Drug Detail:Vonvendi (Von willebrand factor (recombinant) [ von-wil-e-brand-fak-tor ])
Drug Class: Miscellaneous coagulation modifiers
Highlights of Prescribing Information
VONVENDI [von Willebrand factor (recombinant)] lyophilized powder for solution, for intravenous injection
Initial U.S. Approval: 2015
Indications and Usage for Vonvendi
VONVENDI [von Willebrand factor (recombinant)] is a recombinant von Willebrand factor (rVWF) indicated for use in adults (age 18 and older) diagnosed with von Willebrand disease (VWD) for:
- On-demand treatment and control of bleeding episodes. (1)
- Perioperative management of bleeding. (1)
- Routine prophylaxis to reduce the frequency of bleeding episodes in patients with severe Type 3 von Willebrand disease receiving on-demand therapy. (1)
Vonvendi Dosage and Administration
For intravenous use after reconstitution only.
On-Demand Treatment and Control of Bleeding Episodes
- For each bleeding episode, administer the first dose of VONVENDI with an approved recombinant (non-von Willebrand factor containing) factor VIII, if factor VIII baseline levels are below 40% or are unknown. (2.1)
- Initial dose is 40 to 80 International Units (IU) per kg body weight (BW). Adjust the dosage based on the extent and location of bleeding. (2.1)
| Bleeding Episode | Initial Dose | Subsequent Dose |
|---|---|---|
| Minor | 40 to 50 IU/kg | 40 to 50 IU/kg every 8 to 24 hours |
| Major | 50 to 80 IU/kg | 40 to 60 IU/kg every 8 to 24 hours for approximately 2 to 3 days |
Perioperative Management of Bleeding
For Elective Surgical Procedure
- A dose of VONVENDI may be given 12 to 24 hours prior to surgery to allow the endogenous factor VIII levels to increase to at least 30 IU/dL (minor surgery) or 60 IU/dL (major surgery). (2.1)
- Assess FVIII:C levels within 3 hours prior to surgery. If the FVIII:C levels are at or above the recommended minimum target levels, administer a dose of VONVENDI alone within 1 hour prior to the procedure. If the FVIII:C levels are below the recommended minimum target levels, administer recombinant factor VIII in addition to VONVENDI to raise VWF:RCo and FVIII:C. (2.1)
For Emergency Surgery
- Assess baseline VWF:RCo and FVIII:C levels within 3 hours prior to surgery. If not available, use weight-based dosing calculation. (2.1)
- Administer VONVENDI 1 hour before surgery with or without recombinant factor VIII and adjust the dose to raise VWF:RCo and FVIII:C to adequate level. (2.1)
| Type of Surgery | Target Peak Plasma Level | Calculation of rVWF Dose (IU VWF:RCo required) |
|
|---|---|---|---|
| VWF:RCo | FVIII:C | ||
| Minor | 50 to 60 IU/dL | 40 to 50 IU/dL | ∆ VWF:RCo × BW (kg)/IR |
| Major | 100 IU/dL | 80 to 100 IU/dL | ∆ VWF:RCo × BW (kg)/IR |
- Continue to monitor the VWF:RCo and FVIII:C plasma levels after surgical procedure. (2.1)
Routine Prophylaxis to Reduce the Frequency of Bleeding Episodes in Patients with Severe Type 3 VWD Receiving On-Demand Therapy
Initial dosage of VONVENDI is 40 to 60 IU/kg body weight to be administered twice weekly. The dose may be adjusted up to 60 IU/kg twice weekly based on frequency of bleeding episodes. (2.1)
Dosage Forms and Strengths
VONVENDI is available as a lyophilized powder in single-dose vials containing nominally 650 or 1300 international units VWF:RCo. (3)
Contraindications
Do not use in patients who have had life-threatening hypersensitivity reactions to VONVENDI or its components (tri-sodium citrate-dihydrate, glycine, mannitol, trehalose-dihydrate polysorbate 80, and hamster or mouse proteins). (4)
Warnings and Precautions
- Thromboembolic reactions can occur, particularly in patients with risk factors for thrombosis. Monitor for early signs of thrombosis and have prophylaxis measures against thromboembolism instituted according to current recommendations. One out of 100 VWD subjects treated with VONVENDI in clinical trials developed proximal deep vein thrombosis in perioperative period after undergoing total hip replacement surgery. In patients requiring frequent doses of VONVENDI in combination with recombinant factor VIII, monitor plasma levels for FVIII:C because sustained excessive factor VIII plasma levels can increase the risk for thromboembolic events. (5.1)
- Hypersensitivity reactions, including anaphylaxis, may occur. Discontinue VONVENDI if hypersensitivity symptoms occur and administer appropriate emergency treatment. (5.2)
- Inhibitors to von Willebrand factor (VWF) and/or factor VIII can occur. If the expected plasma levels of VWF activity (VWF:RCo) are not attained, or if bleeding is not controlled with an appropriate dose, perform an appropriate assay to determine if an anti-VWF or anti-factor VIII inhibitors are present. (5.3)
Adverse Reactions/Side Effects
- The most common adverse reactions observed (≥2% of subjects) were headache, vomiting, nausea, dizziness, arthralgia, joint injury, vertigo, ALT increased and generalized pruritus. (6.1)
- One subject treated with VONVENDI in perioperative setting developed deep vein thrombosis after undergoing total hip replacement surgery. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals U.S.A., Inc. at 1-877-TAKEDA-7 (1-877-825-3327) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2023
Full Prescribing Information
1. Indications and Usage for Vonvendi
VONVENDI [von Willebrand factor (recombinant)] is a recombinant von Willebrand factor (rVWF) indicated for use in adults (age 18 and older) diagnosed with von Willebrand disease (VWD) for:
- On-demand treatment and control of bleeding episodes.
- Perioperative management of bleeding.
- Routine Prophylaxis to reduce the frequency of bleeding episodes in patients with severe Type 3 VWD receiving on-demand therapy.
2. Vonvendi Dosage and Administration
2.1 Recommended Dosage
For intravenous use after reconstitution only.
- Each vial of VONVENDI is labeled with the actual amount of rVWF activity in International Units (IU), as measured with the Ristocetin cofactor assay (VWF:RCo).
- Individualize dosage and frequency according to clinical judgement and based on the subject's weight, type and severity of the bleeding episodes/surgical intervention and based on monitoring of appropriate clinical and laboratory measures.
- For patients experiencing bleeding, hemostasis cannot be ensured until factor VIII coagulation activity (FVIII:C) has reached 40 IU/deciliter (dL) (i.e., 40% of normal activity). If the patient's baseline plasma FVIII:C level is below 40%, or is unknown, administer an approved recombinant (non-von Willebrand factor containing) factor VIII (rFVIII) with the first infusion of VONVENDI in order to achieve a hemostatic plasma level of FVIII:C. However, if an immediate rise in FVIII:C is not necessary or if the baseline FVIII:C level is sufficient to ensure hemostasis, administer VONVENDI without rFVIII.
- Depending on the patient's baseline FVIII:C level, a single infusion of VONVENDI is expected, in a majority of patients, to lead to an increase in endogenous FVIII:C activity above 40% within 6 hours.
- When repeated infusions are required, monitor factor VIII levels to determine if rFVIII is required with subsequent infusions.
On-Demand Treatment and Control of Bleeding Episodes
Administer initial dose of 40 to 80 IU/kg body weight (BW). Dosing guidelines for treatment of minor and major bleeds are provided in Table 1.
Administer VONVENDI within the designated ranges based on clinical judgment, taking into account severity, site of bleeding, and medical history of the patient. Adjust the dose based on the extent and location of the bleeding episode. Administer subsequent doses as long as clinically required. Monitor appropriate clinical and laboratory measures [see Warnings and Precautions (5.2, 5.3)].
| Bleeding Episodes | Initial Dose* | Subsequent Dose (as clinically required) |
|---|---|---|
|
||
| Minor (e.g., readily managed epistaxis, oral bleeding, menorrhagia) | 40 to 50 IU/kg | 40 to 50 IU/kg every 8 to 24 hours |
| Major† (e.g., severe or refractory epistaxis, menorrhagia, GI bleeding, CNS trauma, hemarthrosis, or traumatic hemorrhage) | 50 to 80 IU/kg | 40 to 60 IU/kg every 8 to 24 hours for approximately 2 to 3 days |
The initial dose of VONVENDI should achieve greater than 60% of von Willebrand factor (VWF) levels (based on VWF:RCo greater than 60 IU/dL) and an infusion of rFVIII should achieve factor VIII levels greater than 40% (FVIII:C greater than 40 IU/dL). In major bleeding episodes, maintain trough levels of VWF:RCo greater than 50% for as long as deemed necessary.
Administer VONVENDI with rFVIII if the FVIII:C level is less than 40%, or is unknown, to control bleeding. The rFVIII dose should be calculated according to the difference between the patient's baseline plasma FVIII:C level, and the desired peak FVIII:C level to achieve an appropriate plasma FVIII:C level based on the approximate mean recovery of 2 (IU/dL)/(IU/kg). Administer the complete dose of VONVENDI followed by rFVIII within 10 minutes.
Perioperative Management of Bleeding
Emergency Surgery
A 12 to 24 hour preoperative dose may not be feasible in subjects requiring emergency surgery. Baseline VWF:RCo and FVIII:C levels should be assessed within 3 hours prior to initiating the surgical procedure if it is feasible. The loading dose (1 hour preoperative dose) can be calculated as the difference in the target peak and baseline plasma VWF:RCo levels divided by the IR. If the IR is not available, assume an IR of 2.0 IU/dL per IU/kg.
If baseline VWF:RCo and FVIII:C is not available, as a general guidance a loading dose (1 hour preoperative dose) of VONVENDI, 40 to 60 IU/kg VWF:RCo, should be administered. Additionally, rFVIII at a dose of 30 to 45 IU/kg may be infused sequentially, preferably within 10 minutes after the VONVENDI infusion in patients whose factor VIII plasma levels already are (or are highly likely to be) less than 40 to 50 IU/dL for minor surgery or 80 to 100 IU/dL for major surgery.
Refer to Table 2 for recommended VWF:RCo and FVIII:C target peak plasma levels and dosing guidelines for perioperative management of bleeding.
| Type of Surgery | VWF:RCo Target Peak Plasma Level | FVIII:C Target Peak Plasma Level* | Calculation of rVWF Dose (to be administered within 1 hour prior to surgery) (IU VWF:RCo required) |
|---|---|---|---|
|
|||
| Minor | 50 to 60 IU/dL | 40 to 50 IU/dL | ∆† VWF:RCo × BW (kg) /IR‡ |
| Major | 100 IU/dL | 80 to 100 IU/dL | ∆† VWF:RCo × BW (kg) /IR‡ |
In the absence of available baseline FVIII:C, VWF:RCo and Incremental Recovery, it is recommended to use body weight-based dosing as outlined below in Table 3.
| Type of Surgery | VWF:RCo (IU VWF:RCo/kg BW) | VWF:RCo Target Peak Plasma Level | FVIII:C (IU FVIII:C/kg BW) | FVIII:C Target Peak Plasma Level |
|---|---|---|---|---|
| Minor | 25 to 30 IU/kg | 50 to 60 IU/dL | 20 to 25 IU/kg | 40 to 50 IU/dL |
| Major | 50 ± 10 IU/kg | 100 IU/dL | 40 to 50 IU/kg | 80 to 100 IU/dL |
Monitor VWF:RCo and FVIII:C plasma levels starting 12 to 24 hours after surgery and at least every 24 hours in the perioperative period to adjust the dosing of VONVENDI or rFVIII levels. VWF:RCo and FVIII:C plasma levels should be monitored and the intra- and postoperative maintenance regimen should be individualized according to the pharmacokinetic (PK) results and intensity and duration of the hemostatic challenge. The frequency of VONVENDI dosing should range between twice a day and every 48 hours.
Refer to Table 4 for recommended VWF:RCo and FVIII:C target trough plasma levels and minimum duration of treatment for subsequent maintenance doses after surgery.
| Type of Surgery | VWF:RCo Target Trough Plasma Level | FVIII:C Target Trough Plasma Level | Minimum Duration of Treatment | Frequency of Dosing | ||
|---|---|---|---|---|---|---|
| up to 72 Hours Post-Surgery | after 72 Hours Post-Surgery | up to 72 Hours Post-Surgery | after 72 Hours Post-Surgery | |||
| Minor | ≥30 IU/dL | - | >30 IU/dL | - | 48 hours | Every 12 to 24 hours to every other day |
| Major | >50 IU/dL | >30 IU/dL | >50 IU/dL | >30 IU/dL | 72 hours | |
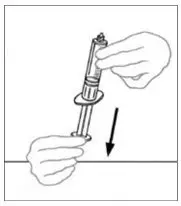
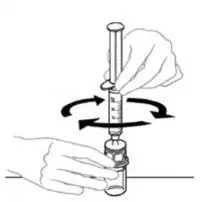
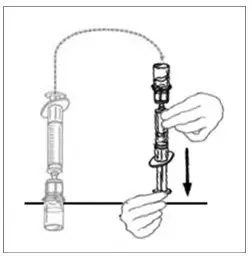
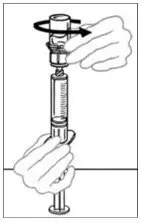
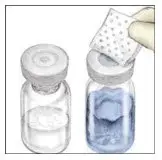
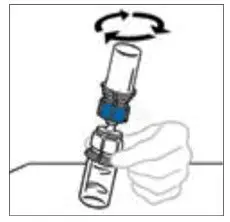
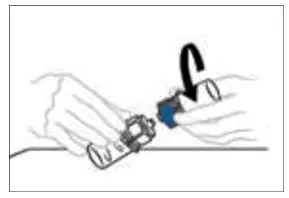
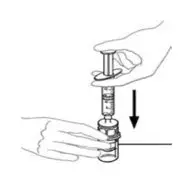
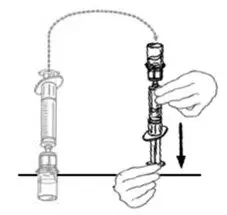
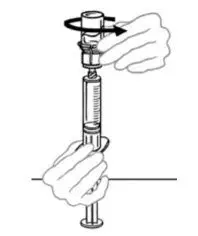
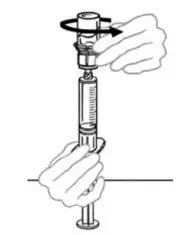
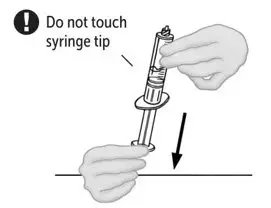
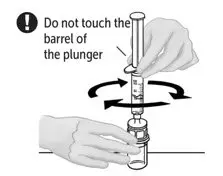
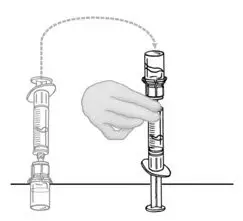
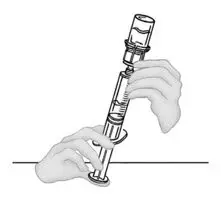
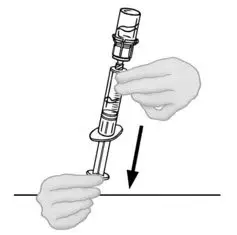
2.2 Preparation and Reconstitution
- Allow VONVENDI and Sterile Water for Injection (diluent) to reach room temperature.
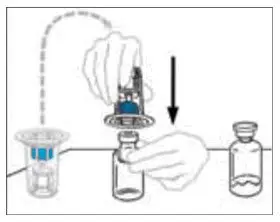
- If the patient requires more than one vial of VONVENDI per injection, reconstitute each vial according to the following instructions.
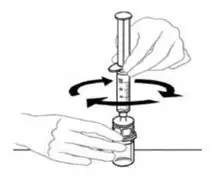
- Some flakes or particles may remain in the reconstituted vial. The filter included in the Mix2Vial device will remove extraneous flakes or particles, and the resulting solution in the syringe should be clear and colorless. Do not use the solution in the syringe if it is cloudy or contains flakes or particles after filtration from the vial into the syringe.
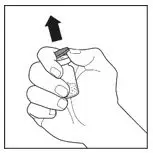

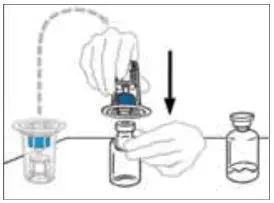
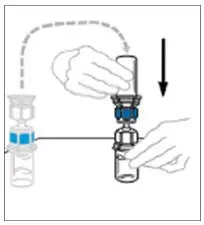
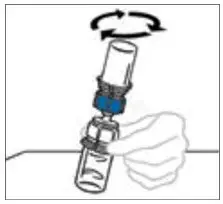
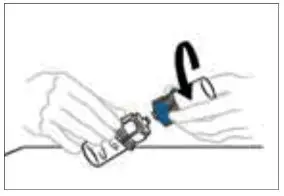
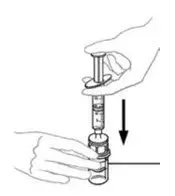
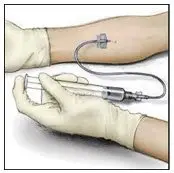
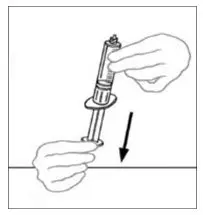
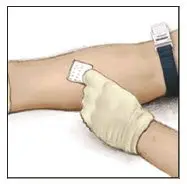
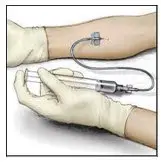
2.3 Administration
For intravenous administration only.
- Administer VONVENDI immediately after reconstitution. If not, store at room temperature not to exceed 25°C (77°F) for up to 3 hours. Discard after 3 hours.
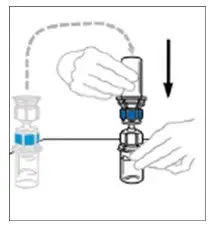
- No more than two vials of VONVENDI may be pooled into a single syringe. Pooling of more than two vials into a syringe may result in formation of filaments, which requires discarding of the solution in the syringe. If a patient is to receive more than one vial of VONVENDI, leave syringe attached to the vial or cover syringe tip with a suitable sterile cap until ready to infuse to reduce risk of contamination.
- Use plastic syringes with this product because proteins in the product tend to stick to the surface of glass syringes.
- Do not mix VONVENDI with other medicinal products.
3. Dosage Forms and Strengths
VONVENDI is available as a non-pyrogenic, white to off-white, lyophilized powder for reconstitution in single-dose vials containing nominally 650 or 1300 IU VWF:RCo/vial.
Each VONVENDI vial is labeled with the number of units of VWF:RCo expressed in IU, which are based on the current World Health Organization (WHO) standard for VWF concentrate.
4. Contraindications
VONVENDI is contraindicated in patients who have had life-threatening hypersensitivity reactions to VONVENDI or constituents of the product (tri-sodium citrate-dihydrate, glycine, mannitol, trehalose-dihydrate, polysorbate 80, and hamster or mouse proteins) [see Description (11)].
5. Warnings and Precautions
5.1 Embolism and Thrombosis
Thromboembolic reactions, including disseminated intravascular coagulation (DIC), venous thrombosis, pulmonary embolism, myocardial infarction, and stroke, can occur, particularly in patients with known risk factors for thrombosis, including low ADAMTS13 levels [see Description (11)]. Monitor for early signs and symptoms of thrombosis such as pain, swelling, discoloration, dyspnea, cough, hemoptysis, and syncope, and have prophylaxis measures against thromboembolism instituted according to current recommendations and standard of care.
In patients requiring frequent doses of VONVENDI in combination with rFVIII, monitor plasma levels for FVIII:C activity because sustained excessive factor VIII plasma levels can increase the risk of thromboembolic complications.
Of the 100 subjects treated with VONVENDI in clinical trials, one subject was diagnosed with deep vein thrombosis, which was revealed by imaging conducted as a part of the hospital's standard of care for high-risk patients, 3 days after total hip replacement surgery while receiving VONVENDI.
5.2 Hypersensitivity Reactions
Hypersensitivity reactions have occurred with VONVENDI. These reactions can include anaphylactic shock, generalized urticaria, angioedema, chest tightness, hypotension, shock, lethargy, nausea, vomiting, paresthesia, pruritus, restlessness, blurred vision, wheezing and/or acute respiratory distress. If signs and symptoms of severe allergic reactions occur, immediately discontinue administration of VONVENDI and provide appropriate supportive care.
VONVENDI contains trace amounts of mouse immunoglobulin G (MuIgG) and hamster proteins less than or equal to 2 ng/IU VONVENDI. Patients treated with this product may develop hypersensitivity reactions to non-human mammalian proteins.
5.3 Neutralizing Antibodies
Neutralizing antibodies (inhibitors) to VWF and/or factor VIII can occur. If the expected plasma levels of VWF activity (VWF:RCo) are not attained, perform an appropriate assay to determine if anti-VWF or anti-factor VIII inhibitors are present. Consider other therapeutic options and direct the patient to a physician with experience in the care of either VWD or hemophilia A.
In patients with high levels of inhibitors to VWF or factor VIII, VONVENDI therapy may not be effective and infusion of this protein may lead to severe hypersensitivity reactions. Since inhibitor antibodies can occur concomitantly with anaphylactic reactions, evaluate patients experiencing an anaphylactic reaction for the presence of inhibitors.
5.4 Monitoring Laboratory Tests
Monitor plasma levels of VWF:RCo and factor VIII activities in patients receiving VONVENDI to avoid sustained excessive VWF and/or factor VIII activity levels, which may increase the risk of thrombotic events, particularly in patients with known clinical or laboratory risk factors.
Monitor for development of VWF and/or factor VIII inhibitors when suspected. Perform appropriate inhibitor assays to determine if VWF and/or factor VIII inhibitors are present if bleeding is not controlled with the expected dose of VONVENDI.
6. Adverse Reactions/Side Effects
The most common adverse reactions observed in greater than or equal to 2% of subjects in clinical trials with VONVENDI (n=100) were headache, vomiting, nausea, dizziness, arthralgia, joint injury, vertigo, ALT increased and generalized pruritus.
One subject treated with VONVENDI in the surgery study for perioperative management of bleeding developed proximal deep vein thrombosis postoperatively [see Warning and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety profile of VONVENDI was evaluated in five prospective, multicenter trials; four were conducted in subjects with VWD (n=100) and one was conducted in subjects with hemophilia A (n=12). The adverse drug reaction (ADR) terms listed below (Table 5) were assessed by the sponsor/company as having a plausible causal relationship to the study medication in three clinical trials (n=80) during which VONVENDI was used for PK assessment, on-demand treatment and perioperative management of bleeding episodes in patients undergoing surgery.
| System Organ Class (SOC) | Adverse Reaction | Number of Subjects (%)†
(n=80) |
|---|---|---|
|
||
| Cardiac Disorders | Tachycardia | 1 (1.25%) |
| Gastrointestinal Disorders | Vomiting | 3 (3.75%) |
| Nausea | 3 (3.75%) | |
| General Disorders and Administration Site Conditions | Infusion site paresthesia | 1 (1.25%) |
| Chest discomfort | 1 (1.25%) | |
| Skin and Subcutaneous Tissues Disorders | Generalized pruritus | 2 (2.50%) |
| Vascular Disorders | Hot flush | 1 (1.25%) |
| Hypertension | 1 (1.25%) | |
| Deep vein thrombosis | 1 (1.25%) | |
| Nervous System Disorders | Dizziness | 3 (3.75%) |
| Vertigo | 2 (2.50%) | |
| Dysgeusia | 1 (1.25%) | |
| Tremor | 1 (1.25%) | |
| Investigations | Heart rate increase | 1 (1.25%) |
| Electrocardiogram T wave inversions | 1 (1.25%) | |
Single patient treated with Vonvendi in a clinical trial developed infusion-related reaction. This patient was previously exposed to Vonvendi without any symptoms. He developed chest discomfort and increased heart rate 3 minutes after the infusion was started. The patient was treated with supportive care and symptoms were resolved in 3 hours.
In the completed prophylaxis clinical study, twenty-two adult subjects of age 18 years and older receiving on-demand therapy or prophylactic therapy with plasma derived VWF prior to study entry received prophylactic treatment with VONVENDI during the study. The adverse drug reaction (ADR) terms are listed below (Table 6).
| System Organ Class (SOC) | Adverse Reaction | Total (N=22) n (%)† |
|---|---|---|
| N=Total number of subjects in the Safety Analysis Set within each column. n=Number of subjects who had at least one event in the category. |
||
|
||
| Nervous System Disorders | Headache‡ | 4 (18.2%) |
| Musculoskeletal and Connective Tissue Disorders | Arthralgia | 3 (13.6%) |
| Investigations | ALT increased | 2 (9.1%) |
| AST increased | 1 (4.5%) | |
| Cardiac Disorders | Supraventricular tachycardia | 1 (4.5%) |
| Ventricular extrasystoles | 1 (4.5%) | |
| Gastrointestinal Disorders | Diarrhea | 1 (4.5%) |
| Skin and Subcutaneous Tissue Disorders | Purpura | 1 (4.5%) |
| Rash pruritic | 1 (4.5%) | |
| General Disorders and Administration Site Conditions | Injection site irritation | 1 (4.5%) |
6.2 Postmarketing Experience
The following adverse drug reactions (ADR) have been identified during post-approval use of VONVENDI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Postmarketing ADRs reported in association with VONVENDI treatment include infusion-related reactions (IRR) which may be clinically manifested by symptoms such as tachycardia, flushing, rash, dyspnea, and blurred vision. In the postmarketing cases reported, the symptoms resolved, and the patients fully recovered within 4 hours after stopping the infusion.
Anaphylactic reaction has been reported with use of Vonvendi in the postmarketing setting.
6.3 Immunogenicity
The immunogenicity of VONVENDI was assessed in clinical trials by assessing the development of neutralizing antibodies against VWF and FVIII, as well as binding antibodies against VWF, Furin, Chinese hamster ovary (CHO) protein and mouse IgG. Of the 100 subjects who received VONVENDI in the clinical trials, one subject who was treated with VONVENDI perioperatively developed treatment-emergent binding antibodies against VWF following a surgery, for whom no adverse events or lack of hemostatic efficacy was reported. No binding antibodies against potential impurities such as rFurin, CHO-protein or mouse IgG developed after treatment with VONVENDI.
Two subjects included in the study had pre-existing high-titer specific binding antibodies against VWF. The high-titer binding anti-VWF antibodies were associated with a significantly decreased VWF:Ag activity post-infusion of either plasma derived VWF (pdVWF) or rVWF and consequently, the decreased activity of VWF:RCo, VWF:CB and FVIII:C. This finding indicates the potential clinical significance of pre-existing binding (non-neutralizing) antibodies: VWD patients previously treated with pdVWF concentrates may be at risk to express a pre-existing binding antibody against VWF prior to first exposure to rVWF which could potentially result in a decreased hemostatic response to rVWF. Such patients could be managed clinically by administration of higher doses of rVWF based on the PK data for each individual patient.
8. Use In Specific Populations
8.2 Lactation
Risk Summary
There is no information regarding the presence of VONVENDI in human milk, the effects on the breastfed infant, or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VONVENDI and any potential adverse effects on the breastfed infant from VONVENDI or from the underlying maternal condition.
11. Vonvendi Description
VONVENDI is a purified rVWF expressed in Chinese Hamster Ovary (CHO) cells. VONVENDI is produced and formulated without the addition of any exogenous raw materials of human or animal origin in the cell culture, purification, or formulation of the final product. Proteins present in the final container product other than rVWF are trace quantities of mouse immunoglobulin (IgG, from the immunoaffinity purification), host cell (i.e., CHO) protein, rFurin (used to further process rVWF), and recombinant factor VIII (rFVIII).
Von Willebrand factor is a large multimeric glycoprotein that is normally found in plasma, and stored as ultra-large multimers in alpha-granules of platelets and intracellular organelles known as Weibel-Palade bodies, prior to secretion into the blood.1 Once the VWF is released to the blood stream and in contact with ADAMTS13 (a proteolytic enzyme in blood), it is cleaved to smaller sizes that can be detected with SDS agarose gels as multimer bands, representing the various species of VWF within the circulation. VONVENDI is rVWF that contains ultra-large multimers in addition to all of the multimers found in plasma as it is not exposed to proteolysis by ADAMTS13 during the manufacturing process.
VONVENDI is formulated as a sterile, non-pyrogenic, white to off-white friable powder for intravenous injection after reconstitution. VONVENDI in a single-dose vial contains nominally 650 or 1300 IU VWF:RCo.
The product contains no preservative. When reconstituted with the provided Sterile Water for Injection the final solution contains the following stabilizers and excipients (Table 7) in targeted amounts:
| Stabilizer and Excipient | Targeted Concentration for Nominal Strengths (650, 1300 IU) |
|---|---|
| Tri-Sodium Citrate-dihydrate | 15 mM |
| Glycine | 15 mM |
| Mannitol | 20 g/L |
| Trehalose-dihydrate | 10 g/L |
| Polysorbate 80 | 0.1 g/L |
Each vial of VONVENDI is labeled with the specific number of units of VWF:RCo expressed in IU, which are based on the current World Health Organization (WHO) standard for VWF concentrate. After reconstitution of the lyophilized powder and filtration/withdrawal into a syringe, all dosage strengths yield a clear, colorless solution, free from particles.
12. Vonvendi - Clinical Pharmacology
12.1 Mechanism of Action
In VWD patients, VONVENDI acts 1) to promote hemostasis by mediating platelet adhesion to damaged vascular sub-endothelial matrix (e.g., collagen) and platelet aggregation, and 2) as a carrier protein for factor VIII, protecting it from rapid proteolysis. The adhesive activity of VWF depends on the size of its multimers, with larger multimers being the most effective in supporting interactions with collagen and platelet receptors.1 The binding capacity and affinity of VONVENDI to factor VIII in plasma is comparable to that of endogenous VWF, allowing for VONVENDI to reduce factor VIII clearance.2
12.2 Pharmacodynamics
The PD of VWF following prophylactic treatment with VONVENDI were investigated in a clinical trial. In Prior OD adult subjects with Type 3 VWD, a single infusion of VONVENDI led to an increase of FVIII:C with peak levels observed approximately 24 hours post-infusion (mean [SD] dose: 50.2 [3.33] IU/kg). After 1-year repeat infusions of 50 ± 10 IU/kg VONVENDI twice weekly, the median (range) pre-dose FVIII:C increased from 2.0 (2.0 to 4.0) IU/dL (baseline after wash-out, N=10) to 10.5 (6.0 to 31.0) IU/dL (Month 12, N=8). The pre-dose FVIII:C (median [range] ≥10.5 [1.0 to 148] IU/dL) were observed over the prophylactic visits at Months 1, 2, 3, 6, 9 and 12. At Month 12, the mean (SD) Cmax and AUC(0-96 hours) of FVIII:C were 106 (37.2) IU/dL and 5962 (2671) IU*h/dL (N=8), respectively.
12.3 Pharmacokinetics
The PK profile of VONVENDI was determined based on data analyses from two clinical trials by assessment of VWF:RCo, VWF:Ag, and VWF:CB following a single dose. Subjects were evaluated in the non-bleeding state.
Table 8 below summarizes the PK parameters of VONVENDI after infusions of 50 IU/kg (PK50) or 80 IU/kg VWF:RCo (PK80) VONVENDI.
| Parameter (unit) | PK50 VONVENDI with ADVATE* | PK50 VONVENDI | PK80 VONVENDI |
|---|---|---|---|
| Mean (SD) Min; Max | Mean (SD) Min; Max | Mean (SD) Min; Max |
|
| AUC(0-inf) = area under plasma concentration-time curve from time 0 hour to infinite time post-infusion; IR = incremental recovery; CL = clearance; t1/2 = half-life. | |||
|
|||
| T1/2 | 19.3 (10.99) | 22.6 (5.34) | 19.1 (4.32) |
| (h) | 10.8; 51.2 | 17.0; 37.2 | 11.8; 28.0 |
| CL | 0.04 (0.028) | 0.02 (0.005) | 0.03 (0.009) |
| ([dL/kg]/h) | 0.01; 0.16 | 0.02; 0.04 | 0.02; 0.05 |
| IR | 1.7 (0.62) | 1.9 (0.41) | 2.0 (0.39) |
| ([IU/dL]/([IU/kg]) | 1.0; 3.6 | 1.2; 2.7 | 1.4; 2.9 |
| AUC0-inf | 1541.4 (554.31) | 2105.4 (427.51) | 2939.0 (732.72) |
| (IU*h/dL) | 173.8; 2862.0 | 1334.0; 2813.3 | 1507.8; 4121.1 |
| AUC0-inf/Dose | 33.4 (13.87) | 42.1 (8.31) | 36.8 (8.97) |
| ([IU*h/dL]/[IU/kg]) | 6.4; 70.4 | 27.8; 54.8 | 18.8; 50.4 |
The PK of VWF following prophylactic treatment with VONVENDI were investigated in a clinical trial. Steady state PK parameters of VWF:RCo are presented in Table 9 for subjects with Type 3 VWD who were previously treated on-demand (OD) with any VWF product prior to study entry (Prior OD group) and subjects who were previously treated prophylactically with a plasma derived VWF product (Switch group) prior to study enrollment. The pharmacokinetics of VWF following single and multiple dosing (at Month 12) were similar in the Prior OD group.
| Parameter (unit) | Prior OD Group N=8 Mean (SD) Min; Max | Switch Group N=6 Mean (SD) Min; Max |
|---|---|---|
| AUC(0-96 hours) = area under plasma concentration-time curve from time 0 to 96 hours post-infusion; Cmax = maximum plasma concentration; IR = incremental recovery; CL = clearance; t1/2 = half-life; dose regimens for subjects with Type 3 VWD in Prior OD group were 41 to 56 IU/kg and for subjects with Type 3 VWD in Switch group were 24 to 63 IU/kg. | ||
|
||
| T1/2 | 16.5 (4.13)* | 14.1 (6.13)† |
| (h) | 10.9; 22.4 | 9.4; 22.9 |
| CL | 0.04 (0.012)* | 0.04 (0.014)† |
| ([dL/kg]/h) | 0.02; 0.05 | 0.02, 0.06 |
| IR at Cmax | 1.8 (0.58) | 1.8 (0.25) |
| ([IU/dL] / [IU/kg]) | 0.9, 2.7 | 1.6, 2.2 |
| Cmax | 86.4 (34.2) | 90.6 (33.7) |
| (IU/dL) | 41.6, 149.0 | 46.7, 141.0 |
| Cmax/Dose | 1.8 (0.58) | 1.8 (0.25) |
| ([IU/dL]/[IU/kg]) | 0.9, 2.7 | 1.6; 2.2 |
| AUC(0-96 hours) | 1199 (760) | 1662 (677)‡ |
| (IU*h/dL) | 460, 2940 | 1230, 2440 |
| AUC(0- 96 hours)/Dose | 24.3 (13.40) | 27.5 (9.74)‡ |
| ([IU*h/dL]/[IU/kg]) | 9.8, 53.0 | 21.7, 38.7 |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In vitro and in vivo genotoxicity studies indicated no mutagenic potential for VONVENDI. Long-term animal studies to assess the carcinogenic potential of VONVENDI were not performed. Animal studies evaluating the developmental and reproductive toxicity of VONVENDI were not conducted.
14. Clinical Studies
On-Demand Treatment and Control of Bleeding Episodes
Hemostatic efficacy of VONVENDI was assessed in a multicenter, open-label trial investigating different dosing strategies with and without rFVIII for on-demand treatment and control of bleeding episodes in adults (age 18 years and older) diagnosed with von Willebrand disease. In this trial, all subjects requiring rFVIII received ADVATE [Antihemophilic factor (recombinant)].
Bleeding episodes were treated initially with an infusion of VONVENDI and ADVATE at a ratio of 1.3:1 respectively (i.e., 30% more VONVENDI than ADVATE), and subsequently with VONVENDI with or without ADVATE, based on FVIII:C levels. The aim of the initial dose of VONVENDI with ADVATE was to achieve target plasma levels of greater than 60 IU/dL (60%) VWF:RCo and greater than 40 IU/dL (40%) of FVIII:C.
A total of 193 bleeding episodes were reported in 22/37 subjects exposed to VONVENDI for on-demand treatment. Demographic and baseline characteristics are listed in Table 10.
| Parameter | Category | Exposed Subjects n=37 n (%) | Treated Subjects n=22 n (%) |
|---|---|---|---|
| Gender | Male | 17 (45.9) | 10 (45.5) |
| Female | 20 (54.1) | 12 (54.5) | |
| Age | Median (years) | 37.0 | 28.0 |
| Race | Caucasian | 32 (86.5) | 20 (90.9) |
| Asian | 5 (13.5) | 2 (9.1) | |
| Ethnicity | Hispanic or Latino | 2 (5.4) | 2 (9.1) |
| Not Hispanic or Latino | 35 (94.6) | 20 (90.9) | |
| VWD Type | 1 | 2 (5.4) | 0 (0.0) |
| 2A | 5 (13.5) | 4 (18.2) | |
| 2B | 0 (0.0) | 0 (0.0) | |
| 2M | 0 (0.0) | 0 (0.0) | |
| 2N | 1 (2.7) | 1 (4.5) | |
| 3 | 29 (78.4) | 17 (77.3) |
The primary efficacy endpoint was the number of subjects with treatment success for control of bleeding episodes. Treatment success was defined as a mean efficacy rating score of less than 2.5 for all bleeding episodes in a subject treated with VONVENDI (with or without ADVATE) during the trial period. The efficacy rating was assessed using a pre-specified 4-point rating scale comparing the prospectively estimated number of infusions needed to treat the bleeding episodes as assessed by the investigator to the actual number of infusions administered. The definitions for each of the 4-point rating scales are provided in Table 11.
| Rating | Minor and Moderate Bleeding Events | Major Bleeding Events |
|---|---|---|
| Excellent (= 1) | Actual number of infusions ≤ estimated number of infusions required to treat that bleeding episode. No additional VWF coagulation factor containing product required. | Actual number of infusions ≤ estimated number of infusions required to treat that bleeding episode. No additional VWF coagulation factor containing product required. |
| Good (= 2) | 1 to 2 infusions greater than estimated required to control that bleeding episode. No additional VWF coagulation factor containing product required. | <1.5× infusions greater than estimated required to control that bleeding episode. No additional VWF coagulation factor containing product required. |
| Moderate (= 3) | 3 or more infusions greater than estimated used to control that bleeding event. No additional VWF coagulation factor containing product required. | ≥1.5× more infusions greater than estimated used to control that bleeding event. No additional VWF coagulation factor containing product required. |
| None (= 4) | Severe uncontrolled bleeding or intensity of bleeding not changed. Additional VWF coagulation factor containing product required. | Severe uncontrolled bleeding or intensity of bleeding not changed. Additional VWF coagulation factor containing product required. |
Secondary efficacy measures were the number of treated bleeding episodes with an efficacy rating of 'excellent' or 'good', the number of infusions and number of units of VONVENDI, administered with or without ADVATE, per bleeding episode.
The primary efficacy assessment excluded subjects with GI bleeds (n=2), and subjects in whom the number of infusions to control a bleeding episode was estimated retrospectively (n=2). The rate of subjects (n=18) with treatment success was 100% (95% CI, 81.5 to 100). Sensitivity analyses of treatment success for bleeding episodes including GI bleeds and those bleeding episodes for which the investigator had to make retrospective assessment of the number of infusions required (n=22: 17 with type 3 VWD, 4 with type 2A VWD and 1 with type 2N VWD) confirmed the primary analysis, with a 100% treatment success rate for each scenario.
All bleeding episodes treated with VONVENDI and ADVATE or VONVENDI alone were controlled with an efficacy rating of excellent (96.9%) or good (3.1%). Control of bleeding episodes was consistent across all degrees of severity.
For an overview of hemostatic efficacy by bleeding severity and number of infusions required to treat a bleeding episode, refer to Table 12.
| Number of Infusions per Bleed | Severity of Bleeding Episodes | ||||
|---|---|---|---|---|---|
| Minor n (%) n=122 | Moderate n (%) n=61 | Major/Severe n (%) n=7 | Unknown n (%) n=2 | All n (%) n=192 |
|
|
|||||
| 1 | 113 (92.6%) | 41 (67.2%) | 1 (14.3%) | 2 (100%) | 157 (81.8%) |
| 2 | 8 (6.6%) | 13 (21.3%) | 4 (57.1%) | 0 (0.0) | 25 (13.0%) |
| 3 | 1 (0.8%) | 6 (9.8%) | 2 (28.6%) | 0 (0.0) | 9 (4.7%) |
| 4 | 0 (0.0) | 1 (1.6%) | 0 (0.0) | 0 (0.0) | 1 (0.5%) |
| Median | 1 | 1 | 2 | 1 | 1 |
| Range | 1 to 3 | 1 to 4 | 1 to 3 | 1 to 1 | 1 to 4 |
The median cumulative dose of VONVENDI administered per bleeding episode (with or without ADVATE) was 48.2 IU/kg (90% CI, 43.9 to 50.2) IU/kg. In relation to bleeding severity, the median cumulative dose to treat a bleeding episode was 43.3 (range, 25.2 to 158.2) IU/kg for minor bleeding episodes (n=122), 52.7 (range, 23.8 to 184.9) IU/kg for moderate bleeding episodes (n=61), 100 (range, 57.5 to 135) IU/kg for major bleeding episodes (n=7).
Table 13 summarizes data obtained for number of infusions and efficacy rating per bleeding episode by location.
| Bleeding Episodes by Location (n) | Median Number of Infusions (Range) | Rating (%) |
|---|---|---|
| Joint (n=59) | 1 (1 to 3) | Excellent (96.6%) |
| Good (3.4%) | ||
| GI (n=6) | 1 (1 to 2) | Excellent (83.3%) |
| Good (16.7%) | ||
| Mucosal: Genital Tract Female (n=32) | 1 (1 to 2) | Excellent (96.9%) |
| Good (3.1%) | ||
| Mucosal: Nasopharyngeal (n=42) | 1 (1 to 2) | Excellent (97.6%) |
| Good (2.4%) | ||
| Mucosal: Mouth and Oral Cavity (n=26) | 1 (1 to 4) | Excellent (100%) |
| Good (0%) |
Perioperative Management of Bleeding
Hemostatic efficacy of VONVENDI was assessed in a prospective, open-label, multicenter trial to evaluate efficacy and safety of VONVENDI with or without ADVATE in elective surgical procedures in adults (age 18 years and older) diagnosed with severe VWD and the subjects were followed for 14 days after surgery.
A total of 15 VWD subjects completed the trial and 93% of the subjects were less than 65 years old (range, 20 to 70 years), of whom 53.3% were females and 53% (8/15) were type 3 VWD patients. Out of 15 subjects, 10 subjects underwent major surgeries and five subjects underwent minor surgeries.
Major surgeries included orthopedic surgeries: total hip replacement, total knee replacement, knee endoprosthesis, ankle prosthesis, anterior cruciate ligament surgery and meniscectomy. Other major surgeries included laparoscopic cholecystectomy, laparoscopic cystectomy and complex dental extractions. Minor surgeries/procedures included nasopharyngoscopy, dental extractions, colonoscopy and radioisotope synovectomy.
All subjects were administered a 12 to 24 hour preoperative dose of 40 to 60 IU/kg of VONVENDI to increase the factor VIII levels to target levels. Within 3 hours prior to surgery, the subjects' FVIII:C levels were assessed to ensure that target of 30 IU/dL for minor surgeries and 60 IU/dL for major surgeries was achieved. Within 1 hour prior to surgery, subjects received a dose of VONVENDI. ADVATE (recombinant Factor VIII) was administered based on FVIII:C levels performed 3 hours prior to surgery. VWF and factor VIII Incremental recovery were used to guide the initial and subsequent doses.
Six of the 10 subjects undergoing major surgery received protocol-specified loading dose. It should be noted that the protocol-specified loading dose was based on VWF:RCo levels assessed prior to the 12 to 24 hour preoperative dose. Four of 10 subjects undergoing major surgery and four out of five subjects undergoing minor surgery received a loading dose of VONVENDI based on VWF:RCo assessed prior to loading dose and after administration of the 12 to 24 hour preoperative dose. Unlike the protocol-specified loading dose based on the levels assessed prior to the preoperative dose between 12 to 24 hours of the surgery, the loading doses in these eight subjects were calculated based on VWF:RCo levels after a preoperative dose, and were therefore lower doses than protocol-specified loading dose. No differences in safety or efficacy were noted between the two groups.
The primary outcome measure was the overall hemostatic efficacy assessed 24 hours after the last perioperative VONVENDI infusion or at completion of study visit whichever occurred earlier using a 4-point ordinal efficacy scale outlined in Table 14 ("excellent", "good", "moderate" and "none") based on estimated expected versus actual blood loss, transfusion requirements and postoperative bleeding and oozing. A rating of excellent or good was required to declare the outcome a success.
| Rating | Overall Assessment of Hemostatic Efficacy 24 Hours after Last Perioperative IP Infusion or at Day 14 Completion Visit (Whatever Occurs Earlier) |
|---|---|
| Excellent (1) | Intra- and postoperative hemostasis achieved with rVWF with or without ADVATE was as good or better than that expected for the type of surgical procedure performed in a hemostatically normal subject. |
| Good (2) | Intra- and postoperative hemostasis achieved with rVWF with or without ADVATE was probably as good as that expected for the type of surgical procedure performed in a hemostatically normal subject. |
| Moderate (3) | Intra- and postoperative hemostasis with rVWF with or without ADVATE was clearly less than optimal for the type of procedure performed but was maintained without the need to change the rVWF concentrate. |
| None (4) | Subject experienced uncontrolled bleeding that was the result of inadequate therapeutic response despite proper dosing, necessitating a change of rVWF concentrate. |
Overall hemostatic efficacy for major and minor surgeries was 100% (15/15) with a 90% confidence interval of 81.9% to 100%. It was excellent for 60% of surgeries and good for 40% of surgeries.
Intraoperative hemostatic efficacy was a secondary endpoint. For major and minor surgeries, it was 100% with a 90% confidence interval of 81.9% to 100%. It was excellent for 73.3% of surgeries and good for 26.7% of surgeries.
For details regarding hemostatic efficacy for minor and major surgery, see Table 15.
| Type of Surgery | Excellent 9/15 (60%) | Good 6/15 (40%) | Moderate 0/15 (0.0%) | Total N=15 |
|---|---|---|---|---|
| Minor | 4 | 1 | 0 | 5 |
| Major | 5 | 5 | 0 | 10 |
Dosing was individualized based on incremental recovery results performed before surgery.
Mean total 12 to 24 preoperative dose was 50.9 IU/kg (median 55.0 IU/kg; range, 36.1 to 59.9 IU/kg).
Mean total loading dose (1 hour preoperative dose) per infusion was 38.6 IU/kg (median 35.8 IU/kg; range, 8.0 to 82.7 IU/kg). Major surgeries required a mean loading dose of 42.8 IU/kg (median 37.6 IU/kg; range, 15.7 to 82.7 IU/kg) in comparison with a mean loading dose of 30.2 IU/kg (median 34.2 IU/kg; range, 8.0 to 46.4 IU/kg) for minor surgeries.
For subjects treated with VONVENDI (with or without ADVATE), the median total postoperative dose within the first 7 days after surgery was 114.2 IU/kg with a range of 23.8 to 318.9 IU/kg (n=13) and 76.2 IU/kg with a range of 23.8 to 214.4 IU/kg for the next 7 postoperative days (n=8).
Routine Prophylaxis to Reduce the Frequency of Bleeding Episodes in Patients with Severe Type 3 VWD Receiving On-Demand Therapy
VONVENDI was studied in a prospective, single arm, open-label, international multicenter study to evaluate efficacy, safety, PK and pharmacodynamics (PD) of prophylactic treatment in reducing the frequency of bleeding episodes in adult subjects (age 18 years and older) diagnosed with VWD.
Subjects were enrolled into two treatment arms based on the treatment they received for management of bleeding prior to study entry consisting of a Prior OD group in which subjects received on-demand (OD) treatment only and the Switch group in which subjects had received prophylactic treatment with plasma derived VWF (pdVWF). Efficacy was assessed based on the median annualized bleeding rate for all bleeds, spontaneous bleeds and joint bleeds and was based on descriptive statistics.
Twenty-two efficacy evaluable subjects received a median of 92.5 doses, 12 of these subjects had previously received on demand treatment (Prior OD group) and 10 subjects previously received prophylactic treatment with pdVWF (Switch group) prior to enrolling into this study. Nine subjects in the OD group and 8 subjects in the Switch group completed the study which required 12 months of treatment.
Median age of subjects in the study was 34 years (18 - 77 years), 54.5% were male and 96% of subjects were white and 86.4% were non-Hispanic or Latino. Of the 12 subjects in the Prior OD group, one had severe Type 1, one had severe Type 2 VWD, and ten had severe Type 3 VWD. Of the 10 subjects in the Switch group, one had severe Type 1, one had severe Type 2 VWD, and eight had severe Type 3 VWD.
Subjects in the Prior OD group began treatment at 50 ± 10 IU/kg per infusion twice weekly. One subject in the Prior OD group required dose adjustments for management of breakthrough bleeding. The majority of subjects (9/10) with Type 3 VWD in the Prior OD group received twice weekly dosing with Vonvendi and had a median maximum dose of 55.9 (IU/kg).
The study did not adequately demonstrate efficacy of Vonvendi as prophylactic treatment to reduce bleeding events in subjects with Type 3 VWD receiving prophylactic therapy prior to study entry (Switch group).
The efficacy parameters of Vonvendi as prophylactic treatment in subjects with Type 3 VWD receiving on-demand treatment prior to study entry is provided in Table 16. The median percentage change of ABRs from historical to on-study were: for all bleeds -54.7%, for spontaneous bleeds -75.9%, and for joint bleeds -100.0%.
| Type or Site of Bleeding Event | Number of BEs Historical/On-Study | Historical Median ABR*
(Min, Max) | On-Study Median ABR*
(Min, Max) |
|---|---|---|---|
|
|||
| All Bleeds† | 201/38 | 5.0 (3.0, 159.0) | 2.3 (0, 157.9) |
| Spontaneous Bleeds‡ | 195/33 | 3.5 (3.0, 158.0) | 1.0 (0.0, 157.9) |
| Joint Bleeds† | 23/3 | 2.0 (0.0, 7.0) | 0.0 (0.0, 1.9) |
15. References
- Stockschlaeder M, Schneppenheim R, Budde U, Update on von Willebrand factor multimers: focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul Fibrinolysis 2014, 25:206-216.
- Turecek PL, Mitterer A, Matthiessen HP, Gritsch H, Varadi K, Siekmann J, Schnecker K, Plaimauer B, Kaliwoda M, Purtscher M, Woehrer W, Mundt W, Muchitsch EM, Suiter T, Ewenstein BM, Ehrlich HJ, Schwarz HP, Development of a plasma- and albumin-free recombinant von Willebrand factor. Haemastaseologie 2009; 29 (Suppl 1): 32-38.
17. Patient Counseling Information
Advise the patient:
- To read the FDA-approved patient labeling (Patient Information and Instructions for Use).
- About early signs of hypersensitivity reactions, including anaphylactic shock, generalized urticaria, angioedema, chest tightness, hypotension, shock, lethargy, nausea, vomiting, paresthesia, pruritus, restlessness, wheezing and/or acute respiratory distress. Advise patients to discontinue use of the product if these symptoms occur and seek immediate emergency treatment with resuscitative measures.
- To contact their physician or treatment center for further treatment and/or assessment if they experience a lack of clinical response to von Willebrand factor therapy, as this may be a manifestation of an inhibitor.
- To consult with their physicians or healthcare provider prior to travel. While traveling, advise patients to bring an adequate supply of VONVENDI based on their current regimen of treatment.




| VONVENDI
von willebrand factor (recombinant) kit |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| VONVENDI
von willebrand factor (recombinant) kit |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Takeda Pharmaceuticals America, Inc. (039997266) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Baxalta US Inc. | 009471603 | MANUFACTURE(0944-7551, 0944-7553, 0944-7550, 0944-7552) , ANALYSIS(0944-7551, 0944-7553, 0944-7550, 0944-7552) , LABEL(0944-7552, 0944-7550) , PACK(0944-7553, 0944-7551) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Siegfried Hameln GmbH | 315869123 | MANUFACTURE(64764-515, 64764-516) , ANALYSIS(64764-515, 64764-516) , LABEL(64764-515, 64764-516) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Baxalta Manufacturing Sàrl | 482124943 | API MANUFACTURE(0944-7550, 0944-7552) , ANALYSIS(0944-7550, 0944-7552) | |
