Drug Detail:Tudorza pressair (Aclidinium [ a-kli-din-ee-um ])
Drug Class: Anticholinergic bronchodilators
Highlights of Prescribing Information
TUDORZA® PRESSAIR® (aclidinium bromide inhalation powder)
FOR ORAL INHALATION ONLY
Initial U.S. Approval: 2012
Indications and Usage for Tudorza Pressair
TUDORZA PRESSAIR is an anticholinergic indicated for the maintenance treatment of patients with chronic obstructive pulmonary disease (COPD). (1)
Tudorza Pressair Dosage and Administration
For oral inhalation only
- •
- One inhalation of TUDORZA PRESSAIR 400 mcg twice daily. (2)
Dosage Forms and Strengths
- •
- Inhalation powder; The multi-dose device is a dry powder inhaler metering 400 mcg of aclidinium bromide per actuation. (3)
Contraindications
- •
- Severe hypersensitivity to milk proteins. (4)
- •
- Hypersensitivity to any ingredient. (4)
Warnings and Precautions
- •
- Not for acute use: Not for use as a rescue medication. (5.1)
- •
- Paradoxical bronchospasm: Discontinue TUDORZA PRESSAIR and consider other treatments if paradoxical bronchospasm occurs. (5.2)
- •
- Worsening of narrow-angle glaucoma may occur. Use with caution in patients with narrow-angle glaucoma and instruct patients to consult a physician immediately if this occurs. (5.3)
- •
- Worsening of urinary retention may occur. Use with caution in patients with prostatic hyperplasia or bladder-neck obstruction and instruct patients to consult a physician immediately if this occurs. (5.4)
- •
- Immediate hypersensitivity reactions: Discontinue TUDORZA PRESSAIR at once and consider alternatives if immediate hypersensitivity reactions, including angioedema, bronchospasm, or anaphylaxis occur. (5.5)
Adverse Reactions/Side Effects
Most common adverse reactions (≥3% incidence and greater than placebo) are headache, nasopharyngitis and cough. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Anticholinergics: May interact additively with concomitantly used anticholinergic medications. Avoid administrations of TUDORZA PRESSAIR with other anticholinergic-containing drugs. (7.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2021
Full Prescribing Information
1. Indications and Usage for Tudorza Pressair
TUDORZA® PRESSAIR® (aclidinium bromide inhalation powder) is indicated for the maintenance treatment of patients with chronic obstructive pulmonary disease (COPD).
2. Tudorza Pressair Dosage and Administration
The recommended dose of TUDORZA PRESSAIR is one oral inhalation of 400 mcg, twice daily (morning and evening approximately 12 hours apart).
3. Dosage Forms and Strengths
Inhalation Powder. TUDORZA PRESSAIR is a breath-actuated multi-dose dry powder inhaler metering 400 mcg of aclidinium bromide per actuation.
4. Contraindications
The use of TUDORZA PRESSAIR is contraindicated in the following conditions:
- •
- Severe hypersensitivity to milk proteins [see Warnings and Precautions (5.5)]
- •
- Hypersensitivity to aclidinium bromide or any of the excipients [see Warnings and Precautions (5.5)]
5. Warnings and Precautions
5.1 Not for Acute Use
TUDORZA PRESSAIR is intended as a twice-daily maintenance treatment for COPD and is not indicated for the initial treatment of acute episodes of bronchospasm (i.e., rescue therapy).
5.2 Paradoxical Bronchospasm
Inhaled medicines, including TUDORZA PRESSAIR, may cause paradoxical bronchospasm. If this occurs, treatment with TUDORZA PRESSAIR should be stopped and other treatments considered.
5.3 Worsening of Narrow-Angle Glaucoma
TUDORZA PRESSAIR should be used with caution in patients with narrow-angle glaucoma. Prescribers and patients should be alert for signs and symptoms of acute narrow-angle glaucoma (e.g., eye pain or discomfort, blurred vision, visual halos, or colored images in association with red eyes from conjunctival congestion and corneal edema). Instruct patients to consult a physician immediately should any of these signs or symptoms develop.
5.4 Worsening of Urinary Retention
TUDORZA PRESSAIR should be used with caution in patients with urinary retention. Prescribers and patients should be alert for signs and symptoms of prostatic hyperplasia or bladder-neck obstruction (e.g., difficulty passing urine, painful urination). Instruct patients to consult a physician immediately should any of these signs or symptoms develop.
5.5 Immediate Hypersensitivity Reactions
Immediate hypersensitivity reactions, including anaphylaxis, angioedema (including swelling of the lips, tongue, or throat), urticaria, rash, bronchospasm, or itching, have occurred after administration of TUDORZA PRESSAIR. If such a reaction occurs, therapy with TUDORZA PRESSAIR should be stopped at once and alternative treatments should be considered.
6. Adverse Reactions/Side Effects
The following adverse reactions are described in greater detail in other sections:
- •
- Paradoxical bronchospasm [see Warnings and Precautions (5.2)]
- •
- Worsening of narrow-angle glaucoma [see Warnings and Precautions (5.3)]
- •
- Worsening of urinary retention [see Warnings and Precautions (5.4)]
- •
- Immediate hypersensitivity reactions [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
3-Month and 6-Month Trials
TUDORZA PRESSAIR was studied in two 3-month (Trials B and C) and one 6-month (Trial D) placebo-controlled trials in patients with COPD. In these trials, 636 patients were treated with TUDORZA PRESSAIR at the recommended dose of 400 mcg twice daily.
The population had a mean age of 64 years (ranging from 40 to 89 years), with 58% males, 94% Caucasian, and had COPD with a mean pre-bronchodilator forced expiratory volume in one second (FEV1) percent predicted of 48%. Patients with unstable cardiac disease, narrow-angle glaucoma, or symptomatic prostatic hypertrophy or bladder outlet obstruction were excluded from these trials.
Table 1 shows all adverse reactions that occurred with a frequency of greater than or equal to 1% in the TUDORZA PRESSAIR group in the two 3-month and one 6-month placebo-controlled trials where the rates in the TUDORZA PRESSAIR group exceeded placebo.
|
Treatment |
||
|
Adverse Reactions |
TUDORZA PRESSAIR |
Placebo |
|
Preferred Term |
(N=636) |
(N=640) |
|
n (%) |
n (%) |
|
|
Headache |
42 (6.6) |
32 (5.0) |
|
Nasopharyngitis |
35 (5.5) |
25 (3.9) |
|
Cough |
19 (3.0) |
14 (2.2) |
|
Diarrhea |
17 (2.7) |
9 (1.4) |
|
Sinusitis |
11 (1.7) |
5 (0.8) |
|
Rhinitis |
10 (1.6) |
8 (1.2) |
|
Toothache |
7 (1.1) |
5 (0.8) |
|
Fall |
7 (1.1) |
3 (0.5) |
|
Vomiting |
7 (1.1) |
3 (0.5) |
In addition, among the adverse reactions observed in the clinical trials with an incidence of less than 1% were diabetes mellitus, dry mouth, 1st degree AV block, osteoarthritis, cardiac failure, and cardio-respiratory arrest.
Long-term Safety Trials
TUDORZA PRESSAIR was studied in three long-term safety trials, two double blind and one open label, ranging from 40 to 52 weeks in patients with moderate to severe COPD. Two of these trials were extensions of the 3-month trials, and one was a dedicated long-term safety trial. In these trials, 891 patients were treated with TUDORZA PRESSAIR at the recommended dose of 400 mcg twice daily. The demographic and baseline characteristics of the long-term safety trials were similar to those of the placebo-controlled trials. The adverse events reported in the long-term safety trials were similar to those occurring in the placebo-controlled trials of 3 to 6 months. No new safety findings were reported compared to the placebo-controlled trials.
Long-Term Trial of up to 3-Years
In a long-term safety trial with 3630 moderate to very severe COPD patients with previous major cardiac events or cardiovascular risk factors at baseline, the adverse reactions reported with a frequency ≥2% in the TUDORZA PRESSAIR group in which the exposure-adjusted incidence rate exceeds the placebo group were nausea, back pain, cough, hypertension, sinusitis, constipation, arthralgia, anemia, muscle spasms, cardiac failure congestive, cellulitis, and gastroesophageal reflux disease. No other new adverse reactions were identified.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of drug TUDORZA PRESSAIR. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In postmarketing experience with TUDORZA PRESSAIR, immediate hypersensitivity reactions, including anaphylaxis, angioedema (including swelling of the lips, tongue, or throat), urticaria, rash, bronchospasm, or itching have been reported. Additionally, nausea, dysphonia, blurred vision, urinary retention, tachycardia, and stomatitis have been observed.
7. Drug Interactions
In vitro studies suggest limited potential for CYP450-related metabolic drug interactions, thus no formal drug interaction studies have been performed with TUDORZA PRESSAIR [see Clinical Pharmacology (12.3)].
7.1 Sympathomimetics, Methylxanthines, Steroids
In clinical studies, concurrent administration of aclidinium bromide and other drugs commonly used in the treatment of COPD including sympathomimetics (short-acting beta2 agonists), methylxanthines, and oral and inhaled steroids showed no increases in adverse drug reactions.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate and well controlled studies of TUDORZA PRESSAIR in pregnant women to inform drug associated risks.
No adverse developmental effects were seen with inhalation administration of aclidinium bromide to pregnant rats and rabbits during organogenesis at 15 or 20 times, respectively, the maximum recommended human daily inhaled dose (MRHDID). However, reduced pup weights were seen when pregnant rats continued inhalation administration through lactation at 5 times the MRHDID of aclidinium bromide. Adverse developmental effects occurred when rabbits were orally dosed with aclidinium bromide at approximately 1,400 times the MRHDID [see Data].
The estimated background risk of major birth defects and miscarriage of the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in pregnant rats dosed during the period of organogenesis from gestation days 6-17, no evidence of structural alterations was observed at approximately 15 times the MRHDID [based on summed AUCs of aclidinium bromide and its metabolites at inhaled doses less than or equal to 5.0 mg/kg/day]. However, in a pre- and post-natal development study, decreased pup weights were observed when pregnant rats were exposed from gestation day 6 and continuing during the lactation period at approximately 5 times the MRHDID [based on summed AUCs of aclidinium bromide and its metabolites at inhaled doses greater than or equal to 0.2 mg/kg/day]. Maternal toxicity was also observed at inhaled doses greater than or equal to 0.2 mg/kg/day.
In an embryo-fetal development study in pregnant Himalayan rabbits administered inhaled doses of aclidinium bromide during the period of organogenesis from gestation days 6-19, no evidence of structural alterations was observed at approximately 20 times the MRHDID [based on summed AUCs of aclidinium bromide and its metabolites at inhaled doses less than or equal to 3.6 mg/kg/day]. However, in another embryo-fetal development study in pregnant Himalayan rabbits dosed orally from gestation days 6-19, increased incidences of additional liver lobes (3-5%), as compared to 0% in the control group, were observed at approximately 1,400 times the MRHDID [based on summed AUCs of aclidinium bromide and its metabolites at oral doses greater than or equal to 150 mg/kg/day], and decreased fetal body weights were observed at approximately 2,300 times the MRHDID [based on summed AUCs of aclidinium bromide and its metabolites at oral doses greater than or equal to 300 mg/kg/day]. These fetal findings were observed in the presence of maternal toxicity.
8.2 Lactation
Risk Summary
There are no available data on the effects of TUDORZA PRESSAIR or aclidinium bromide on the breastfed child or on milk production or presence in human milk. Aclidinium bromide is present in milk of lactating female rats [see Data]. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for TUDORZA PRESSAIR and any potential adverse effects on the breastfed child from TUDORZA PRESSAIR or from the underlying maternal condition.
Data
In a pharmacokinetic study, levels of radioactivity in milk and plasma in rats were measured after a single intravenous dose of 1 mg/kg of radiolabeled aclidinium bromide on approximately post-natal day 14 [see Use in Specific Populations (8.1)]. The maximum concentration of radioactivity [14C-aclidinium] in milk was measured at 6 hours post-dose and was found to be 10-14 times higher than in plasma.
8.4 Pediatric Use
TUDORZA PRESSAIR is approved for use in the maintenance treatment of bronchospasm associated with COPD. COPD does not normally occur in children. The safety and effectiveness of TUDORZA PRESSAIR in pediatric patients have not been established.
8.5 Geriatric Use
Of the 636 COPD patients exposed to TUDORZA PRESSAIR 400 mcg twice daily for up to 24 weeks in three placebo-controlled clinical trials, 197 were less than 60 years, 272 were greater than or equal to 60 to less than 70 years, and 167 were greater than or equal to 70 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Based on available data for TUDORZA PRESSAIR, no adjustment of dosage in geriatric patients is warranted [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
The pharmacokinetics of TUDORZA PRESSAIR were investigated in subjects with normal renal function and in subjects with mild, moderate, and severe renal impairment [see Clinical Pharmacology (12.3)]. No clinically significant differences in aclidinium pharmacokinetics were noted between these populations. Based on available data for TUDORZA PRESSAIR, no adjustment of dosage in renally impaired subjects is warranted.
11. Tudorza Pressair Description
TUDORZA PRESSAIR consists of a dry powder formulation of aclidinium bromide for oral inhalation only.
Aclidinium bromide, the active component of TUDORZA PRESSAIR is an anticholinergic with specificity for muscarinic receptors. Aclidinium bromide is a synthetic, quaternary ammonium compound, chemically described as 1-azoniabicyclo[2.2.2]octane, 3-[(hydroxydi-2-thienylacetyl)oxy]-1-(3-phenoxypropyl)-, bromide, (3R)-. The structural formula is:
Aclidinium bromide is a white powder with a molecular formula of C26H30NO4S2Br and a molecular mass of 564.56. It is very slightly soluble in water and ethanol and sparingly soluble in methanol.
TUDORZA PRESSAIR is a breath-actuated multi-dose dry powder inhaler. Each actuation of TUDORZA PRESSAIR provides a metered dose of 13 mg of the formulation which contains lactose monohydrate (which may contain milk proteins) as the carrier and 400 mcg of aclidinium bromide (equivalent to 343 mcg of aclidinium). This results in delivery of 375 mcg aclidinium bromide (equivalent to 322 mcg of aclidinium) from the mouthpiece, based on in vitro testing at an average flow rate of 63 L/min with constant volume of 2 L. The amount of drug delivered to the lungs will vary depending on patient factors such as inspiratory flow rate and inspiratory time.
12. Tudorza Pressair - Clinical Pharmacology
12.1 Mechanism of Action
Aclidinium bromide is a long-acting antimuscarinic agent, which is often referred to as an anticholinergic. It has similar affinity to the subtypes of muscarinic receptors M1 to M5. In the airways, it exhibits pharmacological effects through inhibition of M3 receptor at the smooth muscle leading to bronchodilation. The competitive and reversible nature of antagonism was shown with human and animal origin receptors and isolated organ preparations. In preclinical in vitro as well as in vivo studies, prevention of acetylcholine-induced bronchoconstriction effects was dose-dependent and lasted longer than 24 hours. The clinical relevance of these findings is unknown. The bronchodilation following inhalation of aclidinium bromide is predominantly a site-specific effect.
12.2 Pharmacodynamics
Cardiovascular Effects
In a thorough QT Study, 200 mcg and 800 mcg TUDORZA PRESSAIR was administered to healthy volunteers once daily for 3 days; no effects on prolongation of QT interval were observed using QTcF heart-rate correction methods.
Additionally, the effect of TUDORZA PRESSAIR on cardiac rhythm was assessed in 336 COPD patients, 164 patients received aclidinium bromide 400 mcg twice daily and 172 patients received placebo, using 24-hr Holter monitoring. No clinically significant effects on cardiac rhythm were observed.
12.3 Pharmacokinetics
Absorption
The absolute bioavailability of aclidinium bromide is approximately 6% in healthy volunteers. Following twice-daily oral inhalation administration of 400 mcg aclidinium bromide in healthy subjects, peak steady state plasma levels were observed within 10 minutes after inhalation.
Distribution
Aclidinium bromide shows a volume of distribution of approximately 300 L following intravenous administration of 400 mcg in humans.
Metabolism
Clinical pharmacokinetics studies, including a mass balance study, indicate that the major route of metabolism of aclidinium bromide is hydrolysis, which occurs both chemically and enzymatically by esterases. Aclidinium bromide is rapidly and extensively hydrolyzed to its alcohol and dithienylglycolic acid derivatives, neither of which binds to muscarinic receptors and are devoid of pharmacologic activity.
Therefore, due to the low plasma levels achieved at the clinically relevant doses, aclidinium bromide and its metabolites are not expected to alter the disposition of drugs metabolized by the human CYP450 enzymes.
Elimination
Total clearance was approximately 170 L/h after an intravenous dose of aclidinium bromide in young healthy volunteers with an inter-individual variability of 36%. Intravenously administered radiolabeled aclidinium bromide was administered to healthy volunteers and was extensively metabolized with 1% excreted as unchanged aclidinium. Approximately 54% to 65% of the radioactivity was excreted in urine and 20% to 33% of the dose was excreted in feces. The combined results indicated that almost the entire aclidinium bromide dose was eliminated by hydrolysis. After dry powder inhalation, urinary excretion of aclidinium is about 0.09% of the dose and the estimated effective half-life is 5 to 8 hours.
Drug Interactions
Formal drug interaction studies were not performed. In vitro studies using human liver microsomes indicated that aclidinium bromide and its major metabolites do not inhibit CYP450, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, or 4A9/11 at concentrations up to 1,000-fold higher than the maximum plasma concentration that would be expected to be achieved at the therapeutic dose. Therefore, it is unlikely that aclidinium bromide causes CYP450 related drug interactions [see Drug Interactions (7)].
Specific Populations
Elderly Patients
The pharmacokinetic profile of aclidinium bromide and its main metabolites was assessed in 12 elderly COPD patients (aged 70 years or older) compared to a younger cohort of 12 COPD patients (40-59 years) that were administered 400 mcg aclidinium bromide once daily for 3 days via inhalation. No clinically significant differences in systemic exposure (AUC and Cmax) were observed when the two groups were compared. No dosage adjustment is necessary in elderly patients [see Use in Specific Populations (8.5)].
Renal Impairment
The impact of renal disease upon the pharmacokinetics of aclidinium bromide was studied in 18 subjects with mild, moderate, or severe renal impairment. Systemic exposure (AUC and Cmax) to aclidinium bromide and its main metabolites following single doses of 400 mcg aclidinium bromide was similar in renally impaired patients compared with 6 matched healthy control subjects. No dose adjustment is necessary in renally impaired patients [see Use in Specific Populations (8.6)].
Hepatic Impairment
The effects of hepatic impairment on the pharmacokinetics of aclidinium bromide were not studied. However, hepatic insufficiency is not expected to have relevant influence on aclidinium bromide pharmacokinetics, since it is predominantly metabolized by chemical and enzymatic hydrolysis to products that do not bind to muscarinic receptors [see Use in Specific Populations (8.7)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year inhalation studies were conducted in mice and rats to assess the carcinogenic potential of aclidinium bromide. No evidence of tumorigenicity was observed in rats and mice at aclidinium doses up to 0.20 and 2.4 mg/kg/day, respectively [approximately 10 and 80 times the Maximum Recommended Human Daily Inhalation Dose (MRHDID), respectively, based on summed AUCs of aclidinium bromide and its metabolites].
Aclidinium bromide was positive in the in vitro bacterial gene mutation assay and the in vitro thymidine locus mouse lymphoma assay. However, aclidinium bromide was negative in the in vivo mouse micronucleus assay and the in vivo/in vitro unscheduled DNA synthesis assay with rat liver.
Aclidinium bromide impaired several fertility and reproductive performance indices (increased number of days to mate, decreased conception rate, decreased number of corpora lutea, increased pre-implantation loss with consequent decreased number of implantations, and live embryos) in both male and female rats administered inhaled doses greater than or equal to 0.8 mg/kg/day [approximately 15 times the MRHDID based on summed AUCs of aclidinium bromide and its metabolites]. These adverse fertility effects were observed in the presence of paternal toxicity as evidenced by mortality and decreased body weight gain. However, there were no effects on mating index and sperm number and morphology. In the separate fertility assessments (treated males mated with untreated females; treated females mated with untreated males), no effect was observed in male and female rats at inhaled doses of 1.9 and 0.8 mg/kg/day, respectively [approximately 30 and 15 times the MRHDID, respectively, based on summed AUCs of aclidinium bromide and its metabolites].
14. Clinical Studies
14.1 Chronic Obstructive Pulmonary Disease (COPD)
The TUDORZA PRESSAIR clinical development program included a dose-ranging trial (Trial A) for nominal dose selection and three confirmatory lung function trials (Trials B, C, and D). Two additional lung function trials (Trials E and F) of aclidinium bromide alone and as part of a fixed-dose combination product also provided information on the effect of TUDORZA PRESSAIR on the St. George’s Respiratory Questionnaire (SGRQ) total score compared to placebo. A long-term study of up to 3 years (Trial G) evaluated the effect of TUDORZA PRESSAIR on major adverse cardiovascular events and on COPD exacerbations.
Dose-ranging trial
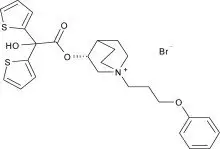
Trial A (NCT01120093) was a randomized, double-blind, placebo-controlled, active-controlled, crossover trial with 7-day treatment periods separated by 5-day washout periods. Trial A enrolled 79 patients who had a clinical diagnosis of COPD were 40 years of age or older, had a history of smoking at least 10 pack-years, had a forced expiratory volume in one second (FEV1) of at least 30% and less than 80% of predicted normal value, and a ratio of FEV1 over forced vital capacity (FEV1/FVC) of less than 0.7. Trial A included TUDORZA PRESSAIR doses of 400 mcg, 200 mcg, and 100 mcg twice daily, formoterol active control, and placebo. Trial A demonstrated that the effect on trough FEV1 and serial FEV1 in patients treated with the TUDORZA PRESSAIR 100 mcg twice daily and 200 mcg twice daily doses was lower compared to patients treated with the TUDORZA PRESSAIR 400 mcg twice daily dose (Figure 1).
Figure 1: Change from baseline in FEV1 Over Time (prior to and after administration of study drug) at Week 1 in Trial A
Confirmatory trials
Trials B (NCT00891462), C (NCT01045161), and D (NCT01001494) were three randomized, double-blind, placebo-controlled trials in patients with COPD. Trials B and C were 3 months in duration, and Trial D was 6 months in duration. These trials enrolled 1,276 patients who had a clinical diagnosis of COPD, including chronic bronchitis and emphysema, were 40 years of age or older, had a history of smoking at least 10 pack-years, had an FEV1 of at least 30% and less than 80% of predicted normal value, and a ratio of FEV1/FVC of less than 0.7; 59% were male, and 93% were Caucasian.
These clinical trials evaluated TUDORZA PRESSAIR 400 mcg twice daily (636 patients) and placebo (640 patients). TUDORZA PRESSAIR 400 mcg resulted in statistically significantly greater bronchodilation as measured by change from baseline in morning pre-dose FEV1 at 12 weeks (the primary efficacy endpoint) compared to placebo in all three trials (Table 2).
|
|||
|
Treatment Arm
|
Baseline |
Change from Baseline
|
Treatment Difference
|
|
Trial B (N=375) | |||
|
Aclidinium 400 mcg |
1.33 |
0.10 (0.01) |
0.12 (0.08, 0.16) |
|
Placebo |
1.38 |
-0.02 (0.02) | |
|
Trial C (N=359) | |||
|
Aclidinium 400 mcg |
1.25 |
0.06 (0.02) |
0.07 (0.03, 0.12) |
|
Placebo |
1.46 |
-0.01 (0.02) | |
|
Trial D* (N=542) | |||
|
Aclidinium 400 mcg |
1.51 |
0.06 (0.02) |
0.11 (0.07, 0.14) |
|
Placebo |
1.50 |
-0.05 (0.02) | |
|
SE=standard error, and LS mean=least square mean. LS mean, and 95% confidence interval were obtained from an ANCOVA model with change from baseline in trough FEV1 as response, with treatment group and sex as factors and baseline trough FEV1 and age as covariates. |
|||
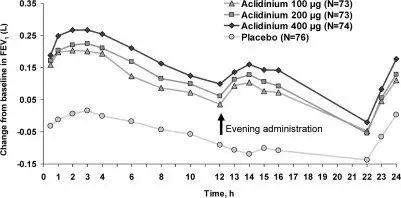
Serial spirometric evaluations were performed throughout daytime hours in a subset of patients in the three trials. The serial FEV1 values over 12 hours for one of the 3-month trials (Trial B) are displayed in Figure 2. Results for the other two placebo-controlled trials were similar to the results for Trial B. Improvement of lung function was maintained for 12 hours after a single dose and was consistent over the 3- or 6-month treatment period.
Figure 2: Mean FEV1 Over Time (prior to and after administration of study drug) on Day 1 and Week 12 in Subset of Patients Participating in the 12 hours Serial Spirometry Substudy for Trial B (a 3-month Placebo-Controlled Study)
Mean peak improvements in FEV1, for TUDORZA PRESSAIR relative to baseline were assessed in all patients in trials B, C, and D after the first dose on Day 1 and were similar at Week 12. In Trials B and D, but not in Trial C, patients treated with TUDORZA PRESSAIR used less daily rescue albuterol during the trial compared to patients treated with placebo.
Trials E (NCT01437397) and F (NCT01462942) were two randomized, double-blind, placebo-controlled trials of an aclidinium bromide-containing fixed- dose combination product and its components compared to placebo in patients with COPD, including chronic bronchitis and emphysema. Trials E and F were 6 months in duration. These trials enrolled 3421 patients who had a clinical diagnosis of COPD, were 40 years of age or older, had a history of smoking at least 10 pack-years, had an FEV1 of at least 30% and less than 80%, and a ratio of FEV1/FVC less than 0.7; 60.5% were male, 94.1% were Caucasian.
The St. George’s Respiratory Questionnaire (SGRQ) was assessed in Trials D, E, and F at 6 months. In Trial D, the SGRQ responder rate (defined as an improvement in score of 4 or more as threshold) was 54.3% in TUDORZA PRESSAIR compared to 39.5% in placebo, with odds ratio of 1.77 (95% CI 1.25, 2.52). In Trial E, the SGRQ responder rate in the TUDORZA PRESSAIR group was 54.5% compared to 38.7% in the placebo group, with odds ratio of 2.18 (95% CI 1.37, 3.48). In Trial F, the SGRQ responder rate in the TUDORZA PRESSAIR group was 53.5% compared to 53.2% in the placebo group, with odds ratio of 0.99 (95% CI 0.6, 1.64).
Long Term Safety and Efficacy Trial of up to 3-years
Trial G (NCT01966107) was a randomized, double-blind, placebo-controlled study of up to 36 months that evaluated the effect of TUDORZA PRESSAIR on major adverse cardiovascular events and COPD exacerbations in patients with moderate to very severe COPD with and without a history of COPD exacerbations.
The trial enrolled 3630 patients with COPD, including chronic bronchitis and emphysema, between 40 and 91 years of age, 58.7% were male and 90.7% were Caucasian, with a mean post-bronchodilator FEV1 of 47.9% of predicted value. All patients had a history of cardiovascular or cerebrovascular disease and/or significant cardiovascular risk factors. All patients had moderate to very severe COPD. 60.1% of patients had at least one prior moderate or severe COPD exacerbation within the past 12 months from the screening visit, and 39.9% had no history of a moderate or severe COPD exacerbation within the past 12 months.
Approximately 48% of enrolled patients had a prior history of at least 1 documented previous CV event; cerebrovascular disease (13.1%), coronary artery disease (35.4%), peripheral vascular disease or history of claudication (13.6%); 63.6% of them were on long-acting β2-adrenergic agonist (LABA or LABA/inhaled corticosteroid (ICS) maintenance therapy at study entry (LABA only 6.3%, LABA/ICS 57.3%)). The majority of patients had moderate (45.1%) or severe (40.2%) airflow obstruction.
The primary safety endpoint was the time to first occurrence of a major adverse cardiovascular event (MACE), defined as any of the following adjudicated events: cardiovascular death, non-fatal myocardial infarction (MI), or non-fatal ischemic stroke. The study was designed to exclude a pre-specified risk margin of 1.8 for the hazard ratio of MACE.
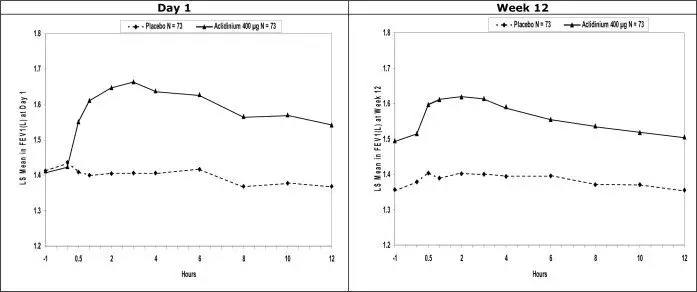
The results of Trial G, including each component event of the primary composite endpoint, are shown in Table 3. The proportion of patients with at least one MACE was 3.9% in the TUDORZA PRESSAIR group compared to 4.2% in the placebo group. The incidence rate of MACE was 2.4 per 100 patient-years on TUDORZA PRESSAIR vs. 2.8 per 100 patient-years on placebo. The estimated hazard ratio of MACE associated with TUDORZA PRESSAIR relative to placebo was 0.89 with a 95% confidence interval of (0.64, 1.23). The upper bound of this confidence interval, 1.23, excluded a risk margin larger than 1.8. TUDORZA PRESSAIR was non-inferior to placebo for risk of major adverse cardiovascular events.
|
|||||
|
TUDORZA PRESSAIR |
Placebo |
Hazard Ratio (95% CI) |
|||
|
N = 1791 |
Total PY = 2828.9 |
N = 1798 |
Total PY = 2748.1 | ||
|
Subjects with Events (%) |
Rate per 100 PY |
Subjects with Events (%) |
Rate per 100 PY | ||
|
MACE |
69 (3.9%) |
2.4 |
76 (4.2%) |
2.8 |
0.89 (0.64, 1.23) |
|
Component Events* | |||||
|
CV Death |
26 (1.5%) |
19 (1.1%) | |||
|
Non-fatal MI |
28 (1.6%) |
38 (2.1%) | |||
|
Non-fatal Ischemic Stroke |
18 (1.0%) |
24 (1.3%) | |||
The study was powered to rule out a hazard ratio of 1.8 in time to first MACE in TUDORZA PRESSAIR-treated patients relative to placebo.
MACE: major adverse cardiovascular event; CV: cardiovascular; MI: myocardial infarction; PY: patient-years; CI: confidence interval
The Kaplan-Meier-based cumulative event probability is presented in Figure 3 for time to first occurrence of the primary MACE composite endpoint by treatment arm.
Figure 3: Estimated Cumulative Incidence of First MACE
Exacerbations
Trial G also evaluated the effect of TUDORZA PRESSAIR 400 mcg BID on COPD exacerbations. The primary efficacy endpoint was the rate of moderate to severe exacerbations during the first year of treatment, defined as worsening of COPD symptoms (dyspnea, cough, sputum) for at least 2 consecutive days that required treatment with antibiotics and/or systemic corticosteroids or resulted in hospitalization or led to death. In total, 54.3% of patients in Trial G completed the first year of treatment, with 9.8% patients treated less than 12 months due to study closure. TUDORZA PRESSAIR demonstrated a statistically significant reduction in the rate of on-study moderate to severe COPD exacerbations during the first year by 17% compared to placebo (rate ratio [RR] 0.83; 95% CI 0.73 to 0.94; p=0.003). TUDORZA PRESSAIR also demonstrated a statistically significant reduction in the rate of on-study hospitalizations due to COPD exacerbation during the first year by 28% compared with placebo (RR 0.72; 95% CI 0.55 to 0.95; p=0.02).
Figure 4: Time to first moderate or severe COPD exacerbation (days), on-study analysis, Kaplan-Meier plot (Full Analysis Set)
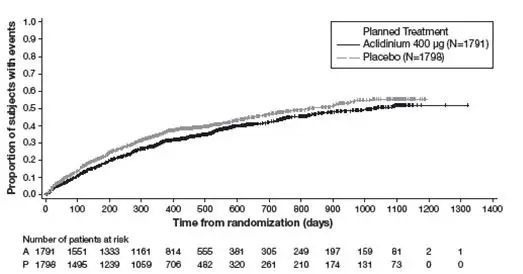
p-value for comparing Aclidinium 400 mcg versus Placebo is based on the Log-rank test stratified by baseline COPD severity and smoking status is p = 0.004.
The Kaplan-Meier curves indicate that the time to first on-study moderate or severe COPD exacerbation was delayed in the aclidinium 400 mcg group compared to the placebo group (see Figure 4). Patients in the aclidinium bromide 400 mcg group had a 15% relative reduction of the risk of an exacerbation (HR 0.85; 95% CI [0.77, 0.95], p=0.004).
16. How is Tudorza Pressair supplied
TUDORZA® PRESSAIR® (aclidinium bromide inhalation powder) 400 mcg is supplied in a sealed bag and is available in 60 metered doses (NDC 0310-0800-60) and 30 metered doses (NDC 0310-0800-39).
The active ingredient is administered using a multi-dose dry powder inhaler, PRESSAIR®, which delivers 60 doses or 30 doses of aclidinium bromide powder for oral inhalation. The PRESSAIR inhaler is a white and green colored device and is comprised of an assembled plastic dosing mechanism with a dose indicator, a drug-product storage unit containing the drug-product formulation, and a mouthpiece covered by a green protective cap.
Store TUDORZA PRESSAIR in a dry place at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature]. Do not store the inhaler on a vibrating surface.
The PRESSAIR inhaler should be stored inside the sealed bag and only be opened immediately before use. Throw away the bag.
Throw away (dispose of) the PRESSAIR inhaler after the marking “0” with a red background shows in the middle of the dose indicator, when the device is empty and locks out, or 45 days after the date you opened the sealed bag that the inhaler comes in, whichever comes first.
Keep out of reach of children.
17. Patient Counseling Information
See FDA-approved Patient Labeling (Patient Information and Instructions for Use)
- •
-
Acute Bronchospasm
Instruct patients that TUDORZA PRESSAIR is a twice daily maintenance bronchodilator and should not be used for immediate relief of breathing problems (i.e., as a rescue medication) [see Warnings and Precautions (5.1)]. - •
-
Paradoxical Bronchospasm
Inform patients that TUDORZA PRESSAIR can cause paradoxical bronchospasm. Advise patients that if paradoxical bronchospasm occurs, patients should discontinue TUDORZA PRESSAIR [see Warnings and Precautions (5.2)]. - •
-
Visual Effects
Eye pain or discomfort, blurred vision, visual halos, or colored images in association with red eyes from conjunctival congestion and corneal edema may be signs of acute narrow-angle glaucoma. Inform patients to consult a physician immediately should any of these signs and symptoms develop. Advise patients that miotic eyedrops alone are not considered to be effective treatment [see Warnings and Precautions (5.3)].
- Inform patients that care must be taken not to allow the powder to enter into the eyes as this may cause blurring of vision and pupil dilation.
- •
-
Urinary Retention
Difficulty passing urine and dysuria may be symptoms of new or worsening prostatic hyperplasia or bladder outlet obstruction. Patients should be instructed to consult a physician immediately should any of these signs or symptoms develop [see Warnings and Precautions (5.4)]. - •
-
Immediate Hypersensitivity Reactions
Inform patients that anaphylaxis, angioedema (including swelling of the lips, tongue, or throat), urticaria, rash, bronchospasm, or itching, may occur after administration of TUDORZA PRESSAIR. Advise patient to immediately discontinue treatment and consult a physician should any of these signs or symptoms develop [see Contraindications (4) and Warnings and Precautions (5.5)]. - •
-
Instructions for Administering TUDORZA PRESSAIR
It is important for patients to understand how to correctly use TUDORZA PRESSAIR.
Inform patients that if they miss a dose, they should take their next dose at the usual time; they should not take 2 doses at one time.
Distributed by:
AstraZeneca Pharmaceuticals LP,
Wilmington, DE 19850
Under license of ALMIRALL, S.A.
TUDORZA® is a registered trademark of ALMIRALL, S.A. and PRESSAIR® is a registered trademark of the AstraZeneca group of companies.
© AstraZeneca 2019
|
Patient Information TUDORZA® PRESSAIR® (TU-door-za PRESS-air) (aclidinium bromide inhalation powder) |
|||
|
Important: For oral inhalation only. Do not get TUDORZA PRESSAIR in your eyes. |
|||
|
What is TUDORZA PRESSAIR?
It is not known if TUDORZA PRESSAIR is safe and effective in children. |
|||
|
Who should not use TUDORZA PRESSAIR? Do not use TUDORZA PRESSAIR if you:
|
|||
|
What should I tell my doctor before using TUDORZA PRESSAIR? Before you use TUDORZA PRESSAIR, tell your doctor about all your medical conditions, including if you:
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines and eyedrops, vitamins, and herbal supplements. TUDORZA PRESSAIR and certain other medicines may interact with each other causing serious side effects. Especially tell your doctor if you take:
Know the medicines you take. Keep a list of them to show your doctor and pharmacist each time you get a new medicine. |
|||
|
How should I use TUDORZA PRESSAIR?
|
|||
|
What are the possible side effects of TUDORZA PRESSAIR? TUDORZA PRESSAIR can cause serious side effects including:
|
|||
|
|
|
|
|
|
|||
|
|
|
|
|
|
|||
|
|
|
|
|
|
If you have any of these symptoms, stop using TUDORZA PRESSAIR and call your doctor or go to the nearest hospital emergency room right away. The most common side effects of TUDORZA PRESSAIR include: |
|||
|
|
|
|
|
|
Tell your doctor if you get any side effect that bothers you or does not go away. These are not all the possible side effects of TUDORZA PRESSAIR. Ask your doctor or pharmacist for more information. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
|
How should I store TUDORZA PRESSAIR?
Keep TUDORZA PRESSAIR and all medicines out of the reach of children. |
|||
|
General information about the safe and effective use of TUDORZA PRESSAIR. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TUDORZA PRESSAIR for a condition for which it was not prescribed. Do not give TUDORZA PRESSAIR to other people even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information about TUDORZA PRESSAIR that is written for health professionals. |
|||
|
What are the ingredients in TUDORZA PRESSAIR? Active ingredient: aclidinium bromide Inactive ingredient: lactose monohydrate For more information, go to www.tudorza.com, or call 1-800-236-9933. |
|||
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: April 2019
|
Instructions for Use TUDORZA® PRESSAIR® (TU-door-za PRESS-air) (aclidinium bromide inhalation powder) |
||||||
|
For oral inhalation only This Instructions for Use contains information on how to use your TUDORZA PRESSAIR inhaler. It is important that you read this information as the TUDORZA PRESSAIR inhaler may work differently from inhalers you have used before. Read this Instructions for Use before you start using TUDORZA PRESSAIR and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment. If you have any questions about how to use your inhaler, ask your doctor, pharmacist, or nurse for help. |
||||||
|
The Instructions for Use is divided into the following sections:
|
||||||
|
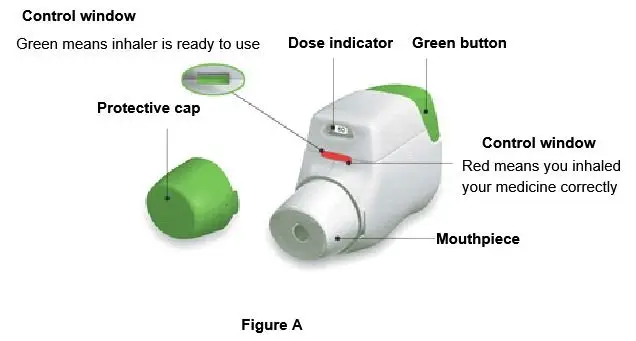
Getting Started Become familiar with the parts of TUDORZA PRESSAIR inhaler (Figure A). |
||||||
|
||||||
|
Before use:
|
||||||
 | ||||||
|
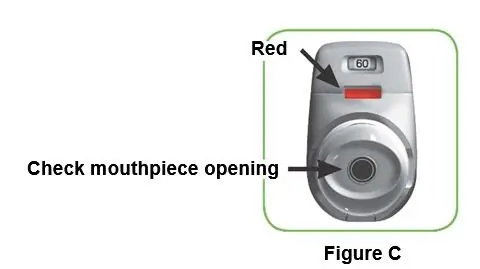
Step 1: Prepare your dose 1.1 Look in the opening of the mouthpiece and make sure nothing is blocking it (Figure C). 1.2 Look at the control window. The control window should be red (Figure C). |
||||||
|
|  | |||||
|
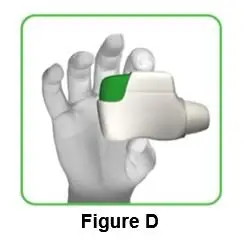
1.3 Hold the inhaler horizontally with the mouthpiece facing you and the green button on top (Figure D). |
||||||
 |
||||||
|
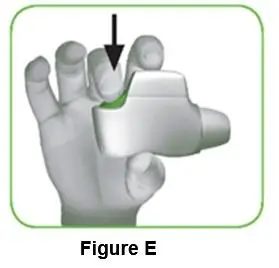
1.4 Press the green button all the way down to load your dose (Figure E). When you press the green button all the way down, the control window changes from red to green. Make sure the green button is on top. Do not tilt the inhaler. |
||||||
 |
||||||
|
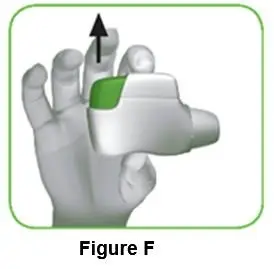
1.5 Release the green button (Figure F). Make sure you release the green button, so the inhaler can work correctly. |
||||||
 |
||||||
|
Stop and Check: 1.6 Make sure the control window is now green (Figure G). |
||||||
 |
||||||
|
What to do if the control window is still red after pressing the button (Figure H). |
||||||
|
 |
||||||
|
The dose is not prepared. Go back to “Step 1: Prepare your dose” and repeat steps 1.1 to 1.6. |
||||||
|
Step 2: Inhale your medicine |
||||||
|
Read steps 2.1 to 2.7 fully before use. Do not tilt the inhaler. |
||||||
|
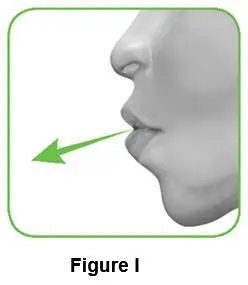
2.1 Hold the inhaler away from your mouth, and breathe out completely. Never breathe out into the inhaler (Figure I). |
||||||
 |
||||||
|
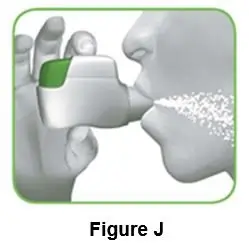
2.2 Hold your head upright, put the mouthpiece between your lips, and close your lips tightly around the mouthpiece (Figure J). |
||||||
|
Do not hold the green button down while inhaling |
||||||
 |
||||||
|
2.3 Take a strong, deep breath through your mouth. Keep breathing in for as long as possible. |
||||||
|
A “click” will let you know that you are inhaling correctly. Keep breathing in as long as possible after you hear the “click”. Some people may not hear the “click”. Use the control window to make sure you have inhaled correctly. |
||||||
|
2.4 Take the inhaler out of your mouth. |
||||||
|
2.5 Hold your breath for as long as possible. |
||||||
|
2.6 Slowly breathe out, away from the inhaler. |
||||||
|
Some people may have a grainy sensation in their mouth, or a slightly sweet or bitter taste. Do not take an extra-dose if you do not taste or feel anything after inhaling. |
||||||
|
Stop and Check: 2.7 Make sure the control window is now red (Figure K). This means you have inhaled your medicine correctly. |
||||||
 |
||||||
|
What to do if the control window is still green after inhalation (Figure L). |
||||||
 |
||||||
|
This means you have not inhaled your medicine correctly. Go back to “Step 2: Inhale your medicine” and repeat steps 2.1 to 2.7. If the control window still does not change to red, you may have forgotten to release the green button before inhaling, or you may not have inhaled strongly enough. If this happens, try again. Make sure you have released the green button, and you have breathed out completely. Then take a strong, deep breath through the mouthpiece. Contact your doctor if the control window is still green after repeated attempts. |
||||||
|
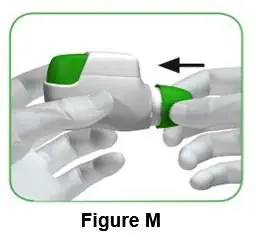
Push the protective cap back onto the mouthpiece after each use (Figure M) to prevent contamination of the inhaler with dust or other materials. You should throw away (dispose of) your inhaler if you lose the cap. |
||||||
 |
||||||
|
For more information about TUDORZA PRESSAIR and a video demonstration on how to use TUDORZA PRESSAIR, go to www.tudorza.com. |
||||||
|
Additional information |
||||||
|
What should I do if I accidentally prepare a dose? Store your inhaler with the protective cap in place until it is time to inhale your medicine, then remove the cap and start at Step 1.6. How does the dose indicator work?
Each time you load a dose by pressing the green button, the dose indicator moves by a small amount towards the next number (50, 40, 30, 20, 10, or 0). |
||||||
 |
||||||
|
When should I get a new inhaler? You should throw away (dispose of) your inhaler if it appears to be damaged or if you lose the protective cap. When a red band appears in the dose indicator, this means you are close to your last dose (Figure N). You should throw away (dispose of) the inhaler and use a new one:
|
||||||
 |
||||||
|
How do I know that my inhaler is empty?
|
||||||
 |
||||||
|
How should I clean the inhaler? Never use water to clean the inhaler, as this may damage your medicine. If you want to clean your inhaler, wipe the outside of the mouthpiece with a dry tissue or paper towel. How should I store TUDORZA PRESSAIR?
Keep TUDORZA PRESSAIR and all medicines out of the reach of children. |
||||||
|
Questions and Answers about your TUDORZA PRESSAIR Inhaler |
||||||
|
Question |
Answer |
|||||
|
Do I need to take extra steps to prepare the inhaler before first use? |
TUDORZA PRESSAIR comes preloaded with medicine and is ready to use. Remove the inhaler from the sealed bag and follow the step-by-step Instructions for Use. |
|||||
|
How do I know if the TUDORZA PRESSAIR inhaler is ready to use before taking each dose? |
The TUDORZA PRESSAIR inhaler is ready to use when the control window on the front of the inhaler is green (Figure G).
|
|||||
|
What if the TUDORZA PRESSAIR inhaler window does not change from red to green? |
The dose is not prepared. Go back to “Step 1: Prepare your dose” and repeat steps 1.1 to 1.6.
|
|||||
|
How do I know that I used TUDORZA PRESSAIR correctly? |
The TUDORZA PRESSAIR inhaler has a helpful feature to let you know that you have inhaled your medicine correctly.
|
|||||
|
What if the TUDORZA PRESSAIR inhaler control window does not change color from green back to red after I inhale? |
This means you may not have inhaled the medicine strongly enough. Go back to “Step 2: Inhale your medicine” and repeat steps 2.1 to 2.7.
|
|||||
|
What if I do not see the dose indicator move after I inhaled? |
|
|||||
|
Can the TUDORZA PRESSAIR inhaler release too much medicine or lose doses of medicine from the inhaler? |
|
|||||
|
This Instructions for Use has been approved by the U.S. Food and Drug Administration. |
||||||
|
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850 Under license of ALMIRALL, S.A. TUDORZA® is a registered trademark of ALMIRALL, S.A. and PRESSAIR® is a registered trademark of the AstraZeneca group of companies. Revised: April 2019 |
||||||

| TUDORZA PRESSAIR
aclidinium bromide powder, metered |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Labeler - AstraZeneca Pharmaceuticals LP (054743190) |
| Registrant - AstraZeneca PLC (230790719) |
