage leads to apoptosis and cell death. Sacituzumab govitecan-hziy decreased tumor growth in mouse xenograft models of triple-negative breast cancer.
12.2 Pharmacodynamics
The TRODELVY exposure-response relationships and pharmacodynamic time course for efficacy have not been fully characterized.
12.3 Pharmacokinetics
The serum pharmacokinetics of sacituzumab govitecan-hziy and SN-38 were evaluated in patients with mBC who received sacituzumab govitecan-hziy as a single agent at a dose of 10 mg/kg. The pharmacokinetic parameters of sacituzumab govitecan-hziy and free SN-38 are presented in Table 10.
| Sacituzumab govitecan-hziy (N=693) | Free SN-38 (N=681) |
|
|---|---|---|
| Cmax: maximum serum concentration from 0–168 hours after the first dose | ||
| AUC0–168: area under serum concentration curve through 168 hours after the first dose | ||
|
||
| Cmax [ng/mL] | 239000 (11%) | 98.0 (45%) |
| AUC0–168 [ng*h/mL] | 5640000 (22%) | 3696 (56%) |
12.5 Pharmacogenomics
SN-38 is metabolized via UGT1A1 [see Clinical Pharmacology (12.3)]. Genetic variants of the UGT1A1 gene such as the UGT1A1*28 allele lead to reduced UGT1A1 enzyme activity. Individuals who are homozygous or heterozygous for the UGT1A1*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia from TRODELVY compared to individuals who are wildtype (*1/*1) [see Warnings and Precautions (5.5)]. Approximately 20% of the Black or African American population, 10% of the White population, and 2% of the East Asian population are homozygous for the UGT1A1*28 allele (*28/*28). Approximately 40% of the Black or African American population, 50% of the White population, and 25% of the East Asian population are heterozygous for the UGT1A1*28 allele (*1/*28). Decreased function alleles other than UGT1A1*28 may be present in certain populations.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with sacituzumab govitecan-hziy.
SN-38 was clastogenic in an in vitro mammalian cell micronucleus test in Chinese hamster ovary cells and was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
Fertility studies with sacituzumab govitecan-hziy have not been conducted. In a repeat-dose toxicity study in cynomolgus monkeys, intravenous administration of sacituzumab govitecan-hziy on Day 1 and Day 4 resulted in endometrial atrophy, uterine hemorrhage, increased follicular atresia of the ovary, and atrophy of vaginal epithelial cells at doses ≥ 60 mg/kg (≥ 6 times the human recommended dose of 10 mg/kg based on body weight).
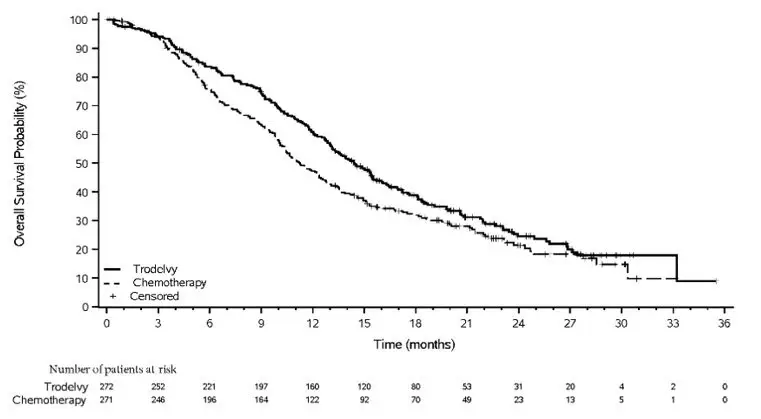
14. Clinical Studies
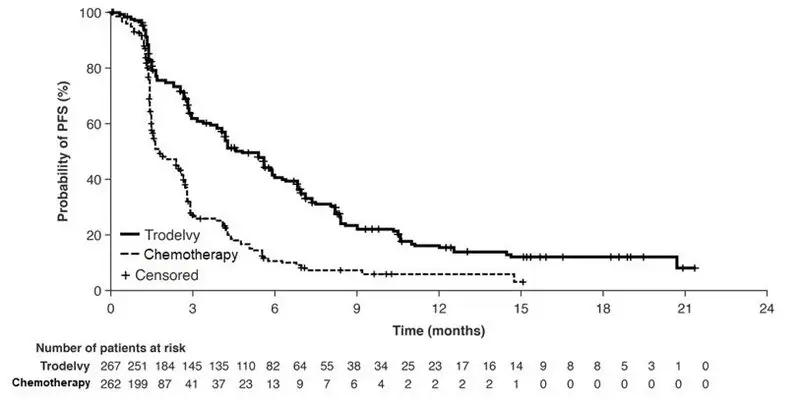
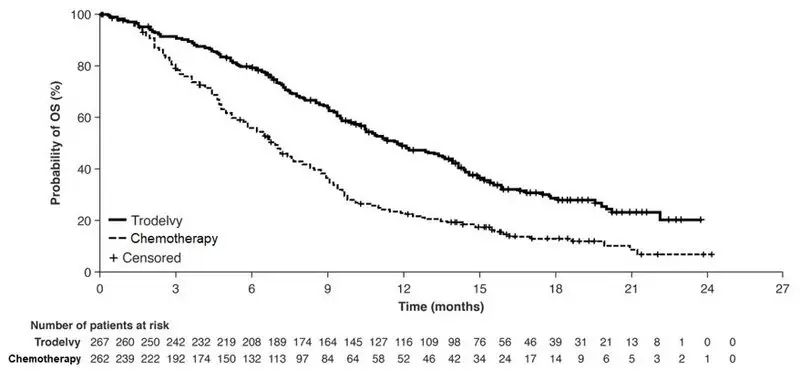
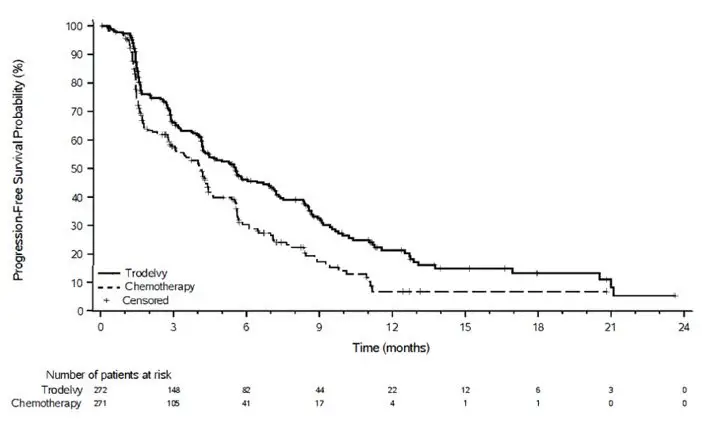
16. How is Trodelvy supplied
TRODELVY (sacituzumab govitecan-hziy) for injection is a sterile, off-white to yellowish lyophilized powder in a single-dose vial. Each TRODELVY vial is individually boxed in a carton:
- NDC 55135-132-01 contains one 180 mg vial
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information)
| The Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 02/2023 | |
| Patient Information
TRODELVY® (troh-DELL-vee) (sacituzumab govitecan-hziy) for injection, for intravenous use |
||
|
||
|
|
|
|
||
| What is TRODELVY?
TRODELVY is a prescription medicine used to treat adults with:
It is not known if TRODELVY is safe and effective in children. |
||
| Do not receive TRODELVY if you have had a severe allergic reaction to TRODELVY. Ask your healthcare provider if you are not sure. | ||
Before receiving TRODELVY, tell your healthcare provider about all of your medical conditions, including if you:
|
||
How will I receive TRODELVY?
|
||
| What are the possible side effects of TRODELVY?
TRODELVY can cause serious side effects, including:
|
||
|
|
|
|
||
|
|
|
| TRODELVY may cause fertility problems in females, which could affect your ability to have a baby. Talk to your healthcare provider if fertility is a concern for you. These are not all of the possible side effects of TRODELVY. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
| General information about the safe and effective use of TRODELVY.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about TRODELVY that is written for health professionals. |
||
| What are the ingredients in TRODELVY? Active ingredient: sacituzumab govitecan-hziy Inactive ingredients: 2-(N-morpholino) ethane sulfonic acid (MES), polysorbate 80 and trehalose dihydrate Manufactured by: Gilead Sciences, Inc., 333 Lakeside Dr., Foster City, CA 94404, USA U.S. License No. 2258 761115-GS-007 For more information about TRODELVY, go to www.TRODELVY.com or call 1-888-983-4668. |
||


| TRODELVY
sacituzumab govitecan powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Gilead Sciences (185049848) |
