Drug Detail:Xgeva (Denosumab [ den-oh-sue-mab ])
Drug Class: Miscellaneous bone resorption inhibitors
Highlights of Prescribing Information
Xgeva (denosumab) injection, for subcutaneous use
Initial U.S. Approval: 2010
Recent Major Changes
| Warnings and Precautions, Hypocalcemia (5.3) | 02/2020 |
Indications and Usage for Xgeva
Xgeva is a RANK ligand (RANKL) inhibitor indicated for:
- Prevention of skeletal-related events in patients with multiple myeloma and in patients with bone metastases from solid tumors. (1.1)
- Treatment of adults and skeletally mature adolescents with giant cell tumor of bone that is unresectable or where surgical resection is likely to result in severe morbidity. (1.2, 14.3)
- Treatment of hypercalcemia of malignancy refractory to bisphosphonate therapy. (1.3)
Xgeva Dosage and Administration
- Xgeva is intended for subcutaneous route only and should not be administered intravenously, intramuscularly, or intradermally. (2.1)
- Multiple Myeloma and Bone Metastasis from Solid Tumors: Administer 120 mg every 4 weeks as a subcutaneous injection in the upper arm, upper thigh, or abdomen. (2.2)
- Giant Cell Tumor of Bone: Administer 120 mg every 4 weeks with additional 120 mg doses on Days 8 and 15 of the first month of therapy. Administer subcutaneously in the upper arm, upper thigh, or abdomen. (2.3)
- Administer calcium and vitamin D as necessary to treat or prevent hypocalcemia. (2.2, 2.3)
- Hypercalcemia of Malignancy: Administer 120 mg every 4 weeks with additional 120 mg doses on Days 8 and 15 of the first month of therapy. Administer subcutaneously in the upper arm, upper thigh, or abdomen. (2.4)
Dosage Forms and Strengths
- Injection: 120 mg/1.7 mL (70 mg/mL) solution in a single-dose vial (3)
Contraindications
- Hypocalcemia (4.1)
- Known clinically significant hypersensitivity to Xgeva (4.2)
Warnings and Precautions
- Same Active Ingredient: Patients receiving Xgeva should not take Prolia®. (5.1)
- Hypersensitivity reactions including anaphylaxis may occur. Discontinue permanently if a clinically significant reaction occurs. (5.2)
- Hypocalcemia: Xgeva can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Correct hypocalcemia prior to initiating Xgeva. Monitor calcium levels during therapy, especially in the first weeks of initiating therapy, and adequately supplement all patients with calcium and vitamin D. (5.3)
- Osteonecrosis of the jaw (ONJ) has been reported in patients receiving Xgeva. Perform an oral examination prior to starting Xgeva. Monitor for symptoms. Avoid invasive dental procedures during treatment with Xgeva. (5.4)
- Atypical femoral fracture: Evaluate patients with thigh or groin pain to rule out a femoral fracture. (5.5)
- Hypercalcemia Following Treatment Discontinuation in Patients with Giant Cell Tumor of Bone and in Patients with Growing Skeletons: Monitor patients for signs and symptoms of hypercalcemia, and manage as clinically appropriate. (5.6, 8.4)
- Multiple Vertebral Fractures (MVF) Following Treatment Discontinuation: When Xgeva treatment is discontinued, evaluate the individual patient’s risk for vertebral fractures. (5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of potential risk to the fetus and to use effective contraception. (5.8, 8.1, 8.3)
Adverse Reactions/Side Effects
- Bone Metastasis from Solid Tumors: Most common adverse reactions (≥ 25%) were fatigue/asthenia, hypophosphatemia, and nausea. (6.1)
- Multiple Myeloma: Most common adverse reactions (≥ 10%) were diarrhea, nausea, anemia, back pain, thrombocytopenia, peripheral edema, hypocalcemia, upper respiratory tract infection, rash, and headache. (6.1)
- Giant Cell Tumor of Bone: Most common adverse reactions (≥ 10%) were arthralgia, headache, nausea, back pain, fatigue, and pain in extremity. (6.1)
- Hypercalcemia of Malignancy: Most common adverse reactions (> 20%) were nausea, dyspnea, decreased appetite, headache, peripheral edema, vomiting, anemia, constipation, and diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Pediatric patients: Recommended only for treatment of skeletally mature adolescents with giant cell tumor of bone. (8.4)
- Renal impairment: Patients with creatinine clearance less than 30 mL/min or receiving dialysis are at risk for hypocalcemia. Adequately supplement with calcium and vitamin D. (8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2020
Full Prescribing Information
1. Indications and Usage for Xgeva
1.1 Multiple Myeloma and Bone Metastasis from Solid Tumors
Xgeva is indicated for the prevention of skeletal-related events in patients with multiple myeloma and in patients with bone metastases from solid tumors.
2. Xgeva Dosage and Administration
2.1 Important Administration Instructions
Xgeva is intended for subcutaneous route only and should not be administered intravenously, intramuscularly, or intradermally.
2.2 Multiple Myeloma and Bone Metastasis from Solid Tumors
The recommended dose of Xgeva is 120 mg administered as a subcutaneous injection every 4 weeks in the upper arm, upper thigh, or abdomen.
Administer calcium and vitamin D as necessary to treat or prevent hypocalcemia [see Warnings and Precautions (5.3)].
2.3 Giant Cell Tumor of Bone
The recommended dose of Xgeva is 120 mg administered every 4 weeks with additional 120 mg doses on Days 8 and 15 of the first month of therapy. Administer subcutaneously in the upper arm, upper thigh, or abdomen.
Administer calcium and vitamin D as necessary to treat or prevent hypocalcemia [see Warnings and Precautions (5.3)].
4. Contraindications
5. Warnings and Precautions
5.1 Drug Products with Same Active Ingredient
Xgeva includes the same active ingredient (denosumab) found in Prolia. Patients receiving Xgeva should not take Prolia.
5.2 Hypersensitivity
Clinically significant hypersensitivity including anaphylaxis has been reported with use of Xgeva. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically significant allergic reaction occurs, initiate appropriate therapy and discontinue Xgeva therapy permanently [see Contraindications (4.2) and Adverse Reactions (6.2)].
5.3 Hypocalcemia
Xgeva can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Correct pre-existing hypocalcemia prior to Xgeva treatment. Monitor calcium levels, throughout Xgeva therapy, especially in the first weeks of initiating therapy, and administer calcium, magnesium, and vitamin D as necessary. Concomitant use of calcimimetics and other drugs that can lower calcium levels may worsen hypocalcemia risk and serum calcium should be closely monitored. Advise patients to contact a healthcare provider for symptoms of hypocalcemia [see Contraindications (4.1), Adverse Reactions (6.1, 6.2), and Patient Counseling Information (17)].
An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/min and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake [see Adverse Reactions (6.1), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
5.4 Osteonecrosis of the Jaw (ONJ)
Osteonecrosis of the jaw (ONJ) has been reported in patients receiving Xgeva, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials in patients with cancer, the incidence of ONJ was higher with longer duration of exposure [see Adverse Reactions (6.1)]. Seventy-nine percent of patients with ONJ had a history of tooth extraction, poor oral hygiene, or use of a dental appliance as a predisposing factor. Other risk factors for the development of ONJ include immunosuppressive therapy, treatment with angiogenesis inhibitors, systemic corticosteroids, diabetes, and gingival infections. Similarly, for Xgeva patients with multiple myeloma that developed ONJ, 58% had a history of invasive dental procedures as a predisposing factor.
Perform an oral examination and appropriate preventive dentistry prior to the initiation of Xgeva and periodically during Xgeva therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with Xgeva. Consider temporary discontinuation of Xgeva therapy if an invasive dental procedure must be performed. There are no data available to suggest the optimal duration of treatment interruption.
Patients who are suspected of having or who develop ONJ while on Xgeva should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition. Clinical judgment of the treating healthcare provider should guide the management plan of each patient based on individual risk/benefit assessment.
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fracture
Atypical femoral fracture has been reported with Xgeva [see Adverse Reactions (6.1)]. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution.
Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture.
During Xgeva treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of Xgeva therapy should be considered, pending a risk/benefit assessment, on an individual basis.
5.6 Hypercalcemia Following Treatment Discontinuation in Patients with Giant Cell Tumor of Bone and in Patients with Growing Skeletons
Clinically significant hypercalcemia requiring hospitalization and complicated by acute renal injury has been reported in Xgeva-treated patients with giant cell tumor of bone and patients with growing skeletons. Hypercalcemia has been reported within the first year after treatment discontinuation. After treatment is discontinued, monitor patients for signs and symptoms of hypercalcemia, assess serum calcium periodically, reevaluate the patient’s calcium and vitamin D supplementation requirements and manage patients as clinically appropriate [see Adverse Reactions (6) and Use in Specific Populations (8.4)].
5.7 Multiple Vertebral Fractures (MVF) Following Treatment Discontinuation
Multiple vertebral fractures (MVF) have been reported following discontinuation of treatment with denosumab. Patients at higher risk for MVF include those with risk factors for or a history of osteoporosis or prior fractures.
When Xgeva treatment is discontinued, evaluate the individual patient’s risk for vertebral fractures [see Patient Counseling Information (17)].
5.8 Embryo-Fetal Toxicity
Based on data from animal studies and its mechanism of action, Xgeva can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of denosumab to cynomolgus monkeys throughout pregnancy at a dose 25-fold higher than the recommended human dose of Xgeva based on body weight resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent peripheral lymph nodes, abnormal bone growth and decreased neonatal growth.
Verify the pregnancy status of females of reproductive potential prior to the initiation of Xgeva. Advise pregnant women and females of reproductive potential that exposure to Xgeva during pregnancy or within 5 months prior to conception can result in fetal harm. Advise females of reproductive potential to use effective contraception during therapy, and for at least 5 months after the last dose of Xgeva [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed below and elsewhere in the labeling:
• Hypersensitivity [see Warnings and Precautions (5.2)]
• Hypocalcemia [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)]
• Osteonecrosis of the Jaw [see Warnings and Precautions (5.4)]
• Atypical Subtrochanteric and Diaphyseal Femoral Fracture [see Warnings and Precautions (5.5)]
• Hypercalcemia following treatment discontinuation in patients with giant cell tumor of bone and in patients with growing skeletons [see Warnings and Precautions (5.6) and Use in Specific Populations (8.4)]
• Multiple vertebral fractures (MVF) following treatment discontinuation [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Bone Metastasis from Solid Tumors
The safety of Xgeva was evaluated in three randomized, double-blind, double-dummy trials [see Clinical Trials (14.1)] in which a total of 2841 patients with bone metastasis from prostate cancer, breast cancer, or other solid tumors, or lytic bony lesions from multiple myeloma received at least one dose of Xgeva. In Studies 20050136, 20050244, and 20050103, patients were randomized to receive either 120 mg of Xgeva every 4 weeks as a subcutaneous injection or 4 mg (dose adjusted for reduced renal function) of zoledronic acid every 4 weeks by intravenous (IV) infusion. Entry criteria included serum calcium (corrected) from 8 to 11.5 mg/dL (2 to 2.9 mmol/L) and creatinine clearance 30 mL/min or greater. Patients who had received IV bisphosphonates were excluded, as were patients with prior history of ONJ or osteomyelitis of the jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure. During the study, serum chemistries including calcium and phosphorus were monitored every 4 weeks. Calcium and vitamin D supplementation was recommended but not required.
The median duration of exposure to Xgeva was 12 months (range: 0.1-41) and median duration on-study was 13 months (range: 0.1-41). Of patients who received Xgeva, 46% were female. Eighty-five percent were White, 5% Hispanic/Latino, 6% Asian, and 3% Black. The median age was 63 years (range: 18-93). Seventy-five percent of patients who received Xgeva received concomitant chemotherapy.
The most common adverse reactions in patients (incidence greater than or equal to 25%) were fatigue/asthenia, hypophosphatemia, and nausea (see Table 1). The most common serious adverse reaction was dyspnea. The most common adverse reactions resulting in discontinuation of Xgeva were osteonecrosis and hypocalcemia.
| Body System | Xgeva
n = 2841 % | Zoledronic Acid
n = 2836 % |
| GASTROINTESTINAL | ||
| Nausea | 31 | 32 |
| Diarrhea | 20 | 19 |
| GENERAL | ||
| Fatigue/Asthenia | 45 | 46 |
| INVESTIGATIONS | ||
| Hypocalcemiab | 18 | 9 |
| Hypophosphatemiab | 32 | 20 |
| NEUROLOGICAL | ||
| Headache | 13 | 14 |
| RESPIRATORY | ||
| Dyspnea | 21 | 18 |
| Cough | 15 | 15 |
a Adverse reactions reported in at least 10% of patients receiving Xgeva in Studies 20050136, 20050244, and 20050103, and meeting one of the following criteria:
|
||
Severe Mineral/Electrolyte Abnormalities
▪ Severe hypocalcemia (corrected serum calcium less than 7 mg/dL or less than 1.75 mmol/L) occurred in 3.1% of patients treated with Xgeva and 1.3% of patients treated with zoledronic acid. Of patients who experienced severe hypocalcemia, 33% experienced 2 or more episodes of severe hypocalcemia and 16% experienced 3 or more episodes [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)].
▪ Severe hypophosphatemia (serum phosphorus less than 2 mg/dL or less than 0.6 mmol/L) occurred in 15.4% of patients treated with Xgeva and 7.4% of patients treated with zoledronic acid.
Osteonecrosis of the Jaw (ONJ)
In the primary treatment phases of Studies 20050136, 20050244, and 20050103, ONJ was confirmed in 1.8% of patients in the Xgeva group (median exposure of 12.0 months; range: 0.1-40.5) and 1.3% of patients in the zoledronic acid group. The trials in patients with breast (Study 20050136) or prostate (Study 20050103) cancer included an Xgeva open-label extension treatment phase where patients were offered Xgeva 120 mg once every 4 weeks (median overall exposure of 14.9 months; range: 0.1-67.2). The patient-year adjusted incidence (number of events per 100 patient years) of confirmed ONJ was 1.1% during the first year of treatment, 3.7% in the second year, and 4.6% per year thereafter. The median time to ONJ was 20.6 months (range: 4-53) [see Warnings and Precautions (5.4)].
In a placebo-controlled clinical trial with an extension treatment phase evaluating Xgeva for the prevention of bone metastases in patients with non-metastatic prostate cancer (a patient population for which Xgeva is not indicated), with longer treatment exposure of up to 7 years, the patient-year adjusted incidence (number of events per 100 patient years) of confirmed ONJ was 1.1% during the first year of treatment, 3.0% in the second year, and 7.1% per year thereafter.
Atypical Subtrochanteric and Diaphyseal Fracture
In the clinical trial program, atypical femoral fracture has been reported in patients treated with Xgeva and the risk increased with longer duration of treatment. Events have occurred during treatment and after treatment was discontinued [see Warnings and Precautions (5.5)].
Multiple Myeloma
The safety of Xgeva was evaluated in an international, randomized (1:1), double-blind, active-controlled trial of patients with newly diagnosed multiple myeloma with treatment through disease progression [see Clinical Trials (14.2)]. In this trial, patients received 120 mg Xgeva every 4 weeks as a subcutaneous injection (n = 850) or 4 mg (dose adjusted for renal function) of zoledronic acid intravenously (IV) every 4 weeks by IV infusion (n = 852). Entry criteria included serum calcium (corrected) from 8 to 11.5 mg/dL (2 to 2.9 mmol/L) and creatinine clearance 30 mL/min or greater. Patients who had received IV bisphosphonates were excluded, as were patients with prior history of ONJ or osteomyelitis of the jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure. During the study, serum chemistries including calcium and phosphorus were monitored every 4 weeks. Calcium and vitamin D supplementation was recommended but not required.
The median duration of exposure to Xgeva was 16 months (range: 1-50) and median duration on-study was 17 months (range: 0-49). Of patients who received Xgeva, 46% were female, 83% percent were White, 13% Asian, 3% Black or African American, and 4% Hispanic/Latino. The median age of the patients randomized to Xgeva was 63 years (range: 29-91) and all patients who received Xgeva received concomitant anti-myeloma chemotherapy.
The adverse reaction profile of Xgeva in patients with multiple myeloma, Study 20090482, was similar to that observed in Studies 20050136, 20050244, and 20050103. The most common adverse reactions (incidence ≥ 10%) were diarrhea (34%), nausea (32%), anemia (22%), back pain (21%), thrombocytopenia (19%), peripheral edema (17%), hypocalcemia (16%), upper respiratory tract infection (15%), rash (14%), and headache (11%). The most common serious adverse reaction (incidence ≥ 5%) was pneumonia (8%). The most common adverse reaction resulting in discontinuation of Xgeva (≥ 1.0%) was osteonecrosis of the jaw.
Hypocalcemia and Hypophosphatemia
Severe hypocalcemia (corrected serum calcium less than 7 mg/dL or less than 1.75 mmol/L) and severe hypophosphatemia (serum phosphorus less than 2 mg/dL or less than 0.6 mmol/L) occurred in 2% and 21% patients treated with Xgeva, respectively.
Osteonecrosis of the Jaw (ONJ)
In the primary treatment phase of Study 20090482, ONJ was confirmed in 4.1% of patients in the Xgeva group (median exposure of 16 months; range: 1-50) and 2.8% of patients in the zoledronic acid group (median 15 months, range: 1-45 months). At the completion of the double-blind treatment phase of Study 20090482, the patient-year adjusted incidence (number of events per 100 patient years) of confirmed ONJ in the Xgeva group (median exposure of 19.4 months; range 1-52) was 2.0% during the first year of treatment, 5.0% in the second year, and 4.5% per year thereafter. The median time to ONJ was 18.7 months (range: 1-44) [see Warnings and Precautions (5.4)].
Giant Cell Tumor of Bone
The safety of Xgeva was evaluated in two single-arm trials (Study 20062004 and Study 20040215) [see Clinical Trials (14.3)] in which a total of 548 adult or skeletally mature adolescent patients with giant cell tumor of bone received at least 1 dose of Xgeva. Patients received 120 mg Xgeva subcutaneously every 4 weeks with additional 120 mg doses on Days 8 and 15 of the first month of therapy. Patients receiving concurrent bisphosphonate therapy were excluded from enrollment in both studies. Patients with prior history of ONJ or osteomyelitis of the jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure were excluded from enrollment in Study 20040215. During the trial, serum chemistries including calcium and phosphorus were monitored every 4 weeks. Calcium and vitamin D supplementation was recommended but not required.
Of the 548 patients who received Xgeva, 467 patients were treated with Xgeva for ≥ 1 year, 323 patients for ≥ 2 years, and 255 patients for ≥ 3 years. The median number of doses received was 33 (range: 4-138 doses) and the median number of months on-study was 60 (range: 0-140 months). Fifty-seven percent of the enrolled patients were women and 82% were White. The median age was 33 years (range: 13-83 years); a total of 19 patients were skeletally mature adolescents (12 to <17 years of age).
The common adverse reaction profile of Xgeva in patients with giant cell tumor of bone was generally similar to that reported in Studies 20050136, 20050244, and 20050103. The most common adverse reactions in patients (incidence ≥ 10%) were arthralgia, back pain, pain in extremity, fatigue, headache, nausea, nasopharyngitis, musculoskeletal pain, toothache, vomiting, hypophosphatemia, constipation, diarrhea, and cough. The most frequent serious adverse reactions were osteonecrosis of the jaw (3.6%), bone giant cell tumor (1.5%), anemia (1.1%), pneumonia (0.9%), and back pain (0.9%). The most frequent adverse reactions resulting in discontinuation of Xgeva was osteonecrosis of the jaw (incidence of 3.6%). The adverse reaction profile appeared similar in skeletally mature adolescents and adults.
Hypocalcemia and Hypophosphatemia
- Moderate to severe hypocalcemia (corrected serum calcium less than 8 mg/dL or less than 2 mmol/L) occurred in 5% of patients treated with Xgeva.
- Severe hypophosphatemia (serum phosphorus less than 2 to 1 mg/dL or less than 0.6 to 0.3 mmol/L) occurred in 20% of patients treated with Xgeva.
Osteonecrosis of the Jaw (ONJ)
In Study 20062004 and Study 20040215, ONJ was confirmed in 6.6% of patients who received Xgeva. [see Warnings and Precautions (5.4)].
Atypical Subtrochanteric and Diaphyseal Fracture
Atypical femoral fracture has been reported with Xgeva and was observed in 0.9% of patients in the pooled safety population [see Warnings and Precautions (5.5)].
Hypercalcemia Following Treatment Discontinuation
In the pooled safety population, 0.7% of patients experienced serious adverse events of hypercalcemia > 30 days following treatment discontinuation that was recurrent in some patients [see Warnings and Precautions (5.6)].
Hypercalcemia of Malignancy
Xgeva was evaluated in an open-label, single-arm trial (Study 20070315) in which 33 patients with hypercalcemia of malignancy (with or without bone metastases) refractory to treatment with intravenous bisphosphonate therapy were enrolled [see Clinical Trials (14.4)].
The adverse reaction profile of Xgeva in patients with hypercalcemia of malignancy was similar to that reported in Studies 20050136, 20050244, 20050103, 20062004, and 20040215. Adverse reactions occurring in greater than 20% of patients were nausea (30%), dyspnea (27%), decreased appetite (24%), headache (24%), peripheral edema (24%), vomiting (24%), anemia (21%), constipation (21%), and diarrhea (21%). The following adverse reactions of Grade 3 or greater severity related to study therapy were reported on-study: fatigue (3%) and infection (6%). Grade 3 laboratory abnormalities included hypomagnesemia (3%), hypokalemia (3%), and hypophosphatemia (76%) of patients. No deaths on-study were related to Xgeva therapy.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of Xgeva. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypocalcemia: Severe symptomatic hypocalcemia, including fatal cases [see Contraindications (4.1) and Warnings and Precautions (5.3)].
- Hypercalcemia: Severe symptomatic hypercalcemia following treatment discontinuation can occur [see Adverse Reactions (6) and Warnings and Precautions (5.6)].
- Hypersensitivity, including anaphylactic reactions [see Contraindications (4.2) and Warnings and Precautions (5.2)].
- Musculoskeletal pain, including severe musculoskeletal pain. Positive re-challenge has been reported.
- Lichenoid drug eruptions (e.g., lichen planus-like reactions).
- Alopecia.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animals and its mechanism of action, Xgeva can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are insufficient data with denosumab use in pregnant women to inform any drug associated risks for adverse developmental outcomes. In utero denosumab exposure from cynomolgus monkeys dosed monthly with denosumab throughout pregnancy at a dose 25-fold higher than the recommended human dose of Xgeva based on body weight resulted in increased fetal loss, stillbirths, and postnatal mortality; and absent lymph nodes, abnormal bone growth, and decreased neonatal growth [see Data].
Apprise pregnant women of the potential risk to the fetus.
The background rate of major birth defects and miscarriage is unknown for the indicated population. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
The effects of denosumab on prenatal development have been studied in both cynomolgus monkeys and genetically engineered mice in which RANK ligand (RANKL) expression was turned off by gene removal (a “knockout mouse”). In cynomolgus monkeys dosed subcutaneously with denosumab throughout pregnancy starting at gestational day 20 and at a pharmacologically active dose 25-fold higher than the recommended human dose of Xgeva based on body weight, there was increased fetal loss during gestation, stillbirths, and postnatal mortality. Other findings in offspring included absence of axillary, inguinal, mandibular, and mesenteric lymph nodes; abnormal bone growth, reduced bone strength, reduced hematopoiesis, dental dysplasia, and tooth malalignment; and decreased neonatal growth. At birth out to one month of age, infants had measurable blood levels of denosumab (22-621% of maternal levels).
Following a recovery period from birth out to 6 months of age, the effects on bone quality and strength returned to normal; there were no adverse effects on tooth eruption, though dental dysplasia was still apparent; axillary and inguinal lymph nodes remained absent, while mandibular and mesenteric lymph nodes were present, though small; and minimal to moderate mineralization in multiple tissues was seen in one recovery animal. There was no evidence of maternal harm prior to labor; adverse maternal effects occurred infrequently during labor. Maternal mammary gland development was normal. There was no fetal NOAEL (no observable adverse effect level) established for this study because only one dose of 50 mg/kg was evaluated. Mammary gland histopathology at 6 months of age was normal in female offspring exposed to denosumab in utero; however, development and lactation have not been fully evaluated.
In RANKL knockout mice, absence of RANKL (the target of denosumab) also caused fetal lymph node agenesis and led to postnatal impairment of dentition and bone growth. Pregnant RANKL knockout mice showed altered maturation of the maternal mammary gland, leading to impaired lactation [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.2)].
8.3 Females and Males of Reproductive Potential
Based on findings in animals and its mechanism of action, Xgeva can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating Xgeva treatment.
Contraception
Females
Advise females of reproductive potential to use effective contraception during therapy, and for at least 5 months after the last dose of Xgeva.
8.4 Pediatric Use
The safety and efficacy of Xgeva have not been established in pediatric patients except in skeletally mature adolescents (aged 12–16 years) with giant cell tumor of bone. Xgeva is recommended only for treatment of skeletally mature adolescents (aged 12–16 years) with giant cell tumor of bone [see Indications and Usage (1.2)]. Clinically significant hypercalcemia after treatment discontinuation has been reported in pediatric patients with growing skeletons who received denosumab for giant cell tumor of bone or for unapproved indications [see Adverse Reactions (6.2) and Warnings and Precautions (5.6)].
Xgeva was studied in an open-label trial that enrolled a subset of 19 adolescent patients (aged 12-16 years) with giant cell tumor of bone who had reached skeletal maturity, defined by at least 1 mature long bone (e.g., closed epiphyseal growth plate of the humerus), and had a body weight ≥ 45 kg [see Indications and Usage (1.2) and Clinical Trials (14.3)]. A total of one of five (20%) evaluable adolescent patients had an objective response by retrospective independent assessment of radiographic response according to modified Response Evaluation Criteria in Solid Tumors (RECIST 1.1). The adverse reaction profile and efficacy results appeared to be similar in skeletally mature adolescents and adults [see Adverse Reactions (6.1) and Clinical Trials (14.3)].
Animal Data
Treatment with Xgeva may impair bone growth in children with open growth plates and may inhibit eruption of dentition. In neonatal rats, inhibition of RANKL (the target of Xgeva therapy) with a construct of osteoprotegerin bound to Fc (OPG-Fc) at doses ≤ 10 mg/kg was associated with inhibition of bone growth and tooth eruption. Adolescent primates treated with denosumab at doses 5 and 25 times (10 and 50 mg/kg dose) higher than the recommended human dose of 120 mg administered once every 4 weeks, based on body weight (mg/kg), had abnormal growth plates, considered to be consistent with the pharmacological activity of denosumab.
Cynomolgus monkeys exposed in utero to denosumab exhibited bone abnormalities, reduced hematopoiesis, tooth malalignment, decreased neonatal growth, and an absence of axillary, inguinal, mandibular, and mesenteric lymph nodes. Some bone abnormalities recovered once exposure was ceased following birth; however, axillary and inguinal lymph nodes remained absent 6 months post-birth [see Use in Specific Populations (8.1)].
8.6 Renal Impairment
Two clinical trials were conducted in patients without cancer and with varying degrees of renal function.
In one study, patients (N = 55) with varying degrees of renal function (ranging from normal through end-stage renal disease requiring dialysis) received a single 60 mg subcutaneous dose of denosumab. In a second study, patients (N = 32) with severe renal dysfunction (creatinine clearance less than 30 mL/min and/or on dialysis) were given two 120 mg subcutaneous doses of denosumab. In both studies, greater risk of developing hypocalcemia was observed with increasing renal impairment, and with inadequate/no calcium supplementation. Hypocalcemia was mild to moderate in severity in 96% of patients. Monitor calcium levels and calcium and vitamin D intake [see Warnings and Precautions (5.3), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)].
12. Xgeva - Clinical Pharmacology
12.1 Mechanism of Action
Xgeva binds to RANKL, a transmembrane or soluble protein essential for the formation, function, and survival of osteoclasts, the cells responsible for bone resorption, thereby modulating calcium release from bone. Increased osteoclast activity, stimulated by RANKL, is a mediator of bone pathology in solid tumors with osseous metastases. Similarly, giant cell tumors of bone consist of stromal cells expressing RANKL and osteoclast-like giant cells expressing RANK receptor, and signaling through the RANK receptor contributes to osteolysis and tumor growth. Xgeva prevents RANKL from activating its receptor, RANK, on the surface of osteoclasts, their precursors, and osteoclast-like giant cells.
12.3 Pharmacokinetics
Following subcutaneous administration, bioavailability was 62%. Denosumab displayed nonlinear pharmacokinetics at doses below 60 mg, but approximately dose-proportional increases in exposure at higher doses.
With multiple subcutaneous doses of 120 mg once every 4 weeks, up to 2.8-fold accumulation in serum denosumab concentrations was observed and steady-state was achieved by 6 months. A mean (± standard deviation) serum steady-state trough concentration of 20.5 (± 13.5) mcg/mL was achieved by 6 months. The mean elimination half-life was 28 days.
In patients with newly diagnosed multiple myeloma who received 120 mg every 4 weeks, denosumab concentrations appear to reach steady-state by month 6. In patients with giant cell tumor of bone, after administration of subcutaneous doses of 120 mg once every 4 weeks with additional 120 mg doses on Days 8 and 15 of the first month of therapy, mean (± standard deviation) serum trough concentrations on Day 8, 15, and one month after the first dose were 19.0 (± 24.1), 31.6 (± 27.3), 36.4 (± 20.6) mcg/mL, respectively. Steady-state was achieved in 3 months after initiation of treatment with a mean serum trough concentration of 23.4 (± 12.1) mcg/mL.
Special Populations
Body Weight: A population pharmacokinetic analysis was performed to evaluate the effects of demographic characteristics. Denosumab clearance and volume of distribution were proportional to body weight. The steady-state exposure following repeat subcutaneous administration of 120 mg every 4 weeks to 45 kg and 120 kg subjects were, respectively, 48% higher and 46% lower than exposure of the typical 66 kg subject.
Age, Gender and Race: The pharmacokinetics of denosumab was not affected by age, gender, and race.
Pediatrics: In skeletally-mature adolescent patients (12 to 16 years of age) with giant cell tumor of bone (GCTB) who received 120 mg every 4 weeks with a 120 mg loading dose on Days 8 and 15, the pharmacokinetics of denosumab were comparable to those observed in adult patients with GCTB.
Hepatic Impairment: No clinical trials have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of denosumab.
Renal Impairment: In clinical trials of 87 patients with varying degrees of renal dysfunction, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics and pharmacodynamics of denosumab [see Use in Specific Populations (8.6)].
Drug Interactions
No formal drug-drug interaction trials have been conducted with Xgeva. There was no evidence that various anticancer treatments affected denosumab systemic exposure and pharmacodynamic effect. Serum denosumab concentrations at 1 and 3 months and reductions in the bone turnover marker uNTx/Cr (urinary N-terminal telopeptide corrected for creatinine) at 3 months were similar in patients with and without prior intravenous bisphosphonate therapy and were not altered by concomitant chemotherapy and/or hormone therapy.
13. Nonclinical Toxicology
13.2 Animal Toxicology and/or Pharmacology
Denosumab is an inhibitor of osteoclastic bone resorption via inhibition of RANKL.
Because the biological activity of denosumab in animals is specific to nonhuman primates, evaluation of genetically engineered (knockout) mice or use of other biological inhibitors of the RANK/RANKL pathway, OPG-Fc and RANK-Fc, provided additional information on the pharmacodynamic properties of denosumab. RANK/RANKL knockout mice exhibited absence of lymph node formation, as well as an absence of lactation due to inhibition of mammary gland maturation (lobulo-alveolar gland development during pregnancy). Neonatal RANK/RANKL knockout mice exhibited reduced bone growth and lack of tooth eruption. A corroborative study in 2-week-old rats given the RANKL inhibitor OPG-Fc also showed reduced bone growth, altered growth plates, and impaired tooth eruption. These changes were partially reversible in this model when dosing with the RANKL inhibitors was discontinued.
14. Clinical Studies
14.1 Bone Metastasis from Solid Tumors
The safety and efficacy of Xgeva for the prevention of skeletal-related events in patients with bone metastases from solid tumors was demonstrated in three international, randomized (1:1), double-blind, active-controlled, noninferiority trials comparing Xgeva with zoledronic acid. In all three trials, patients were randomized to receive 120 mg Xgeva subcutaneously every 4 weeks or 4 mg zoledronic acid intravenously (IV) every 4 weeks (dose adjusted for reduced renal function). Patients with creatinine clearance less than 30 mL/min were excluded. In each trial, the main outcome measure was demonstration of noninferiority of time to first skeletal-related event (SRE) as compared to zoledronic acid. Supportive outcome measures were superiority of time to first SRE and superiority of time to first and subsequent SRE; testing for these outcome measures occurred if the main outcome measure was statistically significant. An SRE was defined as any of the following: pathologic fracture, radiation therapy to bone, surgery to bone, or spinal cord compression.
Study 20050136 (NCT00321464) enrolled 2046 patients with advanced breast cancer and bone metastasis. Randomization was stratified by a history of prior SRE (yes or no), receipt of chemotherapy within 6 weeks prior to randomization (yes or no), prior oral bisphosphonate use (yes or no), and region (Japan or other countries). Forty percent of patients had a previous SRE, 40% received chemotherapy within 6 weeks prior to randomization, 5% received prior oral bisphosphonates, and 7% were enrolled from Japan. Median age was 57 years, 80% of patients were White, and 99% of patients were women. The median number of doses administered was 18 for denosumab and 17 for zoledronic acid.
Study 20050244 (NCT00330759) enrolled 1776 adults with solid tumors other than breast and castrate-resistant prostate cancer with bone metastasis and multiple myeloma. Randomization was stratified by previous SRE (yes or no), systemic anticancer therapy at time of randomization (yes or no), and tumor type (non-small cell lung cancer, myeloma, or other). Eighty-seven percent were receiving systemic anticancer therapy at the time of randomization, 52% had a previous SRE, 64% of patients were men, 87% were White, and the median age was 60 years. A total of 40% of patients had non-small cell lung cancer, 10% had multiple myeloma, 9% had renal cell carcinoma, and 6% had small cell lung cancer. Other tumor types each comprised less than 5% of the enrolled population. The median number of doses administered was 7 for both denosumab and zoledronic acid.
Study 20050103 (NCT00321620) enrolled 1901 men with castrate-resistant prostate cancer and bone metastasis. Randomization was stratified by previous SRE, PSA level (less than 10 ng/mL or 10 ng/mL or greater) and receipt of chemotherapy within 6 weeks prior to randomization (yes or no). Twenty-six percent of patients had a previous SRE, 15% of patients had PSA less than 10 ng/mL, and 14% received chemotherapy within 6 weeks prior to randomization. Median age was 71 years and 86% of patients were White. The median number of doses administered was 13 for denosumab and 11 for zoledronic acid.
Xgeva delayed the time to first SRE following randomization as compared to zoledronic acid in patients with breast or castrate-resistant prostate cancer (CRPC) with osseous metastases (Table 2). In patients with bone metastasis due to other solid tumors or lytic lesions due to multiple myeloma, Xgeva was noninferior to zoledronic acid in delaying the time to first SRE following randomization.
Overall survival and progression-free survival were similar between arms in all three trials.
| Study 20050136
Metastatic Breast Cancer | Study 20050244
Metastatic Solid Tumors or Multiple Myeloma | Study 20050103
Metastatic CRPCa |
|||||||||
| Xgeva
N = 1026 | Zoledronic Acid
N = 1020 | Xgeva
N = 886 | Zoledronic
Acid N = 890 | Xgeva
N = 950 | Zoledronic Acid
N = 951 |
||||||
| First On-study SRE | |||||||||||
| Number of Patients who had SREs (%) | 315 (30.7) | 372 (36.5) | 278 (31.4) | 323 (36.3) | 341 (35.9) | 386 (40.6) | |||||
| Components of First SRE | |||||||||||
| Radiation to Bone | 82 (8.0) | 119 (11.7) | 119 (13.4) | 144 (16.2) | 177 (18.6) | 203 (21.3) | |||||
| Pathological Fracture | 212 (20.7) | 238 (23.3) | 122 (13.8) | 139 (15.6) | 137 (14.4) | 143 (15.0) | |||||
| Surgery to Bone | 12 (1.2) | 8 (0.8) | 13 (1.5) | 19 (2.1) | 1 (0.1) | 4 (0.4) | |||||
| Spinal Cord Compression | 9 (0.9) | 7 (0.7) | 24 (2.7) | 21 (2.4) | 26 (2.7) | 36 (3.8) | |||||
| Median Time to SRE (months) | NRb | 26.4 | 20.5 | 16.3 | 20.7 | 17.1 | |||||
| Hazard Ratio (95% CI) | 0.82 (0.71, 0.95) | 0.84 (0.71, 0.98) | 0.82 (0.71, 0.95) | ||||||||
| Noninferiority p-value | < 0.001 | < 0.001 | < 0.001 | ||||||||
| Superiority p-valuec | 0.010 | 0.060 | 0.008 | ||||||||
| First and Subsequent SREd | |||||||||||
| Mean Number/Patient | 0.46 | 0.60 | 0.44 | 0.49 | 0.52 | 0.61 | |||||
| Rate Ratio (95% CI) | 0.77 (0.66, 0.89) | 0.90 (0.77, 1.04) | 0.82 (0.71, 0.94) | ||||||||
| Superiority p-valuee | 0.001 | 0.145 | 0.009 | ||||||||
| a CRPC = castrate-resistant prostate cancer. b NR = not reached. c Superiority testing performed only after denosumab demonstrated to be noninferior to zoledronic acid within trial. d All skeletal events postrandomization; new events defined by occurrence ≥ 21 days after preceding event. e Adjusted p-values are presented. |
|||||||||||
14.2 Multiple Myeloma
The efficacy of Xgeva for the prevention of skeletal-related events in newly diagnosed multiple myeloma patients with treatment through disease progression, was evaluated in Study 20090482 (NCT01345019), an international, randomized (1:1), double-blind, active-controlled, noninferiority trial comparing Xgeva with zoledronic acid. In this trial, patients were randomized to receive 120 mg Xgeva subcutaneously every 4 weeks or 4 mg zoledronic acid intravenously (IV) every 4 weeks (dose adjusted for reduced renal function). Patients with creatinine clearance less than 30 mL/min were excluded. In this trial, the main efficacy outcome measure was noninferiority of time to first skeletal-related event (SRE). Additional efficacy outcome measures were superiority of time to first SRE, time to first and subsequent SRE, and overall survival. An SRE was defined as any of the following: pathologic fracture, radiation therapy to bone, surgery to bone, or spinal cord compression.
Study 20090482 enrolled 1718 newly diagnosed multiple myeloma patients with bone lesions. Randomization was stratified by a history of prior SRE (yes or no), the anti-myeloma agent being utilized/planned to be utilized in first-line therapy (novel therapy-based or non-novel therapy-based [novel therapies include bortezomib, lenalidomide, or thalidomide]), intent to undergo autologous PBSC transplantation (yes or no), stage at diagnosis (International Staging System I or II or III) and region Japan (yes or no). At study enrollment, 96% of the patients were receiving or planning to receive novel therapy-based first-line anti-myeloma therapy, 55% of the patients intended to undergo autologous PBSC transplantation, 61% of patients had a previous SRE, 32% were at ISS stage I, 38% were at ISS stage II and 29% were at ISS Stage III, and 2% were enrolled from Japan. Median age was 63 years, 82% of patients were White, and 46% of patients were women. The median number of doses administered was 16 for Xgeva and 15 for zoledronic acid.
Xgeva was noninferior to zoledronic acid in delaying the time to first SRE following randomization (HR = 0.98, 95% CI, 0.85-1.14). The results for overall survival (OS) were comparable between Xgeva and zoledronic acid treatment groups with a hazard ratio of 0.90 (95% CI: 0.70, 1.16).
| Study 20090482
Multiple Myeloma |
||
| Xgeva
N = 859 | Zoledronic Acid
N = 859 |
|
| First On-study SRE | ||
| Number of Patients who had SREs (%) | 376 (43.8) | 383 (44.6) |
| Components of First SRE | ||
| Radiation to Bone | 47 (5.5) | 62 (7.2) |
| Pathological Fracture | 342 (39.8) | 338 (39.3) |
| Surgery to Bone | 37 (4.3) | 48 (5.6) |
| Spinal Cord Compression | 6 (0.7) | 4 (0.5) |
| Median Time to SRE (months) (95% CI) | 22.8 (14.7, NEa) | 24 (16.6, 33.3) |
| Hazard Ratio (95% CI) | 0.98 (0.85, 1.14) | |
| a NE = not estimable | ||
14.3 Giant Cell Tumor of Bone
The safety and efficacy of Xgeva for the treatment of giant cell tumor of bone in adults or skeletally mature adolescents were demonstrated in two open-label trials [Study 20040215 (NCT00396279) and Study 20062004 (NCT00680992)] that enrolled patients with histologically confirmed measurable giant cell tumor of bone that was either recurrent, unresectable, or for which planned surgery was likely to result in severe morbidity. Patients received 120 mg Xgeva subcutaneously every 4 weeks with a loading dose on Days 8 and 15 of the first cycle of therapy. Patients who discontinued Xgeva then entered the safety follow-up phase for a minimum of 60 months. Retreatment with Xgeva while in safety follow-up was allowed for patients who initially demonstrated a response to Xgeva (e.g., in the case of recurrent disease).
Study 20040215 was a single-arm, pharmacodynamic, and proof of concept trial conducted in 37 adult patients with unresectable or recurrent giant cell tumor of bone. Patients were required to have histologically confirmed giant cell tumor of bone and radiologic evidence of measurable disease from a computed tomography (CT) or magnetic resonance imaging (MRI) obtained within 28 days prior to study enrollment. Patients enrolled in Study 20040215 underwent CT or MRI assessment of giant cell tumor of bone at baseline and quarterly during Xgeva treatment.
Study 20062004 was a parallel-cohort, proof of concept, and safety trial conducted in 535 adult or skeletally mature adolescent patients with histologically confirmed giant cell tumor of bone and evidence of measurable active disease. Study 20062004 enrolled 19 patients who were 12-16 years of age [see Use in Specific Populations (8.4)]. Patients enrolled into one of three cohorts: Cohort 1 enrolled 268 patients with surgically unsalvageable disease (e.g., sacral or spinal sites of disease, or pulmonary metastases); Cohort 2 enrolled 252 patients with surgically salvageable disease where the investigator determined that the planned surgery was likely to result in severe morbidity (e.g., joint resection, limb amputation, or hemipelvectomy); Cohort 3 enrolled 15 patients who previously participated in Study 20040215. Patients underwent imaging assessment of disease status at intervals determined by their treating physician.
A retrospective interim analysis concluded by an independent review committee evaluated objective response in 187 patients enrolled and treated in Study 20040215 and Study 20062004 for whom baseline and at least one post-baseline radiographic assessment were available (27 of 37 patients enrolled in Study 20040215 and 160 of 270 patients enrolled in Cohorts 1 and 2 of Study 20062004). The primary efficacy outcome measure was objective response rate using Response Evaluation Criteria in Solid Tumors (RECIST) v 1.1.
The overall objective response rate (RECIST 1.1) was 25% (95% CI: 19, 32). All responses were partial responses. The estimated median time to response was 3 months. In the 47 patients with an objective response, the median duration of follow-up was 20 months (range: 2-44 months), and 51% (24/47) had a duration of response lasting at least 8 months. Three patients experienced disease progression following an objective response.
14.4 Hypercalcemia of Malignancy
The safety and efficacy of Xgeva was demonstrated in an open-label, single-arm trial [Study 20070315 (NCT00896454)] that enrolled 33 patients with hypercalcemia of malignancy (with or without bone metastases) refractory to treatment with intravenous bisphosphonate therapy. Patients received Xgeva subcutaneously every 4 weeks with additional 120 mg doses on Days 8 and 15 of the first month of therapy.
In this trial, refractory hypercalcemia of malignancy was defined as an albumin-corrected calcium of > 12.5 mg/dL (3.1 mmol/L) despite treatment with intravenous bisphosphonate therapy in 7-30 days prior to initiation of Xgeva therapy. The primary outcome measure was the proportion of patients achieving a response, defined as corrected serum calcium (CSC) ≤ 11.5 mg/dL (2.9 mmol/L), within 10 days after Xgeva administration. Efficacy data are summarized in Figure 1 and Table 4. Concurrent chemotherapy did not appear to affect response to Xgeva.
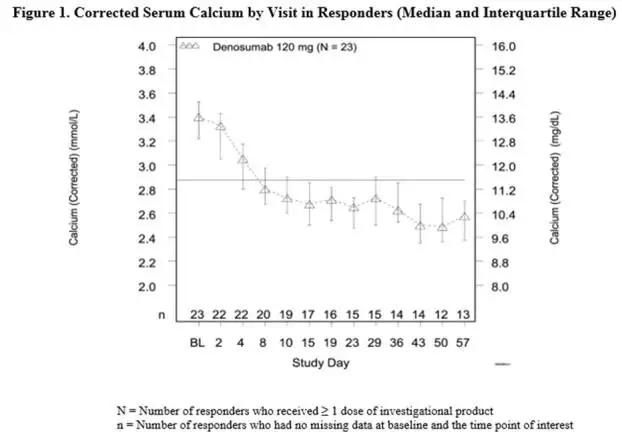
| N = 33 | Proportion (%)
(95% CI) |
|
| All Responders (CSC ≤ 11.5 mg/dL) by Day 10 | 21 | 63.6 |
| (45.1, 79.6) | ||
| All Responders by Day 57 | 23 | 69.7 |
| (51.3, 84.4) | ||
| Complete Responders (CSC ≤ 10.8 mg/dL) by Day 10 | 12 | 36.4 |
| (20.4, 54.9) | ||
| All Complete Responders by Day 57 | 21 | 63.6 |
| (45.1, 79.6) |
Median time to response (CSC ≤ 11.5 mg/dL) was 9 days (95% CI: 8, 19), and the median duration of response was 104 days (95% CI: 7, not estimable). Median time to complete response (CSC ≤ 10.8 mg/dL) was 23 days (95% CI: 9, 36), and the median duration of complete response was 34 days (95% CI: 1, 134).
16. How is Xgeva supplied
Xgeva is supplied in a single-dose vial.
| 120 mg/1.7 mL | 1 vial per carton | NDC 55513-730-01 |
Store Xgeva in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton. Do not freeze. Once removed from the refrigerator, Xgeva must not be exposed to temperatures above 25°C/77°F or direct light and must be used within 14 days. Discard Xgeva if not used within the 14 days. Do not use Xgeva after the expiry date printed on the label.
Protect Xgeva from direct light and heat.
Avoid vigorous shaking of Xgeva.
17. Patient Counseling Information
Drug Products with Same Active Ingredient
Advise patients that denosumab is also marketed as Prolia, and if taking Xgeva, they should not receive Prolia [see Warnings and Precautions (5.1)].
Hypersensitivity
Advise patients to seek prompt medical attention if signs or symptoms of hypersensitivity reactions occur. Advise patients who have had signs or symptoms of systemic hypersensitivity reactions that they should not receive denosumab (Xgeva or Prolia) [see Warnings and Precautions (5.2) and Contraindications (4.2)].
Hypocalcemia
Adequately supplement patients with calcium and vitamin D and instruct them on the importance of maintaining serum calcium levels while receiving Xgeva [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)]. Advise patients to seek prompt medical attention if they develop signs or symptoms of hypocalcemia.
Osteonecrosis of the Jaw
Advise patients to maintain good oral hygiene during treatment with Xgeva and to inform their dentist prior to dental procedures that they are receiving Xgeva. Patients should avoid invasive dental procedures during treatment with Xgeva and inform their healthcare provider or dentist if they experience persistent pain and/or slow healing of the mouth or jaw after dental surgery [see Warnings and Precautions (5.4)].
Atypical Subtrochanteric and Diaphyseal Femoral Fracture
Advise patients to report new or unusual thigh, hip, or groin pain [see Warnings and Precautions (5.5)].
Hypercalcemia Following Treatment Discontinuation in Patients with Giant Cell Tumor of Bone and in Patients with Growing Skeletons
Advise patients to report nausea, vomiting, headache, and decreased alertness following treatment discontinuation [see Warnings and Precautions (5.6) and Use in Specific Populations (8.4)].
Multiple Vertebral Fractures (MVF) Following Treatment Discontinuation
Advise patients that after treatment with Xgeva is stopped there may be an increased risk of having broken bones in the spine especially in patients who have had a fracture or who have had osteoporosis. Advise patients not to interrupt Xgeva therapy without their physician’s advice [see Warnings and Precautions (5.7)].
Embryo-Fetal Toxicity
Advise females of reproductive potential that Xgeva can cause harm to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.8) and Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to use effective contraception during treatment and for at least 5 months after the last dose of Xgeva [see Use in Specific Populations (8.3)].
Xgeva® (denosumab)
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
U.S. License No. 1080
Patent: http://pat.amgen.com/xgeva/
© 2010-2020 Amgen Inc. All rights reserved.
1xxxxx - v20

| XGEVA
denosumab injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Amgen Inc (039976196) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Amgen Manufacturing Ltd | 785800020 | ANALYSIS(55513-730) , MANUFACTURE(55513-730) , PACK(55513-730) , LABEL(55513-730) | |
