Drug Detail:Veklury (Remdesivir [ rem-des-i-veer ])
Drug Class: Purine nucleosides
Highlights of Prescribing Information
VEKLURY® (remdesivir) for injection, for intravenous use
VEKLURY® (remdesivir) injection, for intravenous use
Initial U.S. Approval: 2020
Recent Major Changes
| Indications and Usage (1) | 12/2022 |
| Dosage and Administration | |
| Testing Before Starting and During Treatment with VEKLURY (2.2) | 07/2023 |
| Renal Impairment (2.4) | 07/2023 |
Indications and Usage for Veklury
VEKLURY is a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) nucleotide analog RNA polymerase inhibitor indicated for the treatment of coronavirus disease 2019 (COVID-19) in adults and pediatric patients (28 days of age and older and weighing at least 3 kg) who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. (1)
Veklury Dosage and Administration
- The only approved dosage form of VEKLURY for pediatric patients weighing 3 kg to less than 40 kg is VEKLURY for injection (supplied as 100 mg lyophilized powder in vial). (2.1)
- Testing: In all patients, before starting VEKLURY and during treatment as clinically appropriate, perform hepatic laboratory testing. Assess prothrombin time before starting VEKLURY and monitor as clinically appropriate. (2.2)
- Recommended dosage:
- Adults and pediatric patients weighing at least 40 kg: a single loading dose of VEKLURY 200 mg on Day 1 followed by once-daily maintenance doses of VEKLURY 100 mg from Day 2 via intravenous infusion. (2.3)
- Pediatric patients 28 days of age and older and weighing 3 kg to less than 40 kg: a single loading dose of VEKLURY 5 mg/kg on Day 1 followed by once-daily maintenance doses of VEKLURY 2.5 mg/kg from Day 2 via intravenous infusion. (2.3)
- Hospitalized patients: The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made. (2.3)
- For hospitalized patients requiring invasive mechanical ventilation and/or ECMO, the recommended total treatment duration is 10 days. (2.3)
- For hospitalized patients not requiring invasive mechanical ventilation and/or ECMO, the recommended treatment duration is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days for a total treatment duration of up to 10 days. (2.3)
- Non-hospitalized patients: The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made and within 7 days of symptom onset. (2.3)
- For non-hospitalized patients diagnosed with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, the recommended total treatment duration is 3 days (2.3).
- Renal impairment: No dosage adjustment of VEKLURY is recommended in patients with any degree of renal impairment, including those on dialysis. (2.4)
- Administer VEKLURY via intravenous (IV) infusion over 30 to 120 minutes. (2.5, 2.6)
- Dose preparation and administration: Refer to the full prescribing information for further details for both formulations. (2.5, 2.6)
- Storage of prepared dosages: VEKLURY contains no preservative. (2.7)
Dosage Forms and Strengths
- For injection: 100 mg of remdesivir as a lyophilized powder, in a single-dose vial. (3)
- Injection: 100 mg/20 mL (5 mg/mL) remdesivir, in a single-dose vial. (3)
Contraindications
VEKLURY is contraindicated in patients with a history of clinically significant hypersensitivity reactions to VEKLURY or any components of the product. (4)
Warnings and Precautions
- Hypersensitivity including infusion-related and anaphylactic reactions: Hypersensitivity reactions have been observed during and following administration of VEKLURY. Slower infusion rates, with a maximum infusion time of up to 120 minutes, can be considered to potentially prevent signs and symptoms of hypersensitivity. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate. If signs and symptoms of a clinically significant hypersensitivity reaction occur, immediately discontinue administration of VEKLURY and initiate appropriate treatment. (5.1)
- Increased risk of transaminase elevations: Transaminase elevations have been observed in healthy volunteers and have also been reported in patients with COVID-19 who received VEKLURY. Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate. Consider discontinuing VEKLURY if ALT levels increase to greater than 10 times the upper limit of normal. Discontinue VEKLURY if ALT elevation is accompanied by signs or symptoms of liver inflammation. (5.2)
- Risk of reduced antiviral activity when coadministered with chloroquine phosphate or hydroxychloroquine sulfate: Coadministration of VEKLURY and chloroquine phosphate or hydroxychloroquine sulfate is not recommended based on data from cell culture experiments demonstrating a potential antagonistic effect of chloroquine on the intracellular metabolic activation and antiviral activity of VEKLURY. (5.3)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence greater than or equal to 5%, all grades) observed with treatment with VEKLURY are nausea, ALT increased, and AST increased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 7/2023
Full Prescribing Information
1. Indications and Usage for Veklury
VEKLURY is indicated for the treatment of coronavirus disease 2019 (COVID-19) in adults and pediatric patients (28 days of age and older and weighing at least 3 kg) who are [see Clinical Studies (14)]:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
2. Veklury Dosage and Administration
2.1 Dosage and Administration Overview
- VEKLURY may only be administered in settings in which healthcare providers have immediate access to medications to treat a severe infusion or hypersensitivity reaction, such as anaphylaxis, and the ability to activate the emergency medical system (EMS), as necessary [see Dosage and Administration (2.5, 2.6), Warnings and Precautions (5.1)].
- Administer VEKLURY for the treatment of COVID-19 in adults and pediatric patients (28 days of age and older and weighing at least 3 kg) by intravenous infusion only. Do not administer by any other route.
- There are TWO different formulations of VEKLURY:
-
VEKLURY for injection (supplied as 100 mg lyophilized powder in vial) must be reconstituted with Sterile Water for Injection prior to diluting with 0.9% sodium chloride injection.
- The only approved dosage form of VEKLURY for pediatric patients weighing 3 kg to less than 40 kg is VEKLURY for injection (supplied as 100 mg lyophilized powder in vial).
- VEKLURY injection (supplied as 100 mg/20 mL [5 mg/mL] solution in vial) must be further diluted in 250 mL of 0.9% sodium chloride injection infusion bag.
-
VEKLURY for injection (supplied as 100 mg lyophilized powder in vial) must be reconstituted with Sterile Water for Injection prior to diluting with 0.9% sodium chloride injection.
- There are differences in the way the two formulations are prepared. Carefully follow the product-specific preparation instructions below [see Dosage and Administration (2.5, 2.6)].
2.2 Testing Before Starting and During Treatment with VEKLURY
Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate [see Warnings and Precautions (5.2) and Use in Specific Populations (8.7)].
Determine prothrombin time in all patients before starting VEKLURY and monitor while receiving VEKLURY as clinically appropriate [see Adverse Reactions (6.1)].
2.3 Recommended Dosage in Adults and Pediatric Patients 28 Days of Age and Older and Weighing at Least 3 kg
- The recommended dosage for adults and pediatric patients weighing at least 40 kg is a single loading dose of VEKLURY 200 mg on Day 1 via intravenous infusion followed by once-daily maintenance doses of VEKLURY 100 mg from Day 2 via intravenous infusion.
- The recommended dosage for pediatric patients 28 days of age and older and weighing 3 kg to less than 40 kg is a single loading dose of VEKLURY 5 mg/kg on Day 1 via intravenous infusion followed by once-daily maintenance doses of VEKLURY 2.5 mg/kg from Day 2 via intravenous infusion.
2.4 Renal Impairment
No dosage adjustment of VEKLURY is recommended in patients with any degree of renal impairment, including patients on dialysis. VEKLURY may be administered without regard to the timing of dialysis [see Dosage and Administration (2.3) and Use in Specific Populations (8.4, 8.6)].
2.5 Dosage Preparation and Administration in Adults and Pediatric Patients Weighing at Least 40 kg
There are differences in the way the two formulations are prepared. Carefully follow the product-specific preparation instructions below.
VEKLURY for Injection (Supplied as 100 mg Lyophilized Powder in Vial)
Dilution Instructions
Care should be taken during admixture to prevent inadvertent microbial contamination. As there is no preservative or bacteriostatic agent present in this product, aseptic technique must be used in preparation of the final parenteral solution. It is always recommended to administer intravenous medication immediately after preparation when possible.
- Reconstituted VEKLURY for injection, containing 100 mg/20 mL remdesivir solution, must be further diluted in either a 100 mL or 250 mL 0.9% sodium chloride injection infusion bag. Refer to Table 1 for instructions.
| VEKLURY dose | 0.9% sodium chloride injection infusion bag volume to be used | Volume to be withdrawn and discarded from 0.9% sodium chloride injection infusion bag | Required volume of reconstituted VEKLURY for injection |
|---|---|---|---|
| Loading dose 200 mg (2 vials) | 250 mL | 40 mL | 40 mL (2 × 20 mL) |
| 100 mL | 40 mL | 40 mL (2 × 20 mL) | |
| Maintenance dose 100 mg (1 vial) | 250 mL | 20 mL | 20 mL |
| 100 mL | 20 mL | 20 mL |
- Withdraw and discard the required volume of 0.9% sodium chloride injection from the bag following instructions in Table 1, using an appropriately sized syringe and needle.
- Withdraw the required volume of reconstituted VEKLURY for injection from the VEKLURY vial following instructions in Table 1, using an appropriately sized syringe. Discard any unused portion remaining in the reconstituted vial.
- Transfer the required volume of reconstituted VEKLURY for injection to the selected infusion bag.
- Gently invert the bag 20 times to mix the solution in the bag. Do not shake.
- The prepared infusion solution can be stored for 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) prior to administration.
Administration Instructions
Do not administer the prepared diluted solution simultaneously with any other medication. The compatibility of VEKLURY injection with intravenous solutions and medications other than 0.9% sodium chloride injection, USP is not known. Administer VEKLURY via intravenous infusion over 30 to 120 minutes.
Administration should be under conditions where management of severe hypersensitivity reactions, such as anaphylaxis, is possible. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate [see Warnings and Precautions (5.1)].
Administer the diluted solution with the infusion rate described in Table 2.
| Infusion bag volume | Infusion time | Rate of infusion |
|---|---|---|
| 250 mL | 30 min | 8.33 mL/min |
| 60 min | 4.17 mL/min | |
| 120 min | 2.08 mL/min | |
| 100 mL | 30 min | 3.33 mL/min |
| 60 min | 1.67 mL/min | |
| 120 min | 0.83 mL/min |
VEKLURY Injection (Supplied as 100 mg/20 mL [5 mg/mL] Solution in Vial)
Dilution Instructions
Care should be taken during admixture to prevent inadvertent microbial contamination. As there is no preservative or bacteriostatic agent present in this product, aseptic technique must be used in preparation of the final parenteral solution. It is always recommended to administer intravenous medication immediately after preparation when possible.
- Remove the required number of single-dose vial(s) from storage. Each vial contains 100 mg/20 mL of remdesivir. For each vial:
- Equilibrate to room temperature (20°C to 25°C [68°F to 77°F]). Sealed vials can be stored up to 12 hours at room temperature prior to dilution.
- Inspect the vial to ensure the container closure is free from defects and the solution is free of particulate matter.
- VEKLURY injection must be diluted in an infusion bag containing 250 mL of 0.9% sodium chloride injection only. Refer to Table 3 for instructions.
| VEKLURY dose | 0.9% sodium chloride injection infusion bag volume to be used | Volume to be withdrawn and discarded from 0.9% sodium chloride injection infusion bag | Required volume of VEKLURY injection |
|---|---|---|---|
| Loading dose 200 mg (2 vials) | 250 mL | 40 mL | 40 mL (2 × 20 mL) |
| Maintenance dose 100 mg (1 vial) | 20 mL | 20 mL |
- Withdraw and discard the required volume of 0.9% sodium chloride injection from the bag following instructions in Table 3, using an appropriately sized syringe and needle.
- Withdraw the required volume of VEKLURY injection from the VEKLURY vial following instructions in Table 3, using an appropriately sized syringe.
- Pull the syringe plunger rod back to fill the syringe with approximately 10 mL of air.
- Inject the air into the VEKLURY injection vial above the level of the solution.
- Invert the vial and withdraw the required volume of VEKLURY injection solution into the syringe. The last 5 mL of solution requires more force to withdraw.
- Transfer the required volume of VEKLURY injection to the infusion bag.
- Gently invert the bag 20 times to mix the solution in the bag. Do not shake.
- The prepared infusion solution is stable for 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]).
Administration Instructions
Do not administer the prepared diluted solution simultaneously with any other medication. The compatibility of VEKLURY injection with intravenous solutions and medications other than 0.9% sodium chloride injection, USP is not known. Administer VEKLURY via intravenous infusion over 30 to 120 minutes.
Administration should be under conditions where management of severe hypersensitivity reactions, such as anaphylaxis, is possible. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate [see Warnings and Precautions (5.1)].
Administer the diluted solution with the infusion rate described in Table 4.
| Infusion bag volume | Infusion time | Rate of infusion |
|---|---|---|
| 250 mL | 30 min | 8.33 mL/min |
| 60 min | 4.17 mL/min | |
| 120 min | 2.08 mL/min |
2.6 Dosage Preparation and Administration in Pediatric Patients 28 Days of Age and Older and Weighing 3 kg to Less Than 40 kg
The only approved dosage form of VEKLURY for pediatric patients 28 days of age and older and weighing 3 kg to less than 40 kg is VEKLURY for injection (supplied as 100 mg lyophilized powder in vial). Carefully follow the product-specific preparation instructions below.
Use VEKLURY for Injection (Supplied as 100 mg Lyophilized Powder in Vial) only.
3. Dosage Forms and Strengths
- VEKLURY for injection, 100 mg, available as a sterile, preservative-free white to off-white to yellow lyophilized powder in single-dose vial for reconstitution.
- VEKLURY injection, 100 mg/20 mL (5 mg/mL), available as a clear, colorless to yellow solution, free of visible particles in single-dose vial.
4. Contraindications
VEKLURY is contraindicated in patients with a history of clinically significant hypersensitivity reactions to VEKLURY or any components of the product [see Warnings and Precautions (5.1)].
5. Warnings and Precautions
5.1 Hypersensitivity Including Infusion-related and Anaphylactic Reactions
Hypersensitivity reactions, including infusion-related and anaphylactic reactions, have been observed during and following administration of VEKLURY; most occurred within one hour. Signs and symptoms may include hypotension, hypertension, tachycardia, bradycardia, hypoxia, fever, dyspnea, wheezing, angioedema, rash, nausea, diaphoresis, and shivering. Slower infusion rates, with a maximum infusion time of up to 120 minutes, can be considered to potentially prevent these signs and symptoms. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate. If signs and symptoms of a clinically significant hypersensitivity reaction occur, immediately discontinue administration of VEKLURY and initiate appropriate treatment. The use of VEKLURY is contraindicated in patients with known hypersensitivity to VEKLURY or any components of the product [see Contraindications (4)].
5.2 Increased Risk of Transaminase Elevations
Transaminase elevations have been observed in healthy volunteers who received 200 mg of VEKLURY followed by 100 mg doses for up to 10 days; the transaminase elevations were mild (Grade 1) to moderate (Grade 2) in severity and resolved upon discontinuation of VEKLURY. Transaminase elevations have also been reported in patients with COVID-19 who received VEKLURY [see Adverse Reactions (6.1)]. Because transaminase elevations have been reported as a clinical feature of COVID-19, and the incidence was similar in patients receiving placebo versus VEKLURY in clinical trials of VEKLURY, discerning the contribution of VEKLURY to transaminase elevations in patients with COVID-19 can be challenging.
Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate [see Dosage and Administration (2.1) and Use in Specific Populations (8.7)].
- Consider discontinuing VEKLURY if ALT levels increase to greater than 10 times the upper limit of normal.
- Discontinue VEKLURY if ALT elevation is accompanied by signs or symptoms of liver inflammation.
5.3 Risk of Reduced Antiviral Activity When Coadministered with Chloroquine Phosphate or Hydroxychloroquine Sulfate
Coadministration of VEKLURY and chloroquine phosphate or hydroxychloroquine sulfate is not recommended based on data from cell culture experiments demonstrating a potential antagonistic effect of chloroquine on the intracellular metabolic activation and antiviral activity of VEKLURY [see Drug Interactions (7) and Microbiology (12.4)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in other sections of the labeling:
- Hypersensitivity Including Infusion-related and Anaphylactic Reactions [see Warnings and Precautions (5.1)]
- Increased Risk of Transaminase Elevations [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials in Adult Subjects
The safety of VEKLURY is based on data from four Phase 3 studies in 1,476 hospitalized adult subjects with COVID-19, one Phase 3 study in 279 non-hospitalized adult and pediatric subjects (12 years of age and older weighing at least 40 kg) with mild-to-moderate COVID-19, four Phase 1 studies in 131 healthy adults, and from patients with COVID-19 who received VEKLURY under the Emergency Use Authorization or in a compassionate use program.
Clinical Trials Experience in Adults with COVID-19
NIAID ACTT-1 was a randomized, double-blind, placebo-controlled clinical trial in hospitalized subjects with mild, moderate, and severe COVID-19 treated with VEKLURY (n=532) or placebo (n=516) for up to 10 days. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days [see Clinical Studies (14.1)]. The collection of adverse event data in this trial was limited to severe (Grade 3) or potentially life-threatening (Grade 4) adverse events, serious adverse events, adverse events leading to study drug discontinuation, and moderate (Grade 2) severity or higher hypersensitivity reactions. Rates of adverse reactions (≥ Grade 3), serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 5.
| Types of Adverse Reactions | VEKLURY N=532 n (%) | Placebo N=516 n (%) |
|---|---|---|
|
||
| Adverse reactions, Grades ≥3 | 41 (8%) | 46 (9%) |
| Serious adverse reactions | 2 (0.4%)* | 3 (0.6%) |
| Adverse reactions leading to treatment discontinuation | 11 (2%)† | 15 (3%) |
Study GS-US-540-5773 was a randomized, open-label clinical trial in hospitalized subjects with severe COVID-19 treated with VEKLURY 200 mg on Day 1 and 100 mg once daily for 5 (n=200) or 10 days (n=197). Adverse reactions were reported in 33 (17%) subjects in the 5-day group and 40 (20%) subjects in the 10-day group [see Clinical Studies (14.2)]. The most common adverse reactions occurring in at least 5% of subjects in either the VEKLURY 5-day or 10-day group, respectively, were nausea (5% vs 3%), AST increased (3% vs 6%), and ALT increased (2% vs 7%). Rates of any adverse reactions, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 6.
| Types of Adverse Reactions | VEKLURY 5 Days N=200 n (%) | VEKLURY 10 Days N=197 n (%) |
|---|---|---|
|
||
| Any adverse reaction, all Grades | 33 (17%) | 40 (20%) |
| Serious adverse reactions | 3 (2%)* | 4 (2%)* |
| Adverse reactions leading to treatment discontinuation | 5 (3%)† | 9 (5%)† |
Study GS-US-540-5774 was a randomized, open-label clinical trial in hospitalized subjects with moderate COVID-19 treated with VEKLURY 200 mg on Day 1 and 100 mg daily for 5 (n=191) or 10 days (n=193), or standard of care (SOC) only (n=200) [see Clinical Studies (14.3)]. Adverse reactions were reported in 36 (19%) subjects in the 5-day group and 25 (13%) subjects in the 10-day group. The most common adverse reaction occurring in at least 5% of subjects in the VEKLURY groups was nausea (7% in the 5-day group, 4% in the 10-day group). Rates of any adverse reactions, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 7.
| Types of Adverse Reactions | VEKLURY 5 Days N=191 n (%) | VEKLURY 10 Days N=193 n (%) |
|---|---|---|
|
||
| Any adverse reaction, all Grades | 36 (19%) | 25 (13%) |
| Serious adverse reactions | 1 (<1%)† | 0 |
| Adverse reactions leading to treatment discontinuation | 4 (2%)‡ | 4 (2%)‡ |
Study GS-US-540-9012 was a randomized, double-blind, placebo-controlled clinical trial in subjects who were non-hospitalized, were symptomatic for COVID-19 for ≤7 days, had confirmed SARS-CoV-2 infection, and had at least one risk factor for progression to hospitalization treated with VEKLURY (n=279; 276 adults and 3 pediatric subjects 12 years of age and older weighing at least 40 kg) or placebo (n=283; 278 adults and 5 pediatric subjects 12 years of age and older weighing at least 40 kg) for 3 days. Of the 279 subjects treated with VEKLURY, 227 subjects received at least one dose of VEKLURY at an outpatient facility, 44 subjects received at least one dose of VEKLURY in a home healthcare setting, and 8 subjects received at least one dose of VEKLURY at a skilled nursing facility. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days [see Clinical Studies (14.4)]. Adverse reactions (all grades) were reported in 34 (12%) subjects in the VEKLURY group and 25 (9%) subjects in the placebo group. The most common adverse reaction occurring in at least 5% of subjects in the VEKLURY group was nausea (6%). There were no serious adverse reactions or adverse reactions leading to treatment discontinuation in either treatment group. Safety in subjects who received VEKLURY in a home healthcare setting was comparable to that observed in the overall GS-US-540-9012 study population, but these findings are based on limited data.
Laboratory Abnormalities
Study GS-US-399-5505 was a Phase 1, randomized, blinded, placebo-controlled clinical trial in healthy volunteers administered VEKLURY 200 mg on Day 1 and 100 mg for either 4 days or 9 days. Mild (Grade 1, n=8) to moderate (Grade 2, n=1) elevations in ALT were observed in 9 of 20 subjects receiving 10 days of VEKLURY; the elevations in ALT resolved upon discontinuation of VEKLURY. No subjects (0 of 9) who received 5 days of VEKLURY had graded increases in ALT.
The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 3% of subjects with COVID-19 receiving VEKLURY in Trials NIAID ACTT-1, 5773, and 5774 are presented in Table 8, Table 9, and Table 10, respectively.
| Laboratory Parameter Abnormality* | VEKLURY 10 Days N=532 | Placebo N=516 |
|---|---|---|
|
||
| ALT increased | 3% | 6% |
| AST increased | 6% | 8% |
| Bilirubin increased | 2% | 5% |
| Creatinine clearance decreased† | 18% | 20% |
| Creatinine increased | 15% | 16% |
| eGFR decreased | 18% | 24% |
| Glucose increased | 12% | 13% |
| Hemoglobin decreased | 15% | 22% |
| Lymphocytes decreased | 11% | 18% |
| Prothrombin time increased | 9% | 4% |
| Laboratory Parameter Abnormality* | VEKLURY 5 Days N=200 | VEKLURY 10 Days N=197 |
|---|---|---|
|
||
| ALT increased | 6% | 8% |
| AST increased | 7% | 6% |
| Creatinine clearance decreased† | 10% | 19% |
| Creatinine increased | 5% | 15% |
| Glucose increased | 11% | 8% |
| Hemoglobin decreased | 6% | 8% |
| Laboratory Parameter Abnormality* | VEKLURY 5 Days N=191 | VEKLURY 10 Days N=193 | SOC N=200 |
|---|---|---|---|
| SOC=Standard of care. | |||
|
|||
| ALT increased | 2% | 3% | 8% |
| Creatinine clearance decreased† | 2% | 5% | 8% |
| Glucose increased | 4% | 3% | 2% |
| Hemoglobin decreased | 3% | 1% | 6% |
The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 2% of subjects with COVID-19 receiving VEKLURY in Trial GS-US-540-9012 are presented in Table 11.
| Laboratory Parameter Abnormality* | VEKLURY 3 Days N=279 | Placebo N=283 |
|---|---|---|
|
||
| Creatinine clearance decreased† | 6% | 2% |
| Creatinine increased | 3% | 1% |
| Glucose increased | 6% | 6% |
| Lymphocytes decreased | 2% | 1% |
| Prothrombin time increased | 1% | 2% |
Clinical Trials Experience in Adults with COVID-19 and Renal Impairment
Study GS-US-540-5912 was a randomized, double-blind, placebo-controlled clinical trial in which 163 hospitalized subjects with confirmed COVID-19 and acute kidney injury (AKI; N=60), chronic kidney disease (CKD; eGFR <30 mL/minute/1.73m2; N=44), or end-stage renal disease (ESRD; eGFR <15 mL/minute/1.73m2; N=59) on hemodialysis received VEKLURY for up to 5 days [see Use in Specific Populations (8.6)]. The adverse reactions observed were consistent with those observed in clinical trials of VEKLURY in adults. Adverse reactions (all grades) were reported in 13 (8%) subjects in the VEKLURY group and 3 (4%) subjects in the placebo group. The most common adverse reactions were nausea (1%), abdominal pain (1%), and diarrhea (1%). No subjects experienced serious adverse reactions. One subject permanently discontinued treatment due to an adverse reaction: lipase increased.
The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 3% of subjects with COVID-19 receiving VEKLURY in Trial GS-US-540-5912 are presented in Table 12.
| Laboratory Parameter Abnormality* | VEKLURY 5 Days N=163 | Placebo N=80 |
|---|---|---|
|
||
| Lymphocytes decreased | 27% | 27% |
| Hemoglobin decreased | 25% | 25% |
| Glucose increased | 15% | 19% |
| Uric acid increased | 11% | 4% |
| Creatinine increased | 12% | 14% |
| Albumin decreased | 12% | 10% |
| Lipase increased | 12% | 7% |
| Prothrombin time increased | 11% | 4% |
| Prothrombin INR increased | 7% | 4% |
| AST increased | 6% | 4% |
| Thromboplastin time increased | 5% | 4% |
| ALT increased | 5% | 6% |
| Sodium increased | 3% | 3% |
| Calcium increased | 3% | 0 |
7. Drug Interactions
Due to potential antagonism based on data from cell culture experiments, concomitant use of VEKLURY with chloroquine phosphate or hydroxychloroquine sulfate is not recommended [see Warnings and Precautions (5.3) and Microbiology (12.4)].
Remdesivir and its metabolites are in vitro substrates and/or inhibitors of certain drug metabolizing enzymes and transporters. Based on a drug interaction study conducted with VEKLURY, no clinically significant drug interactions are expected with inducers of cytochrome P450 (CYP) 3A4 or inhibitors of Organic Anion Transporting Polypeptides (OATP) 1B1/1B3, and P-glycoprotein (P-gp) [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.2 Lactation
Risk Summary
A published case report describes the presence of remdesivir and active metabolite GS-441524 in human milk. Available data (n=11) from pharmacovigilance reports do not indicate adverse effects on breastfed infants from exposure to remdesivir and its metabolite through breastmilk. There are no available data on the effects of remdesivir on milk production. In animal studies, remdesivir and metabolites have been detected in the nursing pups of mothers given remdesivir, likely due to the presence of remdesivir in milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VEKLURY and any potential adverse effects on the breastfed child from VEKLURY or from the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
Data
Remdesivir and its metabolites were detected in the plasma of nursing rat pups, likely due to the presence of remdesivir and/or its metabolites in milk, following daily intravenous administration of remdesivir to pregnant rats from Gestation Day 6 to Lactation Day 20. Exposures in nursing pups were approximately 1% that of maternal exposure on Lactation Day 10. The concentration of remdesivir in animal milk does not necessarily predict the concentration of drug in human milk.
8.4 Pediatric Use
The safety and effectiveness of VEKLURY for the treatment of COVID-19 have been established in pediatric patients 28 days of age and older and weighing at least 3 kg, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
Use in this age group is supported by the following:
- trials in adults [see Clinical Studies (14.1, 14.2, 14.3, 14.4, 14.5)]
- an open-label trial (Study 5823) in 53 hospitalized pediatric subjects [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.6)].
Use of VEKLURY in pediatric patients 28 days of age and older and weighing at least 3 kg is supported by Study 5823 where 53 hospitalized pediatric subjects were treated with weight-based VEKLURY for up to 10 days in the following cohorts: subjects ≥12 years and weighing ≥40 kg (n=12); subjects <12 years and weighing ≥40 kg (n=5); subjects ≥28 days and weighing ≥20 to <40 kg (n=12); subjects ≥28 days and weighing ≥12 to <20 kg (n=12); and subjects ≥28 days and weighing ≥3 to <12 kg (n=12). The safety and pharmacokinetic results in pediatric subjects in this group were similar to those in adults [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.6)].
Use of VEKLURY in pediatric patients weighing at least 40 kg is further supported by a clinical trial of VEKLURY in non-hospitalized subjects that included 3 pediatric subjects 12 years and older, and by clinical trials in hospitalized subjects that included 30 adult subjects weighing 40 to 50 kg. The safety in this weight group was comparable to adult subjects weighing greater than 50 kg. Thirty-nine pediatric patients 12 years and older and weighing at least 40 kg received VEKLURY in a compassionate use program in hospitalized subjects; the available clinical data from these patients are limited [see Adverse Reactions (6.1) and Clinical Studies (14)].
Use of VEKLURY in pediatric patients with renal impairment is supported by safety data in adults [see Adverse Reactions (6.1), Use in Specific Populations (8.6)]. Limited data are available regarding the safety of VEKLURY in pediatric patients with mild or moderate renal impairment. No data are available regarding the safety of VEKLURY in pediatric patients with severe renal impairment. In adults with severe renal impairment, including those requiring dialysis, exposures of GS-441524 and GS-704277, the metabolites of remdesivir, and betadex sulfobutyl ether sodium (SBECD) are increased [see Clinical Pharmacology (12.3)]. VEKLURY contains SBECD which, when administered intravenously, is eliminated through glomerular filtration and therefore when administered to pediatric patients with renal immaturity or renal impairment, may result in higher exposure to SBECD.
The safety and effectiveness of VEKLURY have not been established in pediatric patients younger than 28 days of age or weighing less than 3 kg.
8.5 Geriatric Use
Of the 1,062 hospitalized subjects with SARS-CoV-2 infection randomized in ACTT-1, 36% were 65 years or older. Of the 397 hospitalized subjects with SARS-CoV-2 infection randomized in Study GS-US-540-5773, 42% were 65 years or older. Of the 584 hospitalized subjects with SARS-CoV-2 infection randomized in Study GS-US-540-5774, 27% were 65 years or older. Of the 562 non-hospitalized subjects with SARS-CoV-2 infection randomized in Study GS-US-540-9012, 17% were 65 years or older. Reported clinical experience has not identified differences in responses between the elderly and younger patients [see Clinical Studies (14)]. No dosage adjustment is required in patients over the age of 65 years. In general, appropriate caution should be exercised in the administration of VEKLURY and monitoring of elderly patients, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Use of VEKLURY in patients with COVID-19 and renal impairment, including those on dialysis, is supported by safety and pharmacokinetic data from the following:
- a randomized, double-blind, placebo-controlled trial (Study 5912) in adults [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
- an open-label, parallel-group, single-dose trial in subjects with normal renal function and renal impairment (Study 9015) [see Clinical Pharmacology (12.3)].
The pharmacokinetics and safety of VEKLURY in patients with COVID-19 and renal impairment, including those on dialysis, were evaluated in 163 subjects in a randomized, double-blind, placebo-controlled trial, Study GS-US-540-5912 [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
Study GS-US-540-5912 evaluated VEKLURY 200 mg once daily for 1 day followed by VEKLURY 100 mg once daily for 4 days (for a total of up to 5 days of intravenously administered therapy) in 243 hospitalized adult subjects with confirmed COVID-19 and renal impairment. The trial included 90 subjects (37%) with AKI (defined as a 50% increase in serum creatinine within a 48-hour period that was sustained for ≥6 hours despite supportive care), 64 subjects (26%) with CKD (eGFR <30 mL/minute/1.73m2), and 89 subjects (37%) with ESRD (eGFR <15 mL/minute/1.73m2) requiring hemodialysis. Subjects were randomized in a 2:1 manner, stratified by ESRD, high-flow oxygen requirement, and region (US vs ex-US) to receive VEKLURY (n=163) or placebo (n=80), plus standard of care.
At baseline, mean age was 69 years (with 62% of subjects aged 65 or older); 57% of subjects were male, 67% were White, 26% were Black, and 3% were Asian. The most common baseline risk factors were hypertension (89%), diabetes mellitus (79%), and cardiovascular or cerebrovascular disease (51%); the distribution of risk factors was similar between the two treatment groups. A total of 45 subjects (19%) were on high-flow oxygen, 144 (59%) were on low-flow oxygen, and 54 (22%) were on room air at baseline; no subjects were on invasive mechanical ventilation (IMV). A total of 182 subjects (75%) were not on renal replacement therapy, and 31 subjects (13%) had received a COVID-19 vaccine.
The safety results in subjects with COVID-19 and renal impairment, including those on dialysis, were consistent with those observed in clinical trials of VEKLURY in adults [see Adverse Reactions (6.1)]. Study GS-US-540-5912 closed prematurely due to feasibility issues and was underpowered to assess for efficacy because of lower than expected enrollment.
The pharmacokinetics and safety of VEKLURY in subjects with normal renal function and renal impairment, including those on dialysis, were evaluated in 75 subjects (43 subjects with renal impairment plus 32 matched control subjects with normal renal function) in an open-label, parallel-group, single-dose trial, Study GS-US-540-9015 [see Clinical Pharmacology (12.3)].
In studies GS-US-540-5912 and GS-US-540-9015, exposures of GS-441524 and GS-704277, the metabolites of remdesivir, and SBECD are increased in subjects with mild to severe renal impairment, including those requiring dialysis, relative to subjects with normal renal function [see Clinical Pharmacology (12.3)].
No dosage adjustment of VEKLURY is recommended for patients with any degree of renal impairment, including those on dialysis [see Dosage and Administration (2.2, 2.4), Use in Specific Populations (8.4)].
8.7 Hepatic Impairment
The pharmacokinetics of VEKLURY have not been evaluated in patients with hepatic impairment.
Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate [see Dosage and Administration (2.2) and Warnings and Precautions (5.2)].
10. Overdosage
There is no human experience of acute overdosage with VEKLURY. Treatment of overdose with VEKLURY should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with VEKLURY.
11. Veklury Description
VEKLURY contains remdesivir, a SARS-CoV-2 nucleotide analog RNA polymerase inhibitor. The chemical name for remdesivir is 2-ethylbutyl N-{(S)-[2-C-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-2,5-anhydro-d-altrononitril-6-O-yl]phenoxyphosphoryl}-L-alaninate. It has a molecular formula of C27H35N6O8P and a molecular weight of 602.6 g/mol. Remdesivir has the following structural formula:
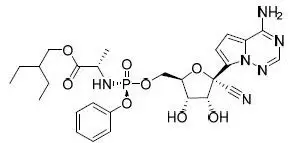
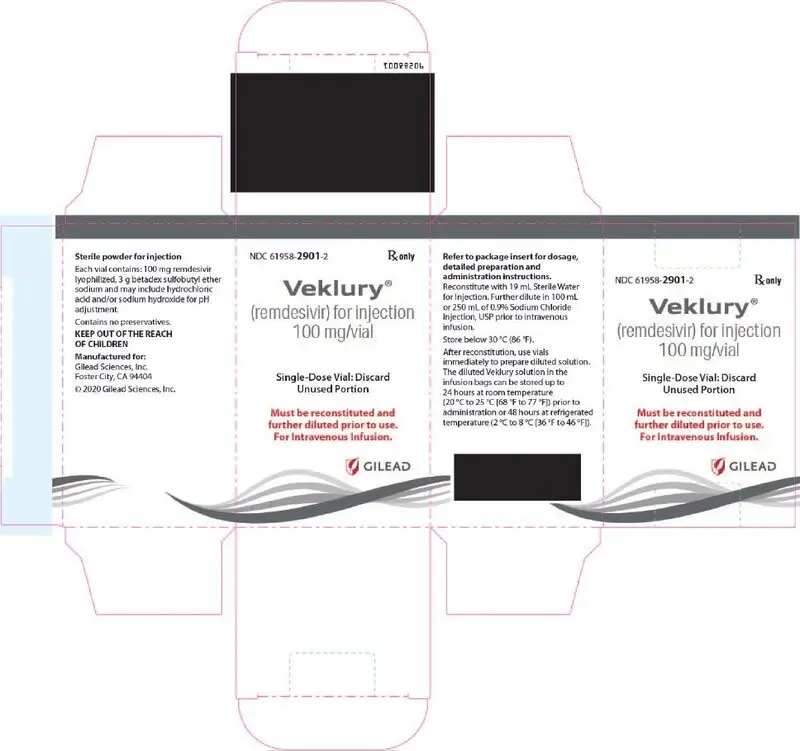
VEKLURY for injection contains 100 mg of remdesivir as a sterile, preservative-free lyophilized white to off-white to yellow powder in a single-dose clear glass vial. It requires reconstitution and then further dilution prior to administration by intravenous infusion [see Dosage and Administration (2.5, 2.6)]. The inactive ingredients are 3 g betadex sulfobutyl ether sodium and may include hydrochloric acid and/or sodium hydroxide for pH adjustment.
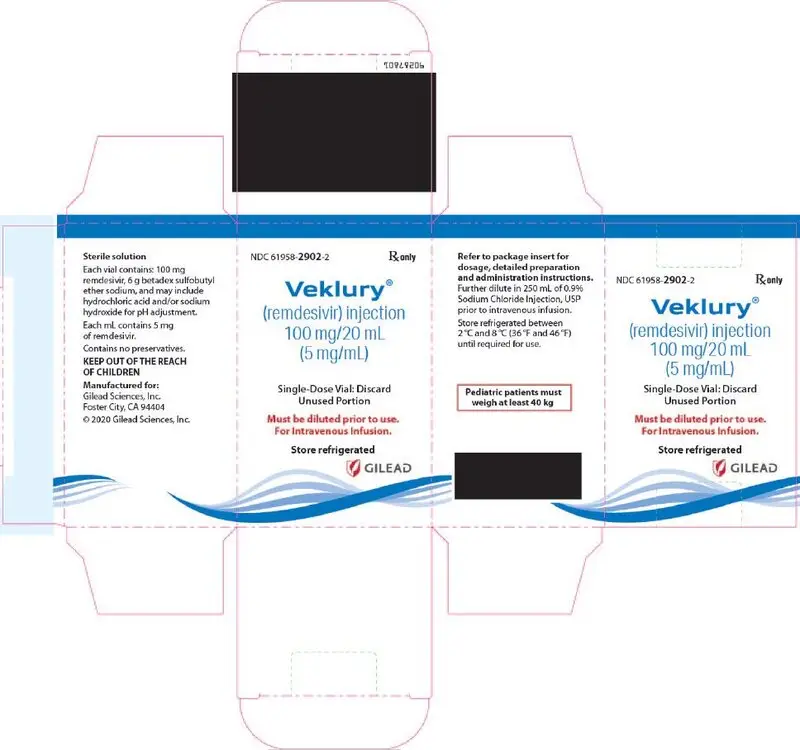
VEKLURY injection contains 100 mg/20 mL (5 mg/mL) of remdesivir as a sterile, preservative-free, clear, colorless to yellow solution in a single-dose clear glass vial. It requires dilution prior to administration by intravenous infusion [see Dosage and Administration (2.5, 2.6)]. The inactive ingredients are 6 g betadex sulfobutyl ether sodium, Water for Injection, USP, and may include hydrochloric acid and/or sodium hydroxide for pH adjustment.
12. Veklury - Clinical Pharmacology
12.1 Mechanism of Action
Remdesivir is an antiviral drug with activity against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [see Microbiology (12.4)].
12.2 Pharmacodynamics
Remdesivir and metabolites exposure-response relationships and the time course of pharmacodynamics response are unknown.
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of remdesivir and metabolites are provided in Table 13. The multiple dose PK parameters of remdesivir and metabolites in adults with COVID-19 are provided in Table 14.
| Remdesivir | GS-441524 | GS-704277 | |
|---|---|---|---|
| ND=not detected | |||
|
|||
| Absorption | |||
| Tmax (h)* | 0.67–0.68 | 1.51–2.00 | 0.75–0.75 |
| Distribution | |||
| % bound to human plasma proteins | 88–93.6† | 2 | 1 |
| Blood-to-plasma ratio | 0.68–1.0 | 1.19 | 0.56 |
| Elimination | |||
| t1/2 (h)‡ | 1 | 27 | 1.3 |
| Metabolism | |||
| Metabolic pathway(s) | CES1 (80%) Cathepsin A (10%) CYP3A (10%) | Not significantly metabolized | HINT1 |
| Excretion | |||
| Major route of elimination | Metabolism | Glomerular filtration and active tubular secretion | Metabolism |
| % of dose excreted in urine§ | 10 | 49 | 2.9 |
| % of dose excreted in feces§ | ND | 0.5 | ND |
| Parameter Mean† (95% CI) | Remdesivir | GS-441524 | GS-704277 |
|---|---|---|---|
| CI=Confidence Interval; ND=Not detectable (at 24 hours post-dose) | |||
|
|||
| Cmax
(nanogram per mL) | 2700 (2440, 2990) | 143 (135, 152) | 198 (180, 218) |
| AUCtau
(nanogram∙h per mL) | 1710 (1480, 1980) | 2410 (2250, 2580) | 392 (348, 442) |
| Ctrough
(nanogram per mL) | ND | 61.5 (56.5, 66.8) | ND |
Specific Populations
Pharmacokinetic differences based on sex, race, age, renal function, and hepatic function on the exposures of remdesivir were evaluated using population pharmacokinetic analysis. Sex and race did not affect the pharmacokinetics of remdesivir and its metabolites (GS-441524 and GS-704277).
Pregnant Individuals
The pharmacokinetics of remdesivir and its circulating metabolites (GS-441524 and GS-704277) were evaluated in pregnant individuals with COVID-19. Exposures (AUCtau, Cmax, and Ctau) of remdesivir and its circulating metabolites during pregnancy were similar to those in non-pregnant individuals (see Table 15).
| Parameter Mean† (90% CI) | Pregnant Individuals (N=21) | Non-Pregnant Individuals (N=22) |
|---|---|---|
| CI=Confidence Interval | ||
|
||
| Remdesivir | ||
| Cmax (nanogram per mL) | 1360 (978, 1890) | 1240 (891, 1720) |
| AUCtau (nanogram∙h per mL) | 1250 (916, 1700)‡ | 1300 (1070, 1590)§ |
| GS-441524 | ||
| Cmax (nanogram per mL) | 113 (102, 126) | 121 (108, 136) |
| AUCtau (nanogram∙h per mL) | 1840 (1630, 2070)¶ | 2050 (1780, 2350)# |
| Ctau (nanogram per mL) | 51.6 (44.7, 59.6)¶ | 57.1 (48.7, 66.9)# |
| GS-704277 | ||
| Cmax (nanogram per mL) | 217 (187, 252) | 213 (188, 240) |
| AUCtau (nanogram∙h per mL) | 454 (406, 508)¶ | 437 (384, 497) |
Patients with Renal Impairment
The pharmacokinetics of remdesivir and its metabolites (GS-441524 and GS-704277) and excipient SBECD were evaluated in healthy subjects, those with mild (eGFR 60–89 mL/minute/1.73m2), moderate (eGFR 30–59 mL/minute/1.73m2), severe (eGFR 15–29 mL/minute/1.73m2) renal impairment, or kidney failure (eGFR <15 mL/minute/1.73m2) on dialysis or not on dialysis following a single dose of up to 100 mg of VEKLURY (see Table 16); and in COVID-19 patients with severely reduced kidney function (AKI [defined as a 50% increase in serum creatinine within a 48-hour period that was sustained for ≥6 hours despite supportive care]; CKD [eGFR <30 mL/minute/1.73m2]; or ESRD [eGFR <15 mL/minute/1.73m2] requiring hemodialysis) receiving VEKLURY 200 mg loading dose on Day 1 followed by 100 mg from Day 2 to Day 5 (see Table 17). Pharmacokinetic exposures of remdesivir were not affected by renal function or timing of VEKLURY administration around dialysis.
Exposures of GS-441524, GS-704277, and SBECD were up to 7.9-fold, 2.8-fold, and 21-fold higher, respectively, in those with renal impairment compared to those with normal renal function (see Table 16 and Table 17). These changes are not considered to be clinically significant [see Adverse Reactions (6.1) and Use in Specific Populations (8.6)].
Remdesivir was not efficiently removed through hemodialysis. Average hemodialysis clearance of GS-441524 and GS-704277 was 149 mL/minute and 92.6 mL/minute, respectively.
| Mean Ratio (90% CI)‡ | 60–89 mL per minute†
N=10 | 30–59 mL per minute†
N=10 | 15–29 mL per minute†
N=10 | <15 mL per minute† | ||
|---|---|---|---|---|---|---|
| Pre-hemodialysis N=6 | Post-hemodialysis N=6 | No dialysis N=3 |
||||
| CI=Confidence Interval | ||||||
|
||||||
| Remdesivir | ||||||
| Cmax | 0.96 (0.71, 1.31) | 1.20 (1.01, 1.42) | 0.97 (0.83, 1.13) | 0.89 (0.67, 1.18) | 1.13 (0.79, 1.60) | 0.94 (0.65, 1.35) |
| AUCinf | 1.00 (0.75, 1.32) | 1.22 (0.98, 1.52) | 0.94 (0.83, 1.07) | 0.80 (0.59, 1.08) | 1.08 (0.72, 1.63) | 0.89 (0.55, 1.43) |
| GS-441524 | ||||||
| Cmax | 1.07 (0.90, 1.26) | 1.44 (1.13, 1.85) | 1.68 (1.28, 2.20) | 2.27 (1.72, 2.99) | 3.07 (2.21, 4.26) | 3.00 (2.63, 3.42) |
| AUCinf | 1.19 (0.97, 1.47) | 2.02 (1.57, 2.62) | 3.26 (2.39, 4.46) | 4.97 (3.65, 6.77) | 6.22 (4.44, 8.71) | 7.87 (6.49, 9.53) |
| GS-704277 | ||||||
| Cmax | 2.25 (1.20, 4.20) | 1.83 (1.34, 2.49) | 1.27 (0.96, 1.68) | 1.43 (1.00, 2.05) | 1.23 (0.84, 1.80) | 1.76 (1.19, 2.61) |
| AUCinf | 1.39 (1.13, 1.71) | 2.01 (1.48, 2.73) | 1.78 (1.27, 2.49) | 2.18 (1.61, 2.95) | 2.06 (1.42, 2.97) | 2.81 (1.79, 4.43) |
| Mean Ratio (90% CI)§ | Remdesivir | GS-441524 | GS-704277 |
|---|---|---|---|
| CI=Confidence Interval; ND=Not detectable (at 24 hours post-dose) | |||
|
|||
| Cmax
(nanogram per mL) | 1.39 (1.25, 1.54) | 4.98 (4.61, 5.38) | 1.84 (1.63, 2.08) |
| AUCtau
(nanogram•h per mL) | 1.79 (1.59, 2.01) | 6.59 (6.05, 7.18) | 3.94 (3.50, 4.43) |
| Ctau
(nanogram per mL) | ND | 5.82 (5.25, 6.45) | ND |
13. Nonclinical Toxicology
13.2 Animal Toxicology and/or Pharmacology
Intravenous administration (slow bolus) of remdesivir to male rhesus monkeys at dosage levels of 5, 10, and 20 mg/kg/day for 7 days resulted, at all dose levels, in increased mean urea nitrogen and increased mean creatinine, renal tubular atrophy, and basophilia and casts.
Intravenous administration (slow bolus) of remdesivir to rats at dosage levels of ≥3 mg/kg/day for up to 4 weeks resulted in findings indicative of kidney injury and/or dysfunction.
Kidney-related effects in rats and monkeys were observed at exposures of the predominant circulating metabolite (GS-441524) that are lower than the exposure in humans at the RHD.
14. Clinical Studies
14.1 Description of Clinical Trials
The efficacy and safety of VEKLURY were evaluated in the trials summarized in Table 20.
| Trial | Population | Trial Arms (N) | Timepoint |
|---|---|---|---|
| COVID-19: coronavirus disease 2019 | |||
|
|||
| NIAID ACTT-1*
(NCT04280705) | Hospitalized with mild/moderate and severe COVID-19 | VEKLURY 10 Days (532) Placebo (516) | 29 Days after Randomization |
| GS-US-540-5773†
(NCT04292899) | Hospitalized with severe COVID-19 | VEKLURY 5 Days (200) VEKLURY 10 Days (197) | Day 14 |
| GS-US-540-5774†
(NCT04292730) | Hospitalized with moderate COVID-19 | VEKLURY 5 Days (191) VEKLURY 10 Days (193) Standard of care (200) | Day 11 |
| GS-US-540-9012*
(NCT04501952) | Non-hospitalized with mild-to-moderate COVID-19 and at high risk for progression to severe disease | VEKLURY 3 Days (279) Placebo (283) | Day 28 |
| GS-US-540-5823 (Cohorts 1–4, 8)‡
(NCT04431453) | Hospitalized pediatric subjects 28 days to <18 years of age and weighing at least 3 kg with COVID-19 | VEKLURY up to 10 Days (53) | Day 10 |
14.2 NIAID ACTT-1 Study in Hospitalized Subjects with Mild/Moderate and Severe COVID-19
A randomized, double-blind, placebo-controlled clinical trial (ACTT-1) of hospitalized adult subjects with confirmed SARS-CoV-2 infection and mild, moderate, or severe COVID-19 compared treatment with VEKLURY for 10 days (n=541) with placebo (n=521). Mild/moderate disease was defined as SpO2 >94% and respiratory rate <24 breaths/minute without supplemental oxygen; severe disease was defined as an SpO2 ≤94% on room air, a respiratory rate ≥24 breaths/minute, an oxygen requirement, or a requirement for mechanical ventilation. Subjects had to have at least one of the following to be enrolled in the trial: radiographic infiltrates by imaging, SpO2 ≤94% on room air, a requirement for supplemental oxygen, or a requirement for mechanical ventilation. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days, for 10 days of treatment via intravenous infusion. Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to the completion of 10 days of treatment.
At baseline, mean age was 59 years (with 36% of subjects aged 65 or older); 64% of subjects were male, 53% were White, 21% were Black, and 13% were Asian; 24% were Hispanic or Latino; 105 subjects had mild/moderate disease (10% in both treatment groups); 957 subjects had severe disease (90% in both treatment groups). Subjects in this trial were unvaccinated. A total of 285 subjects (27%) (n=131 received VEKLURY) were on invasive mechanical ventilation or ECMO. The most common comorbidities were hypertension (51%), obesity (45%), and type 2 diabetes mellitus (31%); the distribution of comorbidities was similar between the two treatment groups.
The primary clinical endpoint was time to recovery within 29 days after randomization. Recovery was defined as discharged from the hospital without limitations on activities, discharged from the hospital with limitations on activities and/or requiring home oxygen, or hospitalized but not requiring supplemental oxygen and no longer requiring ongoing medical care. The median time to recovery was 10 days in the VEKLURY group compared to 15 days in the placebo group (recovery rate ratio 1.29 [95% CI 1.12 to 1.49], p<0.001). Among subjects with mild/moderate disease at enrollment (n=105), the median time to recovery was 5 days in both the VEKLURY and placebo groups (recovery rate ratio 1.22 [95% CI 0.82 to 1.81]). Among subjects with severe disease at enrollment (n=957), the median time to recovery was 11 days in the VEKLURY group compared to 18 days in the placebo group (recovery rate ratio 1.31 [95% CI 1.12 to 1.52]).
A key secondary endpoint was clinical status on Day 15 assessed on an 8-point ordinal scale consisting of the following categories:
- not hospitalized, no limitations on activities;
- not hospitalized, limitation on activities and/or requiring home oxygen;
- hospitalized, not requiring supplemental oxygen - no longer requires ongoing medical care;
- hospitalized, not requiring supplemental oxygen - requiring ongoing medical care (COVID-19 related or otherwise);
- hospitalized, requiring supplemental oxygen;
- hospitalized, on noninvasive ventilation or high-flow oxygen devices;
- hospitalized, on invasive mechanical ventilation or ECMO; and
- death.
Overall, the odds of improvement in the ordinal scale were higher in the VEKLURY group at Day 15 when compared to the placebo group (odds ratio 1.54 [95% CI 1.25 to 1.91]).
Overall, 29-day mortality was 11% for the VEKLURY group vs 15% for the placebo group (hazard ratio 0.73 [95% CI 0.52 to 1.03]).
14.3 Study GS-US-540-5773 in Hospitalized Subjects with Severe COVID-19
A randomized, open-label multi-center clinical trial (Study 5773) in adult subjects with confirmed SARS-CoV-2 infection, an SpO2 of ≤94% on room air, and radiological evidence of pneumonia compared 200 subjects who received VEKLURY for 5 days with 197 subjects who received VEKLURY for 10 days. Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment. Subjects on mechanical ventilation at screening were excluded. All subjects received 200 mg of VEKLURY on Day 1 and 100 mg once daily on subsequent days via intravenous infusion, plus standard of care.
At baseline, the median age of subjects was 61 years (range, 20 to 98 years); 64% were male, 75% were White, 12% were Black, and 12% were Asian; 22% were Hispanic or Latino. More subjects in the 10-day group than the 5-day group required invasive mechanical ventilation or ECMO (5% vs 2%), or high-flow oxygen support (30% vs 25%), at baseline. Subjects in this trial were unvaccinated. Median duration of symptoms and hospitalization prior to first dose of VEKLURY were similar across treatment groups.
The primary endpoint was clinical status on Day 14 assessed on a 7-point ordinal scale consisting of the following categories:
- death;
- hospitalized, receiving invasive mechanical ventilation or ECMO;
- hospitalized, receiving noninvasive ventilation or high-flow oxygen devices;
- hospitalized, requiring low-flow supplemental oxygen;
- hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19);
- hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for remdesivir administration); and
- not hospitalized.
Overall, after adjusting for between-group differences at baseline, subjects receiving a 5-day course of VEKLURY had similar clinical status at Day 14 as those receiving a 10-day course (odds ratio for improvement 0.75 [95% CI 0.51 to 1.12]). There were no statistically significant differences in recovery rates or mortality rates in the 5-day and 10-day groups once adjusted for between-group differences at baseline. All-cause mortality at Day 28 was 12% vs 14% in the 5- and 10-day treatment groups, respectively.
14.4 Study GS-US-540-5774 in Hospitalized Subjects with Moderate COVID-19
A randomized, open-label multi-center clinical trial (Study 5774) of hospitalized adult subjects with confirmed SARS-CoV-2 infection, SpO2 >94% and radiological evidence of pneumonia compared treatment with VEKLURY for 5 days (n=191) and treatment with VEKLURY for 10 days (n=193) with standard of care (n=200). Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days via intravenous infusion.
At baseline, the median age of subjects was 57 years (range, 12 to 95 years); 61% were male, 61% were White, 19% were Black, and 19% were Asian; 18% were Hispanic or Latino. Subjects in this trial were unvaccinated. Baseline clinical status, oxygen support status, and median duration of symptoms and hospitalization prior to first dose of VEKLURY were similar across treatment groups.
The primary endpoint was clinical status on Day 11 assessed on a 7-point ordinal scale consisting of the following categories:
- death;
- hospitalized, receiving invasive mechanical ventilation or ECMO;
- hospitalized, receiving noninvasive ventilation or high-flow oxygen devices;
- hospitalized, requiring low-flow supplemental oxygen;
- hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19);
- hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for remdesivir administration); and
- not hospitalized.
Overall, the odds of improvement in the ordinal scale were higher in the 5-day VEKLURY group at Day 11 when compared to those receiving only standard of care (odds ratio 1.65 [95% CI 1.09 to 2.48], p=0.017). The odds of improvement in clinical status with the 10-day treatment group when compared to those receiving only standard of care were not statistically significant (odds ratio 1.31 [95% CI 0.88 to 1.95]). All-cause mortality at Day 28 was ≤2% in all treatment groups.
14.5 Study GS-US-540-9012 in Non-Hospitalized Subjects with Mild-to-Moderate COVID-19 and at High Risk for Progression to Severe Disease
A randomized, double-blind, placebo-controlled, clinical trial (Study 9012) evaluated VEKLURY 200 mg once daily for 1 day followed by VEKLURY 100 mg once daily for 2 days (for a total of 3 days of intravenously administered therapy) in 554 adult and 8 pediatric subjects (12 years of age and older and weighing at least 40 kg) who were non-hospitalized, had mild-to-moderate COVID-19, were symptomatic for COVID-19 for ≤7 days, had confirmed SARS-CoV-2 infection, and had at least one risk factor for progression to hospitalization. Risk factors for progression to hospitalization included age ≥60 years, obesity (BMI ≥30), chronic lung disease, hypertension, cardiovascular or cerebrovascular disease, diabetes mellitus, immunocompromised state, chronic mild or moderate kidney disease, chronic liver disease, current cancer, and sickle cell disease. Subjects who received, required, or were expected to require supplemental oxygen were excluded from the trial. Subjects were randomized in a 1:1 manner, stratified by residence in a skilled nursing facility (yes/no), age (<60 vs ≥60 years), and region (US vs ex-US) to receive VEKLURY (n=279) or placebo (n=283), plus standard of care.
At baseline, mean age was 50 years (with 30% of subjects aged 60 or older); 52% were male, 80% were White, 8% were Black, and 2% were Asian; 44% were Hispanic or Latino; median body mass index was 30.7 kg/m2. Subjects in this trial were unvaccinated. VEKLURY or placebo was first administered to subjects in outpatient facilities (84%), home healthcare settings (13%), or skilled nursing facilities (3%). The most common comorbidities were diabetes mellitus (62%), obesity (56%), and hypertension (48%). Median (Q1, Q3) duration of symptoms prior to treatment was 5 (3, 6) days; median viral load was 6.3 log10 copies/mL at baseline. The baseline demographics and disease characteristics were well balanced across the VEKLURY and placebo treatment groups.
The primary endpoint was the proportion of subjects with COVID-19 related hospitalization (defined as at least 24 hours of acute care) or all-cause mortality through Day 28. Events occurred in 2 (0.7%) subjects treated with VEKLURY compared to 15 (5.3%) subjects concurrently randomized to placebo (hazard ratio 0.134 [95% CI 0.031 to 0.586]; p=0.0076). No deaths were observed through Day 28.
14.6 Study GS-US-540-5823 in Hospitalized Pediatric Subjects with COVID-19
The primary objectives of this Phase 2/3 single-arm, open-label clinical study (Study GS-US-540-5823) were to evaluate pharmacokinetics and safety of up to 10 days of treatment with VEKLURY in pediatric subjects. A total of 53 pediatric subjects at least 28 days of age and weighing at least 3 kg with confirmed SARS-CoV-2 infection and mild, moderate, or severe COVID-19 was evaluated in five cohorts: subjects ≥12 years and weighing ≥40 kg (n=12); subjects <12 years and weighing ≥40 kg (n=5); subjects ≥28 days and weighing ≥20 to <40 kg (n=12); subjects ≥28 days and weighing ≥12 to <20 kg (n=12); and subjects ≥28 days and weighing ≥3 to <12 kg (n=12). Subjects weighing ≥40 kg received 200 mg of VEKLURY on Day 1 followed by VEKLURY 100 mg once daily on subsequent days; subjects weighing ≥3 kg to <40 kg received VEKLURY 5 mg/kg on Day 1 followed by VEKLURY 2.5 mg/kg once daily on subsequent days. Assessments occurred at the following intervals: Screening; Day 1 (Baseline); Days 2–10, or until discharge, whichever came earlier; Follow-Up on Day 30 (±5). Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to the completion of 10 days of treatment.
At baseline, median age was 7 years (Q1, Q3: 2 years, 12 years); 57% were female, 70% were White, 30% were Black, and 44% were Hispanic or Latino; median weight was 25 kg (range: 4 to 192 kg). Subjects in this trial were unvaccinated. A total of 12 subjects (23%) were on invasive mechanical ventilation, 18 (34%) were on non-invasive ventilation or high-flow oxygen; 10 (19%) were on low-flow oxygen; and 13 (25%) were on room air, at baseline. The overall median (Q1, Q3) duration of symptoms and hospitalization prior to first dose of VEKLURY was 5 (3, 7) days and 1 (1, 3) day, respectively.
The descriptive outcome analyses showed treatment with VEKLURY for up to 10 days resulted in an overall median (Q1, Q3) change from baseline in clinical status (assessed on a 7-point ordinal scale ranging from death [score of 1] to ventilatory support and decreasing levels of oxygen to hospital discharge [score of 7]) of +2.0 (1.0, 4.0) points on Day 10.
Recovery (defined as an improvement from a baseline clinical status score of 2 through 5 to a score of 6 or 7, or an improvement from a baseline score of 6 to a score of 7) was reported for 62% of subjects on Day 10; median (Q1, Q3) time to recovery was 7 (5, 16) days.
Overall, 60% of subjects were discharged by Day 10, and 83% of subjects were discharged by Day 30. Three subjects (6%) died during the study.
16. How is Veklury supplied
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 07/2023 | ||
| PATIENT INFORMATION | |||
| VEKLURY® (VEK-lur-ee) (remdesivir) for injection | VEKLURY® (VEK-lur-ee) (remdesivir) injection |
||
| What is VEKLURY?
VEKLURY is a prescription medicine used for the treatment of coronavirus disease 2019 (COVID-19) in adults and children 28 days of age and older and weighing at least 7 pounds (3 kg) who are:
|
|||
| Do not take VEKLURY if you are allergic to remdesivir or any of the ingredients in VEKLURY. See the end of this leaflet for a complete list of ingredients in VEKLURY. | |||
Before receiving VEKLURY, tell your healthcare provider about all of your medical conditions, including if you:
Especially tell your healthcare provider if you are taking the medicines chloroquine phosphate or hydroxychloroquine sulfate. |
|||
How will I receive VEKLURY?
|
|||
| What are the possible side effects of VEKLURY? VEKLURY may cause serious side effects, including:
|
|||
|
|
||
These are not all of the possible side effects of VEKLURY. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
| General information about the safe and effective use of VEKLURY.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about VEKLURY that is written for healthcare professionals. |
|||
| What are the ingredients in VEKLURY?
Active ingredient: remdesivir Inactive ingredients: VEKLURY for injection: betadex sulfobutyl ether sodium and may include hydrochloric acid and/or sodium hydroxide for pH adjustment. VEKLURY injection: betadex sulfobutyl ether sodium, Water for Injection, USP, and may include hydrochloric acid and/or sodium hydroxide for pH adjustment. Manufactured and distributed by: Gilead Sciences, Inc., Foster City, CA 94404 VEKLURY is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners. © 2023 Gilead Sciences, Inc. All rights reserved. 214787-GS-014 For more information, call 1-800-445-3235 or go to www.VEKLURY.com. |
|||


| VEKLURY
remdesivir injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| VEKLURY
remdesivir injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Gilead Sciences, Inc. (185049848) |
