Drug Detail:Verzenio (Abemaciclib [ a-bem-a-sye-klib ])
Drug Class: CDK 4/6 inhibitors
Highlights of Prescribing Information
VERZENIO® (abemaciclib) tablets, for oral use
Initial U.S. Approval: 2017
Recent Major Changes
| Indications and Usage (1.1, 1.2) | 03/2023 |
| Dosage and Administration, Patient Selection Removed (2) | 03/2023 |
Indications and Usage for Verzenio
VERZENIO® is a kinase inhibitor indicated:
- in combination with endocrine therapy (tamoxifen or an aromatase inhibitor) for the adjuvant treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, node-positive, early breast cancer at high risk of recurrence. (1.1, 14.1)
- in combination with an aromatase inhibitor as initial endocrine-based therapy for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer. (1.2)
- in combination with fulvestrant for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer with disease progression following endocrine therapy. (1.2)
- as monotherapy for the treatment of adult patients with HR-positive, HER2-negative advanced or metastatic breast cancer with disease progression following endocrine therapy and prior chemotherapy in the metastatic setting. (1.2)
Verzenio Dosage and Administration
VERZENIO tablets are taken orally with or without food. (2.1)
- Recommended starting dose in combination with fulvestrant, tamoxifen, or an aromatase inhibitor: 150 mg twice daily. (2.1)
- Recommended starting dose as monotherapy: 200 mg twice daily. (2.1)
- Dosing interruption and/or dose reductions may be required based on individual safety and tolerability. (2.2)
Dosage Forms and Strengths
Tablets: 50 mg, 100 mg, 150 mg, and 200 mg. (3)
Contraindications
None. (4)
Warnings and Precautions
- Diarrhea: VERZENIO can cause severe cases of diarrhea, associated with dehydration and infection. Instruct patients at the first sign of loose stools to initiate antidiarrheal therapy, increase oral fluids, and notify their healthcare provider. (2.2, 5.1)
- Neutropenia: Monitor complete blood counts prior to the start of VERZENIO therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. (2.2, 5.2)
- Interstitial Lung Disease (ILD)/Pneumonitis: Severe and fatal cases of ILD/pneumonitis have been reported. Monitor for clinical symptoms or radiological changes indicative of ILD/pneumonitis. Permanently discontinue VERZENIO in all patients with Grade 3 or 4 ILD or pneumonitis. (2.2, 5.3)
- Hepatotoxicity: Increases in serum transaminase levels have been observed. Perform liver function tests (LFTs) before initiating treatment with VERZENIO. Monitor LFTs every two weeks for the first two months, monthly for the next 2 months, and as clinically indicated. (2.2, 5.4)
- Venous Thromboembolism: Monitor patients for signs and symptoms of thrombosis and pulmonary embolism and treat as medically appropriate. (2.2, 5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.6, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥20%) were diarrhea, neutropenia, nausea, abdominal pain, infections, fatigue, anemia, leukopenia, decreased appetite, vomiting, headache, alopecia, and thrombocytopenia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Eli Lilly and Company at 1-800-LillyRx (1-800-545-5979) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- CYP3A Inhibitors: Avoid concomitant use of ketoconazole. Reduce the VERZENIO dose with concomitant use of other strong and moderate CYP3A inhibitors. (2.2, 7.1)
- CYP3A Inducers: Avoid concomitant use of strong and moderate CYP3A inducers. (7.1)
Use In Specific Populations
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2023
Full Prescribing Information
1. Indications and Usage for Verzenio
1.1 Early Breast Cancer
VERZENIO® (abemaciclib) is indicated:
- in combination with endocrine therapy (tamoxifen or an aromatase inhibitor) for the adjuvant treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, node-positive, early breast cancer at high risk of recurrence [see Clinical Studies (14.1)].
1.2 Advanced or Metastatic Breast Cancer
VERZENIO (abemaciclib) is indicated:
- in combination with an aromatase inhibitor as initial endocrine-based therapy for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer.
- in combination with fulvestrant for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer with disease progression following endocrine therapy.
- as monotherapy for the treatment of adult patients with HR-positive, HER2-negative advanced or metastatic breast cancer with disease progression following endocrine therapy and prior chemotherapy in the metastatic setting.
2. Verzenio Dosage and Administration
2.1 Recommended Dose and Schedule
- When used in combination with fulvestrant, tamoxifen, or an aromatase inhibitor, the recommended dose of VERZENIO is 150 mg taken orally twice daily. Refer to the Full Prescribing Information for the recommended dose of the fulvestrant, tamoxifen, or aromatase inhibitor being used.
- Pre/perimenopausal women and men treated with the combination of VERZENIO plus an aromatase inhibitor should be treated with a gonadotropin-releasing hormone agonist (GnRH) according to current clinical practice standards.
- Pre/perimenopausal women treated with the combination of VERZENIO plus fulvestrant should be treated with a GnRH according to current clinical practice standards
- When used as monotherapy, the recommended dose of VERZENIO is 200 mg taken orally twice daily.
- For early breast cancer, continue VERZENIO until completion of 2 years of treatment or until disease recurrence, or unacceptable toxicity.
- For advanced or metastatic breast cancer, continue treatment until disease progression or unacceptable toxicity.
VERZENIO may be taken with or without food [see Clinical Pharmacology (12.3)].
Instruct patients to take their doses of VERZENIO at approximately the same times every day.
If the patient vomits or misses a dose of VERZENIO, instruct the patient to take the next dose at its scheduled time. Instruct patients to swallow VERZENIO tablets whole and not to chew, crush, or split tablets before swallowing. Instruct patients not to ingest VERZENIO tablets if broken, cracked, or otherwise not intact.
2.2 Dose Modification
Dose Modifications for Adverse Reactions
The recommended VERZENIO dose modifications for adverse reactions are provided in Tables 1-7. Discontinue VERZENIO for patients unable to tolerate 50 mg twice daily.
| Dose Level | VERZENIO Dose
Combination with Fulvestrant, Tamoxifen, or an Aromatase Inhibitor | VERZENIO Dose
for Monotherapy |
| Recommended starting dose | 150 mg twice daily | 200 mg twice daily |
| First dose reduction | 100 mg twice daily | 150 mg twice daily |
| Second dose reduction | 50 mg twice daily | 100 mg twice daily |
| Third dose reduction | not applicable | 50 mg twice daily |
|
Abbreviation: CTCAE = Common Terminology Criteria for Adverse Events. |
|
|
a If blood cell growth factors are required, suspend VERZENIO dose for at least 48 hours after the last dose of blood cell growth factor and until toxicity resolves to ≤Grade 2. Resume at next lower dose unless already performed for the toxicity that led to the use of the growth factor. Growth factor use as per current treatment guidelines. |
|
| Monitor complete blood counts prior to the start of VERZENIO therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. | |
| CTCAE Grade | VERZENIO Dose Modifications |
| Grade 1 or 2 | No dose modification is required. |
| Grade 3 | Suspend dose until toxicity resolves to ≤Grade 2. Dose reduction is not required. |
| Grade 3 recurrent, or Grade 4 | Suspend dose until toxicity resolves to ≤Grade 2. Resume at next lower dose. |
| At the first sign of loose stools, start treatment with antidiarrheal agents and increase intake of oral fluids. | |
| CTCAE Grade | VERZENIO Dose Modifications |
| Grade 1 | No dose modification is required. |
| Grade 2 | If toxicity does not resolve within 24 hours to ≤Grade 1, suspend dose until resolution. No dose reduction is required. |
| Grade 2 that persists or recurs after resuming the same dose despite maximal supportive measures | Suspend dose until toxicity resolves to ≤Grade 1. Resume at next lower dose. |
| Grade 3 or 4 or requires hospitalization | Suspend dose until toxicity resolves to ≤Grade 1. Resume at next lower dose. |
|
Abbreviations: ALT = alanine aminotransferase, AST = aspartate aminotransferase, ULN = upper limit of normal. |
|
| Monitor ALT, AST, and serum bilirubin prior to the start of VERZENIO therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. | |
| CTCAE Grade for ALT and AST | VERZENIO Dose Modifications |
| Grade 1 (>ULN-3.0 x ULN) Grade 2 (>3.0-5.0 x ULN), WITHOUT increase in total bilirubin above 2 x ULN | No dose modification is required. |
| Persistent or Recurrent Grade 2, or Grade 3 (>5.0-20.0 x ULN), WITHOUT increase in total bilirubin above 2 x ULN | Suspend dose until toxicity resolves to baseline or Grade 1. Resume at next lower dose. |
| Elevation in AST and/or ALT >3 x ULN WITH total bilirubin >2 x ULN, in the absence of cholestasis | Discontinue VERZENIO. |
| Grade 4 (>20.0 x ULN) | Discontinue VERZENIO. |
| CTCAE Grade | VERZENIO Dose Modifications |
| Grade 1 or 2 | No dose modification is required. |
| Persistent or recurrent Grade 2 toxicity that does not resolve with maximal supportive measures within 7 days to baseline or Grade 1 | Suspend dose until toxicity resolves to baseline or ≤Grade 1. Resume at next lower dose. |
| Grade 3 or 4 | Discontinue VERZENIO. |
| CTCAE Grade | VERZENIO Dose Modifications |
| Early Breast Cancer | |
| Any Grade | Suspend dose and treat as clinically indicated. Resume VERZENIO when the patient is clinically stable. |
| Advanced or Metastatic Breast Cancer | |
| Grade 1 or 2 | No dose modification is required. |
| Grade 3 or 4 | Suspend dose and treat as clinically indicated. Resume VERZENIO when the patient is clinically stable. |
|
a Excluding diarrhea, hematologic toxicity, hepatotoxicity, ILD/pneumonitis, and VTEs. |
|
| CTCAE Grade | VERZENIO Dose Modifications |
| Grade 1 or 2 | No dose modification is required. |
| Persistent or recurrent Grade 2 toxicity that does not resolve with maximal supportive measures within 7 days to baseline or Grade 1 | Suspend dose until toxicity resolves to baseline or ≤Grade 1. Resume at next lower dose. |
| Grade 3 or 4 | Suspend dose until toxicity resolves to baseline or ≤Grade 1. Resume at next lower dose. |
Refer to the Full Prescribing Information for coadministered fulvestrant, tamoxifen, or an aromatase inhibitor for dose modifications and other relevant safety information.
Dose Modification for Use with Strong and Moderate CYP3A Inhibitors
Avoid concomitant use of the strong CYP3A inhibitor ketoconazole.
With concomitant use of strong CYP3A inhibitors other than ketoconazole, in patients with recommended starting doses of 200 mg twice daily or 150 mg twice daily, reduce the VERZENIO dose to 100 mg twice daily. In patients who have had a dose reduction to 100 mg twice daily due to adverse reactions, further reduce the VERZENIO dose to 50 mg twice daily. If a patient taking VERZENIO discontinues a CYP3A inhibitor, increase the VERZENIO dose (after 3-5 half-lives of the inhibitor) to the dose that was used before starting the strong inhibitor [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
With concomitant use of moderate CYP3A inhibitors, monitor for adverse reactions and consider reducing the VERZENIO dose in 50 mg decrements as demonstrated in Table 1, if necessary.
Dose Modification for Patients with Severe Hepatic Impairment
For patients with severe hepatic impairment (Child Pugh-C), reduce the VERZENIO dosing frequency to once daily [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
Refer to the Full Prescribing Information for the coadministered fulvestrant, tamoxifen, or aromatase inhibitor for dose modification requirements for severe hepatic impairment.
3. Dosage Forms and Strengths
50 mg tablets: oval beige tablet with “Lilly” debossed on one side and “50” on the other side.
100 mg tablets: oval white to practically white tablet with “Lilly” debossed on one side and “100” on the other side.
150 mg tablets: oval yellow tablet with “Lilly” debossed on one side and “150” on the other side.
200 mg tablets: oval beige tablet with “Lilly” debossed on one side and “200” on the other side.
5. Warnings and Precautions
5.1 Diarrhea
Severe diarrhea associated with dehydration and infection occurred in patients treated with VERZENIO.
Across four clinical trials in 3691 patients, diarrhea occurred in 81% to 90% of patients who received VERZENIO. Grade 3 diarrhea occurred in 8% to 20% of patients receiving VERZENIO [see Adverse Reactions (6.1)].
Most patients experienced diarrhea during the first month of VERZENIO treatment. The median time to onset of the first diarrhea event ranged from 6 to 8 days; and the median duration of Grade 2 and Grade 3 diarrhea ranged from 6 to 11 days and 5 to 8 days, respectively. Across trials, 19% to 26% of patients with diarrhea required a VERZENIO dose interruption and 13% to 23% required a dose reduction.
Instruct patients to start antidiarrheal therapy such as loperamide at the first sign of loose stools, increase oral fluids, and notify their healthcare provider for further instructions and appropriate follow up [see Patient Counseling Information (17)]. For Grade 3 or 4 diarrhea, or diarrhea that requires hospitalization, discontinue VERZENIO until toxicity resolves to ≤Grade 1, and then resume VERZENIO at the next lower dose [see Dosage and Administration (2.2)].
5.2 Neutropenia
Neutropenia, including febrile neutropenia and fatal neutropenic sepsis, occurred in patients treated with VERZENIO.
Across four clinical trials in 3691 patients, neutropenia occurred in a 37% to 46% of patients receiving VERZENIO. A Grade ≥3 decrease in neutrophil count (based on laboratory findings) occurred in 19% to 32% of patients receiving VERZENIO. Across trials, the median time to the first episode of Grade ≥3 neutropenia ranged from 29 days to 33 days, and the median duration of Grade ≥3 neutropenia ranged from 11 days to 16 days [see Adverse Reactions (6.1)].
Febrile neutropenia has been reported in <1% of patients exposed to VERZENIO across trials. Two deaths due to neutropenic sepsis were observed in MONARCH 2. Inform patients to promptly report any episodes of fever to their healthcare provider [see Patient Counseling Information (17)].
Monitor complete blood counts prior to the start of VERZENIO therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. Dose interruption, dose reduction, or delay in starting treatment cycles is recommended for patients who develop Grade 3 or 4 neutropenia [see Dosage and Administration (2.2)].
5.3 Interstitial Lung Disease (ILD) or Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD) or pneumonitis can occur in patients treated with VERZENIO and other CDK4/6 inhibitors. In VERZENIO-treated patients in early breast cancer (monarchE, N=2791), 3% of patients experienced ILD or pneumonitis of any grade: 0.4% were Grade 3 or 4 and there was one fatality (0.1%). In VERZENIO-treated patients in advanced or metastatic breast cancer (N=900) (MONARCH 1, MONARCH 2, MONARCH 3), 3.3% of VERZENIO-treated patients had ILD or pneumonitis of any grade: 0.6% had Grade 3 or 4, and 0.4% had fatal outcomes. Additional cases of ILD or pneumonitis have been observed in the postmarketing setting, with fatalities reported [see Adverse Reactions (6.2)].
Monitor patients for pulmonary symptoms indicative of ILD or pneumonitis. Symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams. Infectious, neoplastic, and other causes for such symptoms should be excluded by means of appropriate investigations.
Dose interruption or dose reduction is recommended for patients who develop persistent or recurrent Grade 2 ILD or pneumonitis. Permanently discontinue VERZENIO in all patients with Grade 3 or 4 ILD or pneumonitis [see Dosage and Administration (2.2)].
5.4 Hepatotoxicity
Grade ≥3 ALT (2% to 6%) and AST (2% to 3%) were reported in patients receiving VERZENIO.
Across three clinical trials in 3559 patients (monarchE, MONARCH 2, MONARCH 3), the median time to onset of Grade ≥3 ALT increases ranged from 57 to 87 days and the median time to resolution to Grade <3 was 13 to 14 days. The median time to onset of Grade ≥3 AST increases ranged from 71 to 185 days and the median time to resolution to Grade <3 ranged from 11 to 15 days.
Monitor liver function tests (LFTs) prior to the start of VERZENIO therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. Dose interruption, dose reduction, dose discontinuation, or delay in starting treatment cycles is recommended for patients who develop persistent or recurrent Grade 2, or any Grade 3 or Grade 4 hepatic transaminase elevation [see Dosage and Administration (2.2)].
5.5 Venous Thromboembolism
Across three clinical trials in 3559 patients (monarchE, MONARCH 2, MONARCH 3), venous thromboembolic events were reported in 2% to 5% of patients treated with VERZENIO. Venous thromboembolic events included deep vein thrombosis, pulmonary embolism, pelvic venous thrombosis, cerebral venous sinus thrombosis, subclavian and axillary vein thrombosis, and inferior vena cava thrombosis. In clinical trials, deaths due to venous thromboembolism have been reported in patients treated with VERZENIO.
VERZENIO has not been studied in patients with early breast cancer who had a history of venous thromboembolism. Monitor patients for signs and symptoms of venous thrombosis and pulmonary embolism and treat as medically appropriate. Dose interruption is recommended for early breast cancer patients with any grade venous thromboembolic event and for advanced or metastatic breast cancer patients with a Grade 3 or 4 venous thromboembolic event [see Dosage and Administration (2.2)].
5.6 Embryo-Fetal Toxicity
Based on findings from animal studies and the mechanism of action, VERZENIO can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of abemaciclib to pregnant rats during the period of organogenesis caused teratogenicity and decreased fetal weight at maternal exposures that were similar to the human clinical exposure based on area under the curve (AUC) at the maximum recommended human dose.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with VERZENIO and for 3 weeks after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Diarrhea [see Warnings and Precautions (5.1)].
- Neutropenia [see Warnings and Precautions (5.2)].
- Interstitial Lung Disease (ILD) or Pneumonitis [see Warnings and Precautions (5.3)].
- Hepatotoxicity [see Warnings and Precautions (5.4)].
- Venous Thromboembolism [see Warnings and Precautions (5.5)].
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety population described in the Warnings and Precautions reflect exposure to VERZENIO in 3691 patients from four clinical trials: monarchE, MONARCH 1, MONARCH 2, and MONARCH 3. The safety population includes exposure to VERZENIO as a single agent at 200 mg twice daily in 132 patients in MONARCH 1 and to VERZENIO at 150 mg twice daily in 3559 patients administered in combination with fulvestrant, tamoxifen, or an aromatase inhibitor in monarchE, MONARCH 2, and MONARCH 3. The median duration of exposure ranged from 4.5 months in MONARCH 1 to 24 months in monarchE. The most common adverse reactions (incidence ≥20%) across clinical trials were: diarrhea, neutropenia, nausea, abdominal pain, infections, fatigue, anemia, leukopenia, decreased appetite, vomiting, headache, alopecia, and thrombocytopenia.
Early Breast Cancer
monarchE: VERZENIO in Combination with Tamoxifen or an Aromatase Inhibitor as Adjuvant Treatment
Adult patients with HR-positive, HER2-negative, node-positive early breast cancer at a high risk of recurrence
The safety of VERZENIO was evaluated in monarchE, a study of 5591 adult patients receiving VERZENIO plus endocrine therapy (tamoxifen or an aromatase inhibitor) or endocrine therapy (tamoxifen or an aromatase inhibitor) alone [see Clinical Studies (14.1)]. Patients were randomly assigned to receive 150 mg of VERZENIO orally, twice daily, plus tamoxifen or an aromatase inhibitor, or tamoxifen or an aromatase inhibitor, for two years or until discontinuation criteria were met. The median duration of VERZENIO treatment was 24 months.
The most frequently reported (≥5%) Grade 3 or 4 adverse reactions were neutropenia, leukopenia, diarrhea, and lymphopenia.
Fatal adverse reactions occurred in 0.8% of patients who received VERZENIO plus endocrine therapy (tamoxifen or an aromatase inhibitor), including: cardiac failure (0.1%), cardiac arrest, myocardial infarction, ventricular fibrillation, cerebral hemorrhage, cerebrovascular accident, pneumonitis, hypoxia, diarrhea, and mesenteric artery thrombosis (0.03% each).
Permanent VERZENIO treatment discontinuation due to an adverse reaction was reported in 19% of patients receiving VERZENIO, plus tamoxifen or an aromatase inhibitor. Of the patients receiving tamoxifen or an aromatase inhibitor, 1% permanently discontinued due to an adverse reaction. The most common adverse reactions leading to VERZENIO discontinuations were diarrhea (5%), fatigue (2%), and neutropenia (0.9%).
Dose interruption of VERZENIO due to an adverse reaction occurred in 62% of patients receiving VERZENIO plus tamoxifen or aromatase inhibitors. Adverse reactions leading to VERZENIO dose interruptions in ≥5% of patients were diarrhea (20%), neutropenia (16%), leukopenia (7%), and fatigue (5%).
Dose reductions of VERZENIO due to an adverse reaction occurred in 44% of patients receiving VERZENIO plus endocrine therapy (tamoxifen or an aromatase inhibitor). Adverse reactions leading to VERZENIO dose reductions in ≥5% were diarrhea (17%), neutropenia (8%), and fatigue (5%).
The most common adverse reactions reported (≥20%) in the VERZENIO, plus tamoxifen or an aromatase inhibitor, arm and ≥2% higher than the tamoxifen or an aromatase inhibitor arm were: diarrhea, infections, neutropenia, fatigue, leukopenia, nausea, anemia, and headache. Adverse reactions are shown in Table 8 and laboratory abnormalities are shown in Table 9.
|
a Includes the following fatal adverse reactions: diarrhea (n=1), and infections (n=4) |
||||||
|
b Includes the following fatal adverse reactions: infections (n=5) |
||||||
|
c Includes mouth ulceration, mucosal inflammation, oropharyngeal pain, stomatitis. |
||||||
|
d Includes all reported preferred terms that are part of the Infections and Infestations system organ class. Most common infections (>5%) include upper respiratory tract infection, urinary tract infection, and nasopharyngitis. |
||||||
|
e Includes asthenia, fatigue. |
||||||
|
f Includes exfoliative rash, mucocutaneous rash, rash, rash erythematous, rash follicular, rash generalized, rash macular, rash maculo-papular, rash maculovesicular, rash morbilliform, rash papular, rash papulosquamous, rash pruritic, rash vesicular, vulvovaginal rash. |
||||||
| VERZENIO Plus
Tamoxifen or an Aromatase Inhibitor N=2791 | Tamoxifen or an Aromatase Inhibitor
N=2800 |
|||||
| All Gradesa
% | Grade 3
% | Grade 4
% | All Gradesb
% | Grade 3
% | Grade 4
% |
|
| Gastrointestinal Disorders | ||||||
| Diarrhea | 84 | 8 | 0 | 9 | 0.2 | 0 |
| Nausea | 30 | 0.5 | 0 | 9 | <0.1 | 0 |
| Vomiting | 18 | 0.5 | 0 | 4.6 | 0.1 | 0 |
| Stomatitisc | 14 | 0.1 | 0 | 5 | 0 | 0 |
| Infections and Infestations | ||||||
| Infectionsd | 51 | 4.9 | 0.6 | 39 | 2.7 | 0.1 |
| General Disorders and Administration Site Conditions | ||||||
| Fatiguee | 41 | 2.9 | 0 | 18 | 0.1 | 0 |
| Nervous System Disorders | ||||||
| Headache | 20 | 0.3 | 0 | 15 | 0.2 | 0 |
| Dizziness | 11 | 0.1 | 0 | 7 | <0.1 | 0 |
| Metabolism and Nutrition Disorders | ||||||
| Decreased appetite | 12 | 0.6 | 0 | 2.4 | <0.1 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||||
| Rashf | 11 | 0.4 | 0 | 4.5 | 0 | 0 |
| Alopecia | 11 | 0 | 0 | 2.7 | 0 | 0 |
Clinically relevant adverse reactions in <10% of patients who received VERZENIO in combination with tamoxifen or an aromatase inhibitor in monarchE include:
- Pruritus-9%
- Dyspepsia-8%
- Nail disorder-6% (includes nail bed disorder, nail bed inflammation, nail discoloration, nail disorder, nail dystrophy, nail pigmentation, nail ridging, nail toxicity, onychalgia, onychoclasis, onycholysis, onychomadesis)
- Lacrimation increased-6%
- Dysgeusia-5%
- Interstitial lung disease (ILD)/pneumonitis-3% (includes pneumonitis, radiation pneumonitis, interstitial lung disease, pulmonary fibrosis, organizing pneumonia, radiation fibrosis – lung, lung opacity, sarcoidosis)
- Venous thromboembolic events (VTEs)-3% (includes catheter site thrombosis, cerebral venous thrombosis, deep vein thrombosis, device related thrombosis, embolism, hepatic vein thrombosis, jugular vein occlusion, jugular vein thrombosis, ovarian vein thrombosis, portal vein thrombosis, pulmonary embolism, subclavian vein thrombosis, venous thrombosis limb)
| VERZENIO
Plus Tamoxifen or an Aromatase Inhibitor N=2791 | Tamoxifen or an Aromatase Inhibitor
N=2800 |
|||||
| All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
|
| Creatinine increased | 99 | 0.5 | 0 | 91 | <0.1 | 0 |
| White blood cell decreased | 89 | 19 | <0.1 | 28 | 1.1 | 0 |
| Neutrophil count decreased | 84 | 18 | 0.7 | 23 | 1.6 | 0.3 |
| Anemia | 68 | 1.0 | 0 | 17 | 0.1 | 0 |
| Lymphocyte count decreased | 59 | 13 | 0.2 | 24 | 2.4 | 0.1 |
| Platelet count decreased | 37 | 0.7 | 0.2 | 10 | 0.1 | 0.1 |
| Alanine aminotransferase increased | 37 | 2.5 | <0.1 | 24 | 1.2 | 0 |
| Aspartate aminotransferase increased | 31 | 1.5 | <0.1 | 18 | 0.9 | 0 |
| Hypokalemia | 11 | 1.2 | 0.1 | 3.8 | 0.1 | 0.1 |
Advanced or Metastatic Breast Cancer
MONARCH 3: VERZENIO in Combination with an Aromatase Inhibitor (Anastrozole or Letrozole) as Initial Endocrine-Based Therapy
Postmenopausal Women with HR-positive, HER2-negative locoregionally recurrent or metastatic breast cancer with no prior systemic therapy in this disease setting
The safety of VERZENIO was evaluated in MONARCH 3, a study of 488 women receiving VERZENIO plus an aromatase inhibitor or placebo plus an aromatase inhibitor [see Clinical Studies (14.2)]. Patients were randomly assigned to receive 150 mg of VERZENIO or placebo orally twice daily, plus physician's choice of anastrozole or letrozole once daily. Median duration of treatment was 15.1 months for the VERZENIO arm and 13.9 months for the placebo arm.
The most frequently reported (≥5%) Grade 3 or 4 adverse reactions were neutropenia, diarrhea, leukopenia, increased ALT, and anemia.
Deaths during treatment or during the 30-day follow up, regardless of causality, were reported in 11 cases (3%) of VERZENIO plus an aromatase inhibitor treated patients versus 3 cases (2%) of placebo plus an aromatase inhibitor treated patients. Causes of death for patients receiving VERZENIO plus an aromatase inhibitor included: 3 (0.9%) patient deaths due to underlying disease, 3 (0.9%) due to lung infection, 3 (0.9%) due to VTE, 1 (0.3%) due to pneumonitis, and 1 (0.3%) due to cerebral infarction.
Permanent treatment discontinuation due to an adverse reaction was reported in 13% of patients receiving VERZENIO plus an aromatase inhibitor and in 3% of patients receiving placebo plus an aromatase inhibitor. Adverse reactions leading to permanent discontinuation for patients receiving VERZENIO plus an aromatase inhibitor were diarrhea (2%), ALT increased (2%), infection (1%), venous thromboembolic events (VTE) (1%), neutropenia (0.9%), renal impairment (0.9%), AST increased (0.6%), dyspnea (0.6%), pulmonary fibrosis (0.6%) and anemia, rash, weight decreased and thrombocytopenia (each 0.3%).
Dose interruption of VERZENIO due to an adverse reaction occurred in 56% of patients receiving VERZENIO plus anastrazole or letrozole. Adverse reactions leading to VERZENIO dose interruptions in ≥5% of patients were neutropenia (16%) and diarrhea (15%).
Dose reductions due to an adverse reaction occurred in 43% of patients receiving VERZENIO plus anastrozole or letrozole. Adverse reactions leading to dose reductions in ≥5% of patients were diarrhea and neutropenia. VERZENIO dose reductions due to diarrhea of any grade occurred in 13% of patients receiving VERZENIO plus an aromatase inhibitor compared to 2% of patients receiving placebo plus an aromatase inhibitor. VERZENIO dose reductions due to neutropenia of any grade occurred in 11% of patients receiving VERZENIO plus an aromatase inhibitor compared to 0.6% of patients receiving placebo plus an aromatase inhibitor.
The most common adverse reactions reported (≥20%) in the VERZENIO arm and ≥2% than the placebo arm were: diarrhea, neutropenia, fatigue, infections, nausea, abdominal pain, anemia, vomiting, alopecia, decreased appetite, and leukopenia. Adverse reactions are shown in Table 10 and laboratory abnormalities in Table 11. Diarrhea incidence was greatest during the first month of VERZENIO dosing. The median time to onset of the first diarrhea event was 8 days, and the median durations of diarrhea for Grades 2 and for Grade 3 were 11 days and 8 days, respectively. Most diarrhea events recovered or resolved (88%) with supportive treatment and/or dose reductions [see Dosage and Administration (2.2) and Patient Counseling Information (17)]. Nineteen percent of patients with diarrhea required a dose omission and 13% required a dose reduction. The median time to the first dose reduction due to diarrhea was 38 days.
|
a Includes all reported preferred terms that are part of the Infections and Infestations system organ class. Most common infections (>1%) include upper respiratory tract infection, lung infection, and pharyngitis. |
||||||
| VERZENIO plus
Anastrozole or Letrozole N=327 | Placebo plus
Anastrozole or Letrozole N=161 |
|||||
| All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
|
| Gastrointestinal Disorders | ||||||
| Diarrhea | 81 | 9 | 0 | 30 | 1.2 | 0 |
| Nausea | 39 | 0.9 | 0 | 20 | 1.2 | 0 |
| Abdominal pain | 29 | 1.2 | 0 | 12 | 1.2 | 0 |
| Vomiting | 28 | 1.2 | 0 | 12 | 1.9 | 0 |
| Constipation | 16 | 0.6 | 0 | 12 | 0 | 0 |
| Infections and Infestations | ||||||
| Infectionsa | 39 | 4.0 | 0.9 | 29 | 2.5 | 0.6 |
| General Disorders and Administration Site Conditions | ||||||
| Fatigue | 40 | 1.8 | 0 | 32 | 0 | 0 |
| Influenza like illness | 10 | 0 | 0 | 8 | 0 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||||
| Alopecia | 27 | 0 | 0 | 11 | 0 | 0 |
| Rash | 14 | 0.9 | 0 | 5 | 0 | 0 |
| Pruritus | 13 | 0 | 0 | 9 | 0 | 0 |
| Metabolism and Nutrition Disorders | ||||||
| Decreased appetite | 24 | 1.2 | 0 | 9 | 0.6 | 0 |
| Investigations | ||||||
| Weight decreased | 10 | 0.6 | 0 | 3.1 | 0.6 | 0 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||||||
| Cough | 13 | 0 | 0 | 9 | 0 | 0 |
| Dyspnea | 12 | 0.6 | 0.3 | 6 | 0.6 | 0 |
| Nervous System Disorders | ||||||
| Dizziness | 11 | 0.3 | 0 | 9 | 0 | 0 |
Additional adverse reactions in MONARCH 3 include venous thromboembolic events (deep vein thrombosis, pulmonary embolism, and pelvic venous thrombosis), which were reported in 5% of patients treated with VERZENIO plus anastrozole or letrozole as compared to 0.6% of patients treated with anastrozole or letrozole plus placebo.
| VERZENIO plus
Anastrozole or Letrozole N=327 | Placebo plus
Anastrozole or Letrozole N=161 |
|||||
| Laboratory Abnormality | All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
| Creatinine increased | 98 | 2.2 | 0 | 84 | 0 | 0 |
| White blood cell decreased | 82 | 13 | 0 | 27 | 0.6 | 0 |
| Anemia | 82 | 1.6 | 0 | 28 | 0 | 0 |
| Neutrophil count decreased | 80 | 19 | 2.9 | 21 | 2.6 | 0 |
| Lymphocyte count decreased | 53 | 7 | 0.6 | 26 | 1.9 | 0 |
| Platelet count decreased | 36 | 1.3 | 0.6 | 12 | 0.6 | 0 |
| Alanine aminotransferase increased | 48 | 6 | 0.6 | 25 | 1.9 | 0 |
| Aspartate aminotransferase increased | 37 | 3.8 | 0 | 23 | 0.6 | 0 |
Creatinine Increased
Abemaciclib has been shown to increase serum creatinine due to inhibition of renal tubular secretion transporters, without affecting glomerular function [see Clinical Pharmacology (12.3)]. Across the clinical studies, increases in serum creatinine (mean increase, 0.2-0.3 mg/dL) occurred within the first 28-day cycle of VERZENIO dosing, remained elevated but stable through the treatment period, and were reversible upon treatment discontinuation. Alternative markers such as BUN, cystatin C, or calculated GFR, which are not based on creatinine, may be considered to determine whether renal function is impaired.
MONARCH 2: VERZENIO in Combination with Fulvestrant
Women with HR-positive, HER2-negative advanced or metastatic breast cancer with disease progression on or after prior adjuvant or metastatic endocrine therapy
The safety of VERZENIO (150 mg twice daily) plus fulvestrant (500 mg) versus placebo plus fulvestrant was evaluated in MONARCH 2 [see Clinical Studies (14.2)]. The data described below reflect exposure to VERZENIO in 441 patients with HR-positive, HER2-negative advanced breast cancer who received at least one dose of VERZENIO plus fulvestrant in MONARCH 2.
Median duration of treatment was 12 months for patients receiving VERZENIO plus fulvestrant and 8 months for patients receiving placebo plus fulvestrant.
The most frequently reported (≥5%) Grade 3 or 4 adverse reactions were neutropenia, diarrhea, leukopenia, anemia, and infections.
Deaths during treatment or during the 30-day follow up, regardless of causality, were reported in 18 cases (4%) of VERZENIO plus fulvestrant treated patients versus 10 cases (5%) of placebo plus fulvestrant treated patients. Causes of death for patients receiving VERZENIO plus fulvestrant included: 7 (2%) patient deaths due to underlying disease, 4 (0.9%) due to sepsis, 2 (0.5%) due to pneumonitis, 2 (0.5%) due to hepatotoxicity, and one (0.2%) due to cerebral infarction.
Permanent study treatment discontinuation due to an adverse reaction were reported in 9% of patients receiving VERZENIO plus fulvestrant and in 3% of patients receiving placebo plus fulvestrant. Adverse reactions leading to permanent discontinuation for patients receiving VERZENIO plus fulvestrant were infection (2%), diarrhea (1%), hepatotoxicity (1%), fatigue (0.7%), nausea (0.2%), abdominal pain (0.2%), acute kidney injury (0.2%), and cerebral infarction (0.2%).
Dose interruption of VERZENIO due to an adverse reaction occurred in 52% of patients receiving VERZENIO plus fulvestrant. Adverse reactions leading to VERZENIO dose interruptions in ≥5% of patients were diarrhea (19%) and neutropenia (16%).
Dose reductions due to an adverse reaction occurred in 43% of patients receiving VERZENIO plus fulvestrant. Adverse reactions leading to reductions in ≥5% of patients were diarrhea and neutropenia. VERZENIO dose reductions due to diarrhea of any grade occurred in 19% of patients receiving VERZENIO plus fulvestrant compared to 0.4% of patients receiving placebo and fulvestrant. VERZENIO dose reductions due to neutropenia of any grade occurred in 10% of patients receiving VERZENIO plus fulvestrant compared to no patients receiving placebo plus fulvestrant.
The most common adverse reactions reported (≥20%) in the VERZENIO arm were: diarrhea, fatigue, neutropenia, nausea, infections, abdominal pain, anemia, leukopenia, decreased appetite, vomiting, and headache. Adverse reactions are shown in Table 12 and laboratory abnormalities in Table 13.
|
a Includes abdominal pain, abdominal pain upper, abdominal pain lower, abdominal discomfort, abdominal tenderness. |
||||||
|
b Includes upper respiratory tract infection, urinary tract infection, lung infection, pharyngitis, conjunctivitis, sinusitis, vaginal infection, sepsis. |
||||||
|
c Includes asthenia, fatigue. |
||||||
| VERZENIO plus Fulvestrant
N=441 | Placebo plus Fulvestrant
N=223 |
|||||
| All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
|
| Gastrointestinal Disorders | ||||||
| Diarrhea | 86 | 13 | 0 | 25 | 0.4 | 0 |
| Nausea | 45 | 2.7 | 0 | 23 | 0.9 | 0 |
| Abdominal paina | 35 | 2.5 | 0 | 16 | 0.9 | 0 |
| Vomiting | 26 | 0.9 | 0 | 10 | 1.8 | 0 |
| Stomatitis | 15 | 0.5 | 0 | 10 | 0 | 0 |
| Infections and Infestations | ||||||
| Infectionsb | 43 | 5 | 0.7 | 25 | 3.1 | 0.4 |
| General Disorders and Administration Site Conditions | ||||||
| Fatiguec | 46 | 2.7 | 0 | 32 | 0.4 | 0 |
| Edema peripheral | 12 | 0 | 0 | 7 | 0 | 0 |
| Pyrexia | 11 | 0.5 | 0.2 | 6 | 0.4 | 0 |
| Metabolism and Nutrition Disorders | ||||||
| Decreased appetite | 27 | 1.1 | 0 | 12 | 0.4 | 0 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||||
| Cough | 13 | 0 | 0 | 11 | 0 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||||
| Alopecia | 16 | 0 | 0 | 1.8 | 0 | 0 |
| Pruritus | 13 | 0 | 0 | 6 | 0 | 0 |
| Rash | 11 | 1.1 | 0 | 4.5 | 0 | 0 |
| Nervous System Disorders | ||||||
| Headache | 20 | 0.7 | 0 | 15 | 0.4 | 0 |
| Dysgeusia | 18 | 0 | 0 | 2.7 | 0 | 0 |
| Dizziness | 12 | 0.7 | 0 | 6 | 0 | 0 |
| Investigations | ||||||
| Weight decreased | 10 | 0.2 | 0 | 2.2 | 0.4 | 0 |
Additional adverse reactions in MONARCH 2 include venous thromboembolic events (deep vein thrombosis, pulmonary embolism, cerebral venous sinus thrombosis, subclavian vein thrombosis, axillary vein thrombosis, and DVT inferior vena cava), which were reported in 5% of patients treated with VERZENIO plus fulvestrant as compared to 0.9% of patients treated with fulvestrant plus placebo.
| VERZENIO plus Fulvestrant
N=441 | Placebo plus Fulvestrant
N=223 |
|||||
| All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
|
| Creatinine increased | 98 | 1.2 | 0 | 74 | 0 | 0 |
| White blood cell decreased | 90 | 23 | 0.7 | 33 | 0.9 | 0 |
| Neutrophil count decreased | 87 | 29 | 3.5 | 30 | 3.7 | 0.5 |
| Anemia | 84 | 2.6 | 0 | 34 | 0.5 | 0 |
| Lymphocyte count decreased | 63 | 12 | 0.2 | 32 | 1.8 | 0 |
| Platelet count decreased | 53 | 0.9 | 1.2 | 15 | 0 | 0 |
| Alanine aminotransferase increased | 41 | 3.9 | 0.7 | 32 | 1.4 | 0 |
| Aspartate aminotransferase increased | 37 | 3.9 | 0 | 25 | 3.7 | 0.5 |
Creatinine Increased
Abemaciclib has been shown to increase serum creatinine due to inhibition of renal tubular secretion transporters, without affecting glomerular function [see Clinical Pharmacology (12.3)]. In clinical studies, increases in serum creatinine (mean increase, 0.2-0.3 mg/dL) occurred within the first 28-day cycle of VERZENIO dosing, remained elevated but stable through the treatment period, and were reversible upon treatment discontinuation. Alternative markers such as BUN, cystatin C, or calculated glomerular filtration rate (GFR), which are not based on creatinine, may be considered to determine whether renal function is impaired.
MONARCH 1: VERZENIO Administered as a Monotherapy in Metastatic Breast Cancer
Patients with HR-positive, HER2-negative breast cancer who received prior endocrine therapy and 1-2 chemotherapy regimens in the metastatic setting
The safety of VERZENIO was evaluated in MONARCH 1, a single-arm, open-label, multicenter study in 132 women with measurable HR-positive, HER2-negative metastatic breast cancer [see Clinical Studies (14.2)]. Patients received 200 mg VERZENIO orally twice daily until development of progressive disease or unmanageable toxicity. Median duration of treatment was 4.5 months.
The most frequently reported (≥5%) Grade 3 or 4 adverse reactions were diarrhea, neutropenia, fatigue, and leukopenia.
Deaths due to adverse reactions during treatment or during the 30-day follow up were reported in 2% of patients. Cause of death in these patients was due to infection (2 patients) or pneumonitis (1 patient).
Ten patients (8%) discontinued study treatment from adverse reactions due to (1 patient each), abdominal pain, arterial thrombosis, aspartate aminotransferase (AST) increased, blood creatinine increased, chronic kidney disease, diarrhea, ECG QT prolonged, fatigue, hip fracture, and lymphopenia.
Dose interruption of VERZENIO due to an adverse reaction occurred in 58% of patients. The most frequent (≥5%) adverse reactions leading to dose interruptions were diarrhea (24%), neutropenia (16%), fatigue (10%), vomiting (6%), and nausea (5%).
Forty-nine percent of patients had dose reductions due to an adverse reaction. The most frequent adverse reactions that led to dose reductions were diarrhea (20%), neutropenia (11%), and fatigue (9%).
The most common reported adverse reactions (≥20%) were: diarrhea, fatigue, nausea, decreased appetite, abdominal pain, neutropenia, vomiting, infections, anemia, headache, and thrombocytopenia. Adverse reactions are shown in Table 14 and laboratory abnormalities in Table 15.
|
a Includes asthenia, fatigue. |
|||
| VERZENIO
N=132 |
|||
| All Grades
% | Grade 3
% | Grade 4
% |
|
| Gastrointestinal Disorders | |||
| Diarrhea | 90 | 20 | 0 |
| Nausea | 64 | 4.5 | 0 |
| Abdominal pain | 39 | 2.3 | 0 |
| Vomiting | 35 | 1.5 | 0 |
| Constipation | 17 | 0.8 | 0 |
| Dry mouth | 14 | 0 | 0 |
| Stomatitis | 14 | 0 | 0 |
| Infections and Infestations | |||
| Infections | 31 | 4.5 | 0 |
| General Disorders and Administration Site Conditions | |||
| Fatiguea | 65 | 13 | 0 |
| Pyrexia | 11 | 0 | 0 |
| Metabolism and Nutrition Disorders | |||
| Decreased appetite | 45 | 3.0 | 0 |
| Dehydration | 10 | 2.3 | 0 |
| Respiratory, Thoracic and Mediastinal Disorders | |||
| Cough | 19 | 0 | 0 |
| Musculoskeletal and Connective Tissue Disorders | |||
| Arthralgia | 15 | 0 | 0 |
| Nervous System Disorders | |||
| Headache | 20 | 0 | 0 |
| Dysgeusia | 12 | 0 | 0 |
| Dizziness | 11 | 0 | 0 |
| Skin and Subcutaneous Tissue Disorders | |||
| Alopecia | 12 | 0 | 0 |
| Investigations | |||
| Weight decreased | 14 | 0 | 0 |
| VERZENIO
N=132 |
|||
| All Grades
% | Grade 3
% | Grade 4
% |
|
| Creatinine increased | 99 | 0.8 | 0 |
| White blood cell decreased | 91 | 28 | 0 |
| Neutrophil count decreased | 88 | 22 | 4.6 |
| Anemia | 69 | 0 | 0 |
| Lymphocyte count decreased | 42 | 13 | 0.8 |
| Platelet count decreased | 41 | 2.3 | 0 |
| ALT increased | 31 | 3.1 | 0 |
| AST increased | 30 | 3.8 | 0 |
Creatinine Increased
Abemaciclib has been shown to increase serum creatinine due to inhibition of renal tubular secretion transporters, without affecting glomerular function [see Clinical Pharmacology (12.3)]. In clinical studies, increases in serum creatinine (mean increase, 0.2-0.3 mg/dL) occurred within the first 28-day cycle of VERZENIO dosing, remained elevated but stable through the treatment period, and were reversible upon treatment discontinuation. Alternative markers such as BUN, cystatin C, or calculated GFR, which are not based on creatinine, may be considered to determine whether renal function is impaired.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VERZENIO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory disorders: Interstitial lung disease (ILD)/pneumonitis [see Warnings and Precautions (5.3)].
7. Drug Interactions
7.1 Effect of Other Drugs on VERZENIO
CYP3A Inhibitors
Strong and moderate CYP3A4 inhibitors increased the exposure of abemaciclib plus its active metabolites to a clinically meaningful extent and may lead to increased toxicity.
Ketoconazole
Avoid concomitant use of ketoconazole. Ketoconazole is predicted to increase the AUC of abemaciclib by up to 16-fold [see Clinical Pharmacology (12.3)].
Other Strong CYP3A Inhibitors
In patients with recommended starting doses of 200 mg twice daily or 150 mg twice daily, reduce the VERZENIO dose to 100 mg twice daily with concomitant use of strong CYP3A inhibitors other than ketoconazole. In patients who have had a dose reduction to 100 mg twice daily due to adverse reactions, further reduce the VERZENIO dose to 50 mg twice daily with concomitant use of strong CYP3A inhibitors. If a patient taking VERZENIO discontinues a strong CYP3A inhibitor, increase the VERZENIO dose (after 3-5 half-lives of the inhibitor) to the dose that was used before starting the inhibitor. Patients should avoid grapefruit products [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
Strong and Moderate CYP3A Inducers
Coadministration of strong or moderate CYP3A inducers decreased the plasma concentrations of abemaciclib plus its active metabolites and may lead to reduced activity. Avoid concomitant use of strong or moderate CYP3A inducers and consider alternative agents [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animals and its mechanism of action, VERZENIO can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus. In animal reproduction studies, administration of abemaciclib during organogenesis was teratogenic and caused decreased fetal weight at maternal exposures that were similar to human clinical exposure based on AUC at the maximum recommended human dose (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk in the U.S. general population of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies.
Data
Animal Data
In an embryo-fetal development study, pregnant rats received oral doses of abemaciclib up to 15 mg/kg/day during the period of organogenesis. Doses ≥4 mg/kg/day caused decreased fetal body weights and increased incidence of cardiovascular and skeletal malformations and variations. These findings included absent innominate artery and aortic arch, malpositioned subclavian artery, unossified sternebra, bipartite ossification of thoracic centrum, and rudimentary or nodulated ribs. At 4 mg/kg/day in rats, the maternal systemic exposures were approximately equal to the human exposure (AUC) at the recommended dose.
8.2 Lactation
Risk Summary
There are no data on the presence of abemaciclib in human milk, or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed infants from VERZENIO, advise lactating women not to breastfeed during VERZENIO treatment and for 3 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal studies, VERZENIO can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating treatment with VERZENIO.
8.4 Pediatric Use
The safety and effectiveness of VERZENIO have not been established in pediatric patients.
8.5 Geriatric Use
Of the 2791 VERZENIO-treated patients in monarchE, 15% were 65 years of age or older and 2.7% were 75 years of age or older.
Of the 900 patients who received VERZENIO in MONARCH 1, MONARCH 2, and MONARCH 3, 38% were 65 years of age or older and 10% were 75 years of age or older. The most common adverse reactions (≥5%) Grade 3 or 4 in patients ≥65 years of age across MONARCH 1, 2, and 3 were: neutropenia, diarrhea, fatigue, nausea, dehydration, leukopenia, anemia, infections, and ALT increased.
No overall differences in safety or effectiveness of VERZENIO were observed between these patients and younger patients.
8.6 Renal Impairment
No dosage adjustment is required for patients with mild or moderate renal impairment (CLcr ≥30-89 mL/min, estimated by Cockcroft-Gault [C-G]). The pharmacokinetics of abemaciclib in patients with severe renal impairment (CLcr <30 mL/min, C-G), end stage renal disease, or in patients on dialysis is unknown [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustments are necessary in patients with mild or moderate hepatic impairment (Child-Pugh A or B).
Reduce the dosing frequency when administering VERZENIO to patients with severe hepatic impairment (Child-Pugh C) [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
11. Verzenio Description
Abemaciclib is a kinase inhibitor for oral administration. It is a white to yellow powder with the empirical formula C27H32F2N8 and a molecular weight 506.59.
The chemical name for abemaciclib is 2-Pyrimidinamine, N-[5-[(4-ethyl-1-piperazinyl)methyl]-2-pyridinyl]-5-fluoro-4-[4-fluoro-2-methyl-1-(1-methylethyl)-1H-benzimidazol-6-yl]-. Abemaciclib has the following structure:
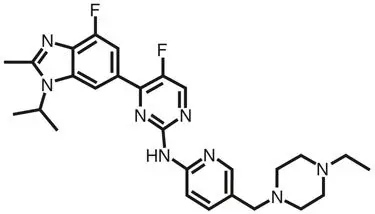
VERZENIO (abemaciclib) tablets are provided as immediate-release oval white, beige, or yellow tablets. Inactive ingredients are as follows: Excipients—microcrystalline cellulose 102, microcrystalline cellulose 101, lactose monohydrate, croscarmellose sodium, sodium stearyl fumarate, silicon dioxide. Color mixture ingredients—polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, iron oxide yellow, and iron oxide red.
12. Verzenio - Clinical Pharmacology
12.1 Mechanism of Action
Abemaciclib is an inhibitor of cyclin-dependent kinases 4 and 6 (CDK4 and CDK6). These kinases are activated upon binding to D-cyclins. In estrogen receptor-positive (ER+) breast cancer cell lines, cyclin D1 and CDK4/6 promote phosphorylation of the retinoblastoma protein (Rb), cell cycle progression, and cell proliferation. In vitro, continuous exposure to abemaciclib inhibited Rb phosphorylation and blocked progression from G1 into S phase of the cell cycle, resulting in senescence and apoptosis. In breast cancer xenograft models, abemaciclib dosed daily without interruption as a single agent or in combination with antiestrogens resulted in reduction of tumor size.
12.3 Pharmacokinetics
The pharmacokinetics of abemaciclib were characterized in patients with solid tumors, including breast cancer, and in healthy subjects.
Following single and repeated twice daily dosing of 50 mg (0.3 times the approved recommended 150 mg dosage) to 200 mg of abemaciclib, the increase in plasma exposure (AUC) and Cmax was approximately dose proportional. Steady state was achieved within 5 days following repeated twice daily dosing, and the estimated geometric mean accumulation ratio was 2.3 (50% CV) and 3.2 (59% CV) based on Cmax and AUC, respectively.
Absorption
The absolute bioavailability of abemaciclib after a single oral dose of 200 mg is 45% (19% CV). The median Tmax of abemaciclib is 8.0 hours (range: 4.1-24.0 hours).
Effect of Food
A high-fat, high-calorie meal (approximately 800 to 1000 calories with 150 calories from protein, 250 calories from carbohydrate, and 500 to 600 calories from fat) administered to healthy subjects increased the AUC of abemaciclib plus its active metabolites by 9% and increased Cmax by 26%.
Distribution
In vitro, abemaciclib was bound to human plasma proteins, serum albumin, and alpha-1-acid glycoprotein in a concentration independent manner from 152 ng/mL to 5066 ng/mL. In a clinical study, the mean (standard deviation, SD) bound fraction was 96.3% (1.1) for abemaciclib, 93.4% (1.3) for M2, 96.8% (0.8) for M18, and 97.8% (0.6) for M20. The geometric mean systemic volume of distribution is approximately 690.3 L (49% CV).
In patients with advanced cancer, including breast cancer, concentrations of abemaciclib and its active metabolites M2 and M20 in cerebrospinal fluid are comparable to unbound plasma concentrations.
Elimination
The geometric mean hepatic clearance (CL) of abemaciclib in patients was 26.0 L/h (51% CV), and the mean plasma elimination half-life for abemaciclib in patients was 18.3 hours (72% CV).
Metabolism
Hepatic metabolism is the main route of clearance for abemaciclib. Abemaciclib is metabolized to several metabolites primarily by cytochrome P450 (CYP) 3A4, with formation of N-desethylabemaciclib (M2) representing the major metabolism pathway. Additional metabolites include hydroxyabemaciclib (M20), hydroxy-N-desethylabemaciclib (M18), and an oxidative metabolite (M1). M2, M18, and M20 are equipotent to abemaciclib and their AUCs accounted for 25%, 13%, and 26% of the total circulating analytes in plasma, respectively.
Specific Populations
Age, Gender, and Body Weight
Based on a population pharmacokinetic analysis in patients with cancer, age (range 24-91 years), gender (134 males and 856 females), and body weight (range 36-175 kg) had no effect on the exposure of abemaciclib.
Patients with Renal Impairment
In a population pharmacokinetic analysis of 990 individuals, in which 381 individuals had mild renal impairment (60 mL/min ≤ CLcr <90 mL/min) and 126 individuals had moderate renal impairment (30 mL/min ≤ CLcr <60 mL/min), mild and moderate renal impairment had no effect on the exposure of abemaciclib [see Use in Specific Populations (8.6)]. The effect of severe renal impairment (CLcr <30 mL/min) on pharmacokinetics of abemaciclib is unknown.
Patients with Hepatic Impairment
Following a single 200 mg oral dose of abemaciclib, the relative potency adjusted unbound AUC0-INF of abemaciclib plus its active metabolites (M2, M18, M20) in plasma increased 1.2-fold in subjects with mild hepatic impairment (Child-Pugh A, n=9), 1.1-fold in subjects with moderate hepatic impairment (Child-Pugh B, n=10), and 2.4-fold in subjects with severe hepatic impairment (Child-Pugh C, n=6) relative to subjects with normal hepatic function (n=10) [see Use in Specific Populations (8.7)]. In subjects with severe hepatic impairment, the mean plasma elimination half-life of abemaciclib increased to 55 hours compared to 24 hours in subjects with normal hepatic function.
Drug Interaction Studies
Effects of Other Drugs on Abemaciclib
Strong CYP3A Inhibitors: Ketoconazole (a strong CYP3A inhibitor) is predicted to increase the AUC of abemaciclib by up to 16-fold.
Coadministration of 500 mg twice daily doses of clarithromycin (a strong CYP3A inhibitor) with a single 50 mg dose of VERZENIO (0.3 times the approved recommended 150 mg dosage) increased the relative potency adjusted unbound AUC0-INF of abemaciclib plus its active metabolites (M2, M18, and M20) by 2.5-fold relative to abemaciclib alone in cancer patients.
Moderate CYP3A Inhibitors: Verapamil and diltiazem (moderate CYP3A inhibitors) are predicted to increase the relative potency adjusted unbound AUC of abemaciclib plus its active metabolites (M2, M18, and M20) by approximately 1.6-fold and 2.4-fold, respectively.
Strong CYP3A Inducers: Coadministration of 600 mg daily doses of rifampin (a strong CYP3A inducer) with a single 200 mg dose of VERZENIO decreased the relative potency adjusted unbound AUC0-INF of abemaciclib plus its active metabolites (M2, M18, and M20) by approximately 70% in healthy subjects.
Moderate CYP3A Inducers: Efavirenz, bosentan, and modafinil (moderate CYP3A inducers) are predicted to decrease the relative potency adjusted unbound AUC of abemaciclib plus its active metabolites (M2, M18, and M20) by 53%, 41%, and 29%, respectively.
Effects of Abemaciclib on Other Drugs
Loperamide: In a clinical drug interaction study in healthy subjects, coadministration of a single 8 mg dose of loperamide with a single 400 mg abemaciclib (2.7 times the approved recommended 150 mg dosage) increased loperamide AUC0-INF by 9% and Cmax by 35% relative to loperamide alone. These increases in loperamide exposure are not considered clinically relevant.
Metformin: In a clinical drug interaction study in healthy subjects, coadministration of a single 1000 mg dose of metformin, a clinically relevant substrate of renal OCT2, MATE1, and MATE2-K transporters, with a single 400 mg dose of abemaciclib (2.7 times the approved recommended 150 mg dosage) increased metformin AUC0-INF by 37% and Cmax by 22% relative to metformin alone. Abemaciclib reduced the renal clearance and renal secretion of metformin by 45% and 62%, respectively, relative to metformin alone, without any effect on glomerular filtration rate (GFR) as measured by iohexol clearance and serum cystatin C.
Endocrine Therapies: In clinical studies in patients with breast cancer, there was no clinically relevant effect of abemaciclib on the pharmacokinetics of fulvestrant, anastrozole, letrozole, exemestane, or tamoxifen.
CYP Metabolic Pathways: In a clinical drug interaction study in patients with cancer, multiple doses of abemaciclib (200 mg twice daily for 7 days) did not result in clinically meaningful changes in the pharmacokinetics of CYP1A2, CYP2C9, CYP2D6 and CYP3A4 substrates. Abemaciclib is a substrate of CYP3A4, and time-dependent changes in pharmacokinetics of abemaciclib as a result of autoinhibition of its metabolism were not observed.
In Vitro Studies
Transporter Systems: Abemaciclib and its major active metabolites inhibit the renal transporters OCT2, MATE1, and MATE2-K at concentrations achievable at the approved recommended dosage. The observed serum creatinine increase in clinical studies with abemaciclib is likely due to inhibition of tubular secretion of creatinine via OCT2, MATE1, and MATE2-K [see Adverse Effects (6.1)]. Abemaciclib and its major metabolites at clinically relevant concentrations do not inhibit the hepatic uptake transporters OCT1, OATP1B1, and OATP1B3 or the renal uptake transporters OAT1 and OAT3.
Abemaciclib is a substrate of P-gp and BCRP. Abemaciclib and its major active metabolites, M2 and M20, are not substrates of hepatic uptake transporters OCT1, organic anion transporting polypeptide 1B1 (OATP1B1), or OATP1B3.
Abemaciclib inhibits P-gp and BCRP. The clinical consequences of this finding on sensitive P-gp and BCRP substrates are unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Abemaciclib was assessed for carcinogenicity in a 2-year rat study. Abemaciclib was not carcinogenic in male and female rats at oral doses up to 3 mg/kg/day (approximately 1 time the exposure at the maximum recommended human dose based on AUC).
Abemaciclib and its active human metabolites M2 and M20 were not mutagenic in a bacterial reverse mutation (Ames) assay or clastogenic in an in vitro chromosomal aberration assay in Chinese hamster ovary cells or human peripheral blood lymphocytes. Abemaciclib, M2, and M20 were not clastogenic in an in vivo rat bone marrow micronucleus assay.
Abemaciclib may impair fertility in males of reproductive potential. In repeat-dose toxicity studies up to 3-months duration, abemaciclib-related findings in the testis, epididymis, prostate, and seminal vesicle at doses ≥10 mg/kg/day in rats and ≥0.3 mg/kg/day in dogs included decreased organ weights, intratubular cellular debris, hypospermia, tubular dilatation, atrophy, and degeneration/necrosis. These doses in rats and dogs resulted in approximately 2 and 0.02 times, respectively, the exposure (AUC) in humans at the maximum recommended human dose. In a rat male fertility study, abemaciclib had no effects on mating and fertility at oral doses up to 10 mg/kg/day (approximately 2 times the exposure at the maximum recommended human dose based on AUC).
In a rat female fertility and early embryonic development study, abemaciclib did not affect mating and fertility at doses up to 20 mg/kg/day (approximately 3 times the exposure at the maximum recommended human dose based on AUC).
13.2 Animal Toxicology and/or Pharmacology
In repeat-dose toxicity studies up to 6-months duration, oral administration of abemaciclib resulted in retinal atrophy of the eyes in mice at a dose of 150 mg/kg/day (approximately 10 times the exposure at the maximum recommended human dose based on AUC) and in rats at a dose of 30 mg/kg/day (approximately 5 times the exposure at the maximum recommended human dose based on AUC). In a 2-year rat carcinogenicity study, oral administration of abemaciclib resulted in retinal atrophy in the eyes at doses ≥0.3 mg/kg/day (approximately 0.05 times the exposure at the maximum recommended human dose based on AUC).
14. Clinical Studies
14.1 Early Breast Cancer
VERZENIO in Combination with Standard Endocrine Therapy (monarchE)
Patients with HR-positive, HER2-negative, node-positive early breast cancer at high risk of recurrence
monarchE (NCT03155997) was a randomized (1:1), open-label, two cohort, multicenter study in adult women and men with HR-positive, HER2-negative, node-positive, resected, early breast cancer with clinical and pathological features consistent with a high risk of disease recurrence. To be enrolled, patients had to have HR-positive HER2-negative early breast cancer with tumor involvement in at least 1 axillary lymph node (pALN) and to be enrolled in cohort 1 had to have either:
- ≥4 pALN or
- 1-3 pALN and at least one of:
- –
- tumor grade 3 or
- –
- tumor size ≥50 mm
Patients enrolled in cohort 2 could not have met the eligibility criteria for cohort 1. To be enrolled in cohort 2, patients had to have 1-3 pALN and Ki-67 score ≥20%. Breast tumor samples were tested at central sites using the Ki-67 IHC MIB-1 pharmDx (Dako Omnis) assay to establish if the Ki-67 score was ≥20%.
Patients were randomized to receive 2 years of VERZENIO plus physician's choice of standard endocrine therapy or standard endocrine therapy alone. Randomization to treatment was stratified by prior treatment (neoadjuvant chemotherapy versus adjuvant chemotherapy versus no chemotherapy); menopausal status (premenopausal versus postmenopausal); and region (North America/Europe versus Asia versus other). Men were stratified as postmenopausal. After the end of the study treatment period, standard adjuvant endocrine therapy was continued for a duration of at least 5 years if deemed medically appropriate.
The major efficacy outcome measure was invasive disease–free survival (IDFS). IDFS was defined as the time from randomization to the first occurrence of: ipsilateral invasive breast tumor recurrence, regional invasive breast cancer recurrence, distant recurrence, contralateral invasive breast cancer, second primary non-breast invasive cancer, or death attributable to any cause. Overall survival (OS) was an additional outcome measure.
A statistically significant difference in IDFS was observed in the intent-to-treat (ITT) population which was primarily attributed to the patients treated in cohort 1. While the OS data in cohort 2 remains immature, more deaths were observed among those receiving VERZENIO plus standard endocrine therapy compared to those receiving standard endocrine therapy alone (10/253 vs. 5/264).
Of 5637 patients randomized, 5120 (91%) were randomized in cohort 1. Patient median age was 51 years (range, 22-89 years), 99% were women, 70% were White, 24% were Asian, 1.7% were Black or African American, 2.1% were American Indian or Alaska Native, and 0.1% were Native Hawaiian or Other Pacific Islander. Forty-three percent of patients were premenopausal. Most patients received prior chemotherapy (37% neoadjuvant, 59% adjuvant) and prior radiotherapy (96%). Sixty-five percent of the patients had 4 or more positive lymph nodes with 22% having ≥10 positive lymph nodes, 41% had Grade 3 tumor, and 24% had pathological tumor size ≥50 mm. The majority of patients (99%) had estrogen receptor positive disease and 87% had progesterone receptor positive disease. Initial endocrine therapy received by patients included letrozole (39%), tamoxifen (31%), anastrozole (22%), or exemestane (8%).
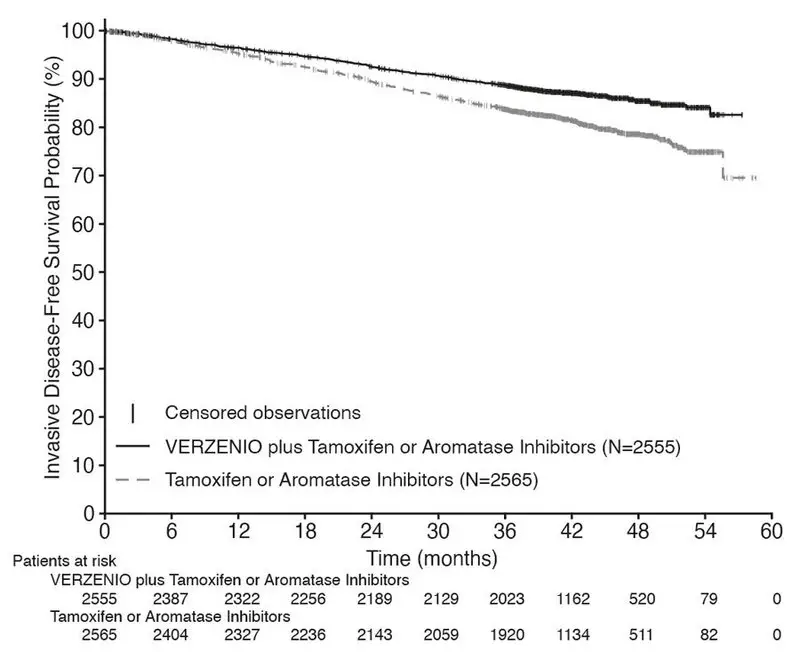
Efficacy results for cohort 1 are summarized in Table 16 and Figure 1. At the time of OS interim analysis 2, OS was immature and a total of 315 (6%) of patients had died in cohort 1.
|
Abbreviation: CI = confidence interval. |
||
| VERZENIO Plus
Tamoxifen or an Aromatase Inhibitor N=2555 | Tamoxifen or an Aromatase Inhibitor
N=2565 |
|
| Invasive Disease–Free Survival (IDFS) | ||
| Number of patients with an event (n, %) | 317 (12.4) | 474 (18.5) |
| Hazard ratio (95% CI) | 0.653 (0.567, 0.753) | |
| IDFS at 48 months (%, 95% CI) | 85.5 (83.8, 87.0) | 78.6 (76.7, 80.4) |
14.2 Advanced or Metastatic Breast Cancer
VERZENIO in Combination with an Aromatase Inhibitor (Anastrozole or Letrozole) (MONARCH 3)
Postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer with no prior systemic therapy in this disease setting
MONARCH 3 (NCT02246621) was a randomized (2:1), double-blinded, placebo-controlled, multicenter study in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer in combination with a nonsteroidal aromatase inhibitor as initial endocrine-based therapy, including patients not previously treated with systemic therapy for breast cancer.
Randomization was stratified by disease site (visceral, bone only, or other) and by prior (neo)adjuvant endocrine therapy (aromatase inhibitor versus other versus no prior endocrine therapy). A total of 493 patients were randomized to receive 150 mg VERZENIO or placebo orally twice daily, plus physician's choice of letrozole (80% of patients) or anastrozole (20% of patients). Patient median age was 63 years (range, 32-88 years) and the majority were White (58%) or Asian (30%). A total of 51% had received prior systemic therapy and 39% of patients had received chemotherapy, 53% had visceral disease, and 22% had bone-only disease.
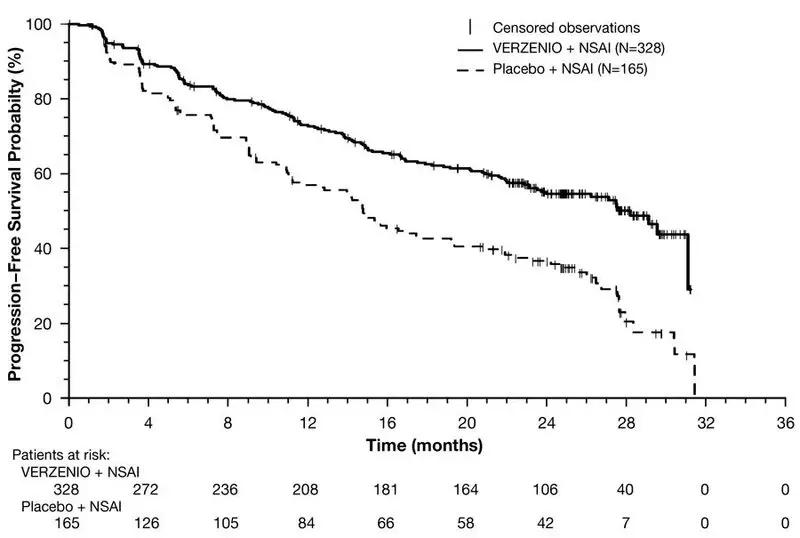
Efficacy results are summarized in Table 17 and Figure 2. PFS was evaluated according to RECIST version 1.1 and PFS assessment based on a blinded independent radiologic review was consistent with the investigator assessment. Consistent results were observed across patient stratification subgroups of disease site and prior (neo)adjuvant endocrine therapy. At the time of the PFS analysis, 19% of patients had died, and OS data were immature.
|
Abbreviations: CI = confidence interval, NR = not reached. |
||
|
a Complete response + partial response. |
||
|
b Based upon confirmed responses. |
||
| VERZENIO plus
Anastrozole or Letrozole | Placebo plus
Anastrozole or Letrozole |
|
| Progression-Free Survival | N=328 | N=165 |
| Number of patients with an event (n, %) | 138 (42.1) | 108 (65.5) |
| Median (months, 95% CI) | 28.2 (23.5, NR) | 14.8 (11.2, 19.2) |
| Hazard ratio (95% CI) | 0.540 (0.418, 0.698) | |
| p-value | <0.0001 | |
| Objective Response for Patients with Measurable Disease | N=267 | N=132 |
| Objective response ratea,b (n, %) | 148 (55.4) | 53 (40.2) |
| 95% CI | 49.5, 61.4 | 31.8, 48.5 |
VERZENIO in Combination with Fulvestrant (MONARCH 2)
Patients with HR-positive, HER2-negative advanced or metastatic breast cancer with disease progression on or after prior adjuvant or metastatic endocrine therapy
MONARCH 2 (NCT02107703) was a randomized, placebo-controlled, multicenter study in women with HR-positive, HER2-negative metastatic breast cancer in combination with fulvestrant in patients with disease progression following endocrine therapy who had not received chemotherapy in the metastatic setting. Randomization was stratified by disease site (visceral, bone only, or other) and by sensitivity to prior endocrine therapy (primary or secondary resistance). Primary endocrine therapy resistance was defined as relapse while on the first 2 years of adjuvant endocrine therapy or progressive disease within the first 6 months of first line endocrine therapy for metastatic breast cancer. A total of 669 patients were randomized to receive VERZENIO or placebo orally twice daily plus intramuscular injection of 500 mg fulvestrant on days 1 and 15 of cycle 1 and then on day 1 of cycle 2 and beyond (28-day cycles). Pre/perimenopausal women were enrolled in the study and received the gonadotropin-releasing hormone agonist goserelin for at least 4 weeks prior to and for the duration of MONARCH 2. Patients remained on continuous treatment until development of progressive disease or unmanageable toxicity.
Patient median age was 60 years (range, 32-91 years), and 37% of patients were older than 65. The majority were White (56%), and 99% of patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Twenty percent (20%) of patients had de novo metastatic disease, 27% had bone-only disease, and 56% had visceral disease. Twenty-five percent (25%) of patients had primary endocrine therapy resistance. Seventeen percent (17%) of patients were pre- or perimenopausal.
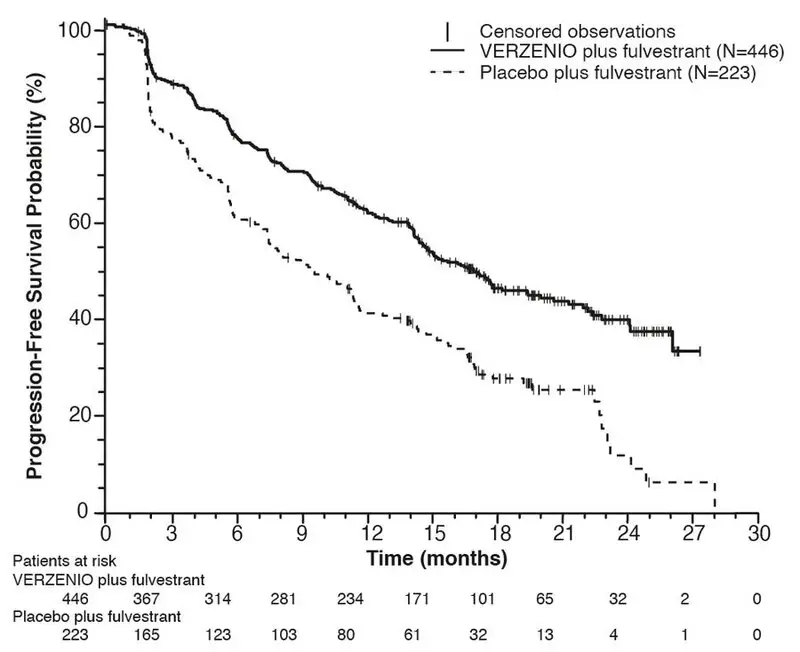
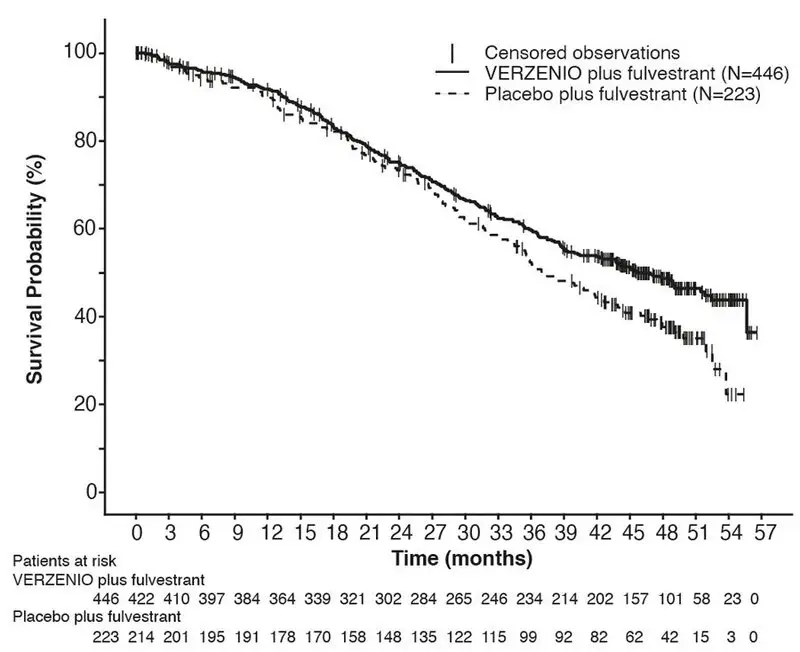
The efficacy results from the MONARCH 2 study are summarized in Table 18, Figure 3, and Figure 4. PFS assessment based on a blinded independent radiologic review was consistent with the investigator assessment. Consistent results were observed across patient stratification subgroups of disease site and endocrine therapy resistance for PFS and OS.
|
Abbreviation: CI = confidence interval, OS = overall survival. |
||
|
a Stratified by disease site (visceral metastases vs. bone-only metastases vs. other) and endocrine therapy resistance (primary resistance vs. secondary resistance) |
||
|
b Data from a pre-specified interim analysis (77% of the number of events needed for the planned final analysis) with the p-value compared with the allocated alpha of 0.021. |
||
|
c Complete response + partial response. |
||
| VERZENIO plus Fulvestrant | Placebo plus Fulvestrant | |
| Progression-Free Survival
(Investigator Assessment) | N=446 | N=223 |
| Number of patients with an event (n, %) | 222 (49.8) | 157 (70.4) |
| Median (months, 95% CI) | 16.4 (14.4, 19.3) | 9.3 (7.4, 12.7) |
| Hazard ratio (95% CI)a | 0.553 (0.449, 0.681) | |
| p-valuea | p<.0001 | |
| Overall Survivalb | ||
| Number of deaths (n, %) | 211 (47.3) | 127 (57.0) |
| Median OS in months (95% CI) | 46.7 (39.2, 52.2) | 37.3 (34.4, 43.2) |
| Hazard ratio (95% CI)a | 0.757 (0.606, 0.945) | |
| p-valuea | p=.0137 | |
| Objective Response for Patients with Measurable Disease | N=318 | N=164 |
| Objective response ratec (n, %) | 153 (48.1) | 35 (21.3) |
| 95% CI | 42.6, 53.6 | 15.1, 27.6 |
VERZENIO Administered as a Monotherapy in Metastatic Breast Cancer (MONARCH 1)
Patients with HR-positive, HER2-negative breast cancer who received prior endocrine therapy and 1-2 chemotherapy regimens in the metastatic setting
MONARCH 1 (NCT02102490) was a single-arm, open-label, multicenter study in women with measurable HR-positive, HER2-negative metastatic breast cancer whose disease progressed during or after endocrine therapy, had received a taxane in any setting, and who received 1 or 2 prior chemotherapy regimens in the metastatic setting. A total of 132 patients received 200 mg VERZENIO orally twice daily on a continuous schedule until development of progressive disease or unmanageable toxicity.
Patient median age was 58 years (range, 36-89 years), and the majority of patients were White (85%). Patients had an Eastern Cooperative Oncology Group performance status of 0 (55% of patients) or 1 (45%). The median duration of metastatic disease was 27.6 months. Ninety percent (90%) of patients had visceral metastases, and 51% of patients had 3 or more sites of metastatic disease. Fifty-one percent (51%) of patients had had one line of chemotherapy in the metastatic setting. Sixty-nine percent (69%) of patients had received a taxane-based regimen in the metastatic setting and 55% had received capecitabine in the metastatic setting. Table 19 provides the efficacy results from MONARCH 1.
|
Abbreviations: CI = confidence interval, NR = not reached. |
||
|
a All responses were partial responses. |
||
|
b Based upon confirmed responses. |
||
| VERZENIO 200 mg
N=132 |
||
| Investigator Assessed | Independent Review | |
| Objective Response Ratea,b, n (%) | 26 (19.7) | 23 (17.4) |
| 95% CI (%) | 13.3, 27.5 | 11.4, 25.0 |
| Median Duration of Response | 8.6 months | 7.2 months |
| 95% CI (%) | 5.8, 10.2 | 5.6, NR |
16. How is Verzenio supplied
How Supplied
VERZENIO 50 mg tablets are oval beige tablet with “Lilly” debossed on one side and “50” on the other side.
VERZENIO 100 mg tablet are oval white to practically white tablet with “Lilly” debossed on one side and “100” on the other side.
VERZENIO 150 mg tablets are oval yellow tablet with “Lilly” debossed on one side and “150” on the other side.
VERZENIO 200 mg tablets are oval beige tablet with “Lilly” debossed on one side and “200” on the other side.
VERZENIO tablets are supplied in 7-day dose pack configurations as follows:
|
| NDC 0002-6216-54 |
|
| NDC 0002-5337-54 |
|
| NDC 0002-4815-54 |
|
| NDC 0002-4483-54 |
17. Patient Counseling Information
Advise patients to read the FDA-approved Patient Information.
Diarrhea
VERZENIO may cause diarrhea, which may be severe in some cases [see Warnings and Precautions (5.1)].
- Early identification and intervention is critical for the optimal management of diarrhea. Instruct patients that at the first sign of loose stools, they should start antidiarrheal therapy (for example, loperamide) and notify their healthcare provider for further instructions and appropriate follow up.
- Encourage patients to increase oral fluids.
- If diarrhea does not resolve with antidiarrheal therapy within 24 hours to ≤Grade 1, suspend VERZENIO dosing [see Dosage and Administration (2.2)].
Neutropenia
Advise patients of the possibility of developing neutropenia and to immediately contact their healthcare provider should they develop a fever, particularly in association with any signs of infection [see Warnings and Precautions (5.2)].
Interstitial Lung Disease/Pneumonitis
Advise patients to immediately report new or worsening respiratory symptoms [see Warnings and Precautions (5.3)].
Hepatotoxicity
Inform patients of the signs and symptoms of hepatotoxicity. Advise patients to contact their healthcare provider immediately for signs or symptoms of hepatotoxicity [see Warnings and Precautions (5.4)].
Venous Thromboembolism
Advise patients to immediately report any signs or symptoms of thromboembolism such as pain or swelling in an extremity, shortness of breath, chest pain, tachypnea, and tachycardia [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during VERZENIO treatment and for 3 weeks after the last dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise lactating women not to breastfeed during VERZENIO treatment and for at least 3 weeks after the last dose [see Use in Specific Populations (8.2)].
Infertility
Inform males of reproductive potential that VERZENIO may impair fertility [see Use in Specific Populations (8.3)].
Drug Interactions
- Inform patients to avoid concomitant use of ketoconazole. Dose reduction may be required for other strong CYP3A inhibitors or for moderate CYP3A inhibitors [see Dosage and Administration (2.2) and Drug Interactions (7)].
- Grapefruit may interact with VERZENIO. Advise patients not to consume grapefruit products while on treatment with VERZENIO.
- Advise patients to avoid concomitant use of strong and moderate CYP3A inducers and to consider alternative agents [see Dosage and Administration (2.2) and Drug Interactions (7)].
- Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Dosage and Administration (2.2) and Drug Interactions (7)].
Dosing
- Instruct patients to take the doses of VERZENIO at approximately the same times every day and to swallow whole (do not chew, crush, or split them prior to swallowing) [see Dosage and Administration (2.1)].
- If patient vomits or misses a dose, advise the patient to take the next prescribed dose at the usual time [see Dosage and Administration (2.1)].
- Advise the patient that VERZENIO may be taken with or without food [see Dosage and Administration 2.1)].
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
VER-0008-USPI-20230303
|
This Patient Information has been approved by the U.S. Food and Drug Administration. |
Revised: March-2023 |
|
| PATIENT INFORMATION
VERZENIO® (ver-ZEN-ee-oh) (abemaciclib) tablets |
||
|
||
|
|
|
|
||
|
|
|
| See “What are the possible side effects of VERZENIO?” for more information about side effects. | ||
| What is VERZENIO?
VERZENIO is a prescription medicine used:
|
||
| When VERZENIO is used in combination with fulvestrant, tamoxifen, or an aromatase inhibitor, also read the Patient Information for the prescribed product. Ask your healthcare provider if you are not sure. It is not known if VERZENIO is safe and effective in children. |
||
Before taking VERZENIO, tell your healthcare provider about all of your medical conditions, including if you:
|
||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. VERZENIO may affect the way other medicines work, and other medicines may affect how VERZENIO works, causing serious side effects. Especially tell your healthcare provider if you take a medicine that contains ketoconazole. Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine. |
||
How should I take VERZENIO?
|
||
What should I avoid during treatment with VERZENIO?
|
||
|
What are the possible side effects of VERZENIO?
|
||
| The most common side effects of VERZENIO include: | ||
|
|
|
| VERZENIO may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if this is a concern for you. These are not all the possible side effects of VERZENIO. For more information, ask your healthcare provider or pharmacist. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store VERZENIO?
|
||
| Keep VERZENIO and all medicines out of the reach of children. | ||
| General information about the safe and effective use of VERZENIO.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VERZENIO for a condition for which it was not prescribed. Do not give VERZENIO to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for more information about VERZENIO that is written for health professionals. |
||
| What are the ingredients in VERZENIO?
Active ingredient: abemaciclib Inactive ingredients: microcrystalline cellulose 102, microcrystalline cellulose 101, lactose monohydrate, croscarmellose sodium, sodium stearyl fumarate, silicon dioxide. Color mixture ingredients: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, iron oxide yellow, iron oxide red. Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA Copyright © 2017, 2023, Eli Lilly and Company. All rights reserved. VER-0005-PPI-20230303 For more information, go to www.verzenio.com or call 1-800-545-5979. |
||
PACKAGE CARTON – VERZENIO 50 mg 14ct
14 tablets
NDC 0002-4483-54
Rx only
Verzenio®
(abemaciclib) tablets
50 mg dose
7-Day Supply
This 50 mg Dose Pack contains fourteen 50 mg tablets.
www.verzenio.com
LIFT TO OPEN
Lilly

PACKAGE CARTON – VERZENIO 100 mg 14ct
14 tablets
NDC 0002-4815-54
Rx only
Verzenio®
(abemaciclib) tablets
100 mg dose
7-Day Supply
This 100 mg Dose Pack contains fourteen 100 mg tablets.
www.verzenio.com
LIFT TO OPEN
Lilly


| VERZENIO
abemaciclib tablet |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| VERZENIO
abemaciclib tablet |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| VERZENIO
abemaciclib tablet |
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
| VERZENIO
abemaciclib tablet |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Labeler - Eli Lilly and Company (006421325) |
