Drug Detail:Zortress (Everolimus (zortress) [ e-ver-oh-li-mus ])
Drug Class: MTOR inhibitors Selective immunosuppressants
Highlights of Prescribing Information
ZORTRESS (everolimus) tablets, for oral use
Initial U.S. Approval: 2009
WARNING: MALIGNANCIES AND SERIOUS INFECTIONS, KIDNEY GRAFT THROMBOSIS; NEPHROTOXICITY; AND MORTALITY IN HEART TRANSPLANTATION
See full prescribing information for complete boxed warning.
-
Only physicians experienced in immunosuppressive therapy and management of transplant patients should use Zortress (5.1)
-
Increased susceptibility to infection and the possible development of malignancies may result from immunosuppression (5.2, 5.3)
-
Increased incidence of kidney graft thrombosis (5.4)
-
Reduced doses of cyclosporine are required for use in combination with Zortress in order to reduce nephrotoxicity (2.4, 2.5, 5.6, 12.7, 12.8)
- Increased mortality in a heart transplant clinical trial. Use in heart transplantation is not recommended (5.7)
Indications and Usage for Zortress
• Zortress is an mTOR inhibitor immunosuppressant indicated for the prophylaxis of organ rejection in adult patients:
• Kidney Transplant: at low-moderate immunologic risk. Use in combination with basiliximab, cyclosporine (reduced doses) and corticosteroids (1.1)
• Liver Transplant: Administer no earlier than 30 days posttransplant. Use in combination with tacrolimus (reduced doses) and corticosteroids (1.2, 5.5)
Limitations of Use (1.3)
Safety and efficacy have not been established in the following:
• Kidney transplant patients at high immunologic risk (1.3)
• Recipients of transplanted organs other than kidney or liver (1.3, 5.7)
• Pediatric patients (less than 18 years) (1.3)
Zortress Dosage and Administration
• Kidney Transplantation: starting oral dose of 0.75 mg twice daily as soon as possible after transplantation (2.1)
• Liver Transplantation: starting oral dose of 1 mg twice daily starting 30 days after transplantation (2.2)
• Monitor everolimus Concentrations: Adjust maintenance dose to achieve trough concentrations within the 3 to 8 ng/mL target range using LC/MS/MS assay method (2.1, 2.2, 2.3)
• Administer consistently with or without food at the same time as cyclosporine or tacrolimus (2.6, 12.3)
• Mild Hepatic Impairment: Reduce initial daily dose by one-third (2.7)
• Moderate or Severe Hepatic Impairment: Reduce initial daily dose by one-half (2.7, 12.6)
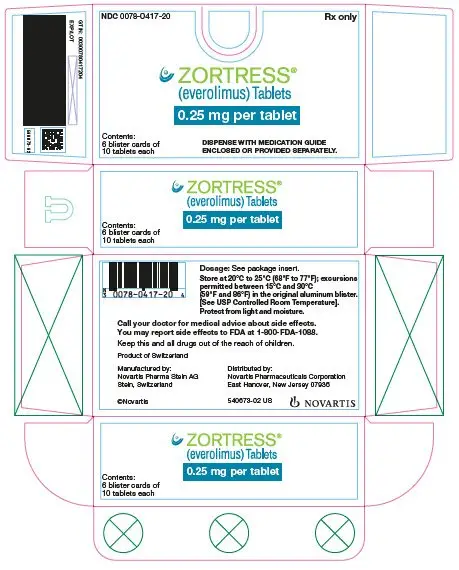
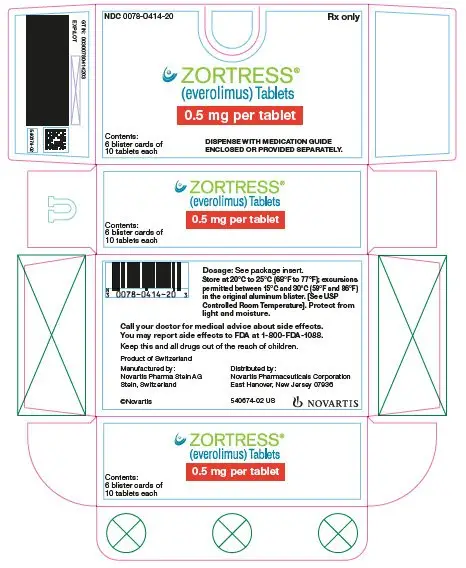
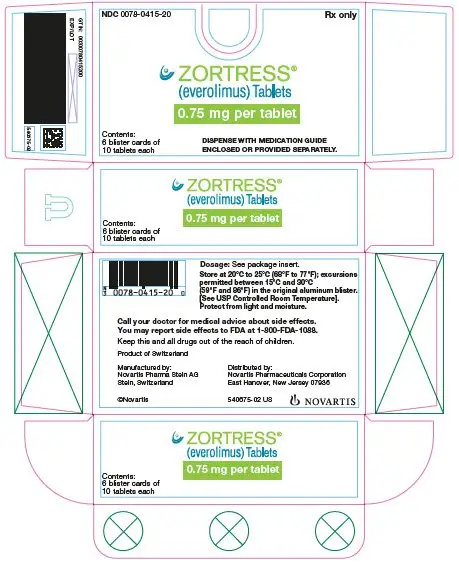
Dosage Forms and Strengths
Zortress is available as 0.25 mg, 0.5 mg, 0.75 mg, and 1 mg tablets (3)
Contraindications
- Hypersensitivity to everolimus, sirolimus, or to components of the drug product (4)
Warnings and Precautions
• Angioedema [increased risk with concomitant angiotensin converting enzyme (ACE inhibitors)]: Monitor for symptoms and treat promptly (5.8)
• Delayed Wound Healing/Fluid Accumulation: Monitor symptoms; treat promptly to minimize complications (5.9)
• Interstitial Lung Disease (ILD)/Non-Infectious Pneumonitis: Monitor for symptoms or radiologic changes; manage by dose reduction or discontinuation until symptoms resolve; consider use of corticosteroids (5.10)
• Hyperlipidemia (elevations of serum cholesterol and triglycerides): Monitor and consider anti-lipid therapy (5.11)
• Proteinuria (increased risk with higher trough concentrations): Monitor urine protein (5.12)
• Polyoma Virus Infections (activation of latent viral infections; BK-virus associated nephropathy): Consider reducing immunosuppression (5.13)
• TMA/TTP/HUS (concomitant use with cyclosporine may increase risk): Monitor for hematological changes or symptoms (5.15)
• New Onset Diabetes After Transplantation: Monitor serum glucose (5.16)
• Male Infertility: Azospermia or oligospermia may occur (5.18, 13.1)
• Immunizations: Avoid live vaccines (5.19)
• Embryo-Fetal Toxicity: Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment with Zortress and for 8 weeks after final dose (5.17, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions were as follows:
- Kidney Transplantation (incidence greater than or equal to 20%): peripheral edema, constipation, hypertension, nausea, anemia, UTI, and hyperlipidemia (6.1)
- Liver Transplantation (incidence greater than 10%): diarrhea, headache, peripheral edema, hypertension, nausea, pyrexia, abdominal pain, leukopenia, and hypercholesterolemia (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Strong-moderate CYP3A4 inhibitors (e.g., cyclosporine, ketoconazole, erythromycin, verapamil) and CYP3A4 inducers (e.g., rifampin) may affect everolimus concentrations (7.1). Consider Zortress dose adjustment (5.14)
Use In Specific Populations
- Pregnancy: Based on animal data may cause maternal and fetal harm (8.1)
- Lactation: Breastfeeding not recommended (8.2)
- Females and Males of Reproductive Potential: May impair fertility (8.1, 8.3, 13.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2021
Full Prescribing Information
1. Indications and Usage for Zortress
1.1 Prophylaxis of Organ Rejection in Kidney Transplantation
Zortress is indicated for the prophylaxis of organ rejection in adult patients at low-moderate immunologic risk receiving a kidney transplant [see Clinical Studies (14.1)]. Zortress is to be administered in combination with basiliximab induction and concurrently with reduced doses of cyclosporine and with corticosteroids. Therapeutic drug monitoring (TDM) of everolimus and cyclosporine is recommended for all patients receiving these products [see Dosage and Administration (2.2, 2.3)].
1.2 Prophylaxis of Organ Rejection in Liver Transplantation
Zortress is indicated for the prophylaxis of allograft rejection in adult patients receiving a liver transplant. Zortress is to be administered no earlier than 30 days posttransplant concurrently in combination with reduced doses of tacrolimus and with corticosteroids [see Warnings and Precautions (5.5), Clinical Studies (14.2)]. TDM of everolimus and tacrolimus is recommended for all patients receiving these products [see Dosage and Administration (2.3, 2.5)].
1.3 Limitations of Use
The safety and efficacy of Zortress has not been established in the following populations:
- Kidney transplant patients at high immunologic risk
- Recipients of transplanted organs other than kidney and liver [see Warnings and Precautions (5.7)]
- Pediatric patients (less than 18 years).
2. Zortress Dosage and Administration
Patients receiving Zortress may require dose adjustments based on everolimus blood concentrations achieved, tolerability, individual response, change in concomitant medications and the clinical situation. Optimally, dose adjustments of Zortress should be based on trough concentrations obtained 4 or 5 days after a previous dosing change. Dose adjustment is required if the trough concentration is below 3 ng/mL. The total daily dose of Zortress should be doubled using the available tablet strengths (0.25 mg, 0.5 mg, 0.75 mg, or 1 mg). Dose adjustment is also required if the trough concentration is greater than 8 ng/mL on 2 consecutive measures; the dose of Zortress should be decreased by 0.25 mg twice daily [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
2.1 Dosage in Adult Kidney Transplant Patients
An initial Zortress dose of 0.75 mg orally twice daily (1.5 mg per day) is recommended for adult kidney transplant patients in combination with reduced dose cyclosporine, administered as soon as possible after transplantation [see Dosage and Administration (2.3, 2.4), Clinical Studies (14.1)].
Oral prednisone should be initiated once oral medication is tolerated. Steroid doses may be further tapered on an individualized basis depending on the clinical status of patient and function of graft.
2.2 Dosage in Adult Liver Transplant Patients
Start Zortress at least 30 days posttransplant. An initial dose of 1 mg orally twice daily (2 mg per day) is recommended for adult liver transplant patients in combination with reduced dose tacrolimus [see Dosage and Administration (2.3, 2.5), Clinical Studies (14.2)].
Steroid doses may be further tapered on an individualized basis depending on the clinical status of patient and function of graft.
2.3 Therapeutic Drug-Monitoring (TDM) - Everolimus
Routine everolimus whole blood therapeutic drug concentration monitoring is recommended for all patients. The recommended everolimus therapeutic range is 3 to 8 ng/mL [see Clinical Pharmacology (12.7)]. Careful attention should be made to clinical signs and symptoms, tissue biopsies, and laboratory parameters. It is important to monitor everolimus blood concentrations, in patients with hepatic impairment, during concomitant administration of CYP3A4 inducers or inhibitors, when switching cyclosporine formulations and/or when cyclosporine dosing is reduced according to recommended target concentrations [see Clinical Pharmacology (12.7, 12.8)].
There is an interaction of cyclosporine on everolimus, and consequently, everolimus concentrations may decrease if cyclosporine exposure is reduced. There is little to no pharmacokinetic interaction of tacrolimus on everolimus, and thus, everolimus concentrations do not decrease if the tacrolimus exposure is reduced [see Drug Interactions (7.2)].
The everolimus recommended therapeutic range of 3 to 8 ng/mL is based on an LC/MS/MS assay method. Currently in clinical practice, everolimus whole blood trough concentrations may be measured by chromatographic or immunoassay methodologies. Because the measured everolimus whole blood trough concentrations depend on the assay used, individual patient sample concentration values from different assays may not be interchangeable. Consideration of assay results must be made with knowledge of the specific assay used. Therefore, communication should be maintained with the laboratory performing the assay.
2.4 Therapeutic Drug-Monitoring (TDM) - Cyclosporine in Kidney Transplant Patients
Both cyclosporine doses and the target range for whole blood trough concentrations should be reduced, when given in a regimen with Zortress, in order to minimize the risk of nephrotoxicity [see Warnings and Precautions (5.6), Drug Interactions (7.2), Clinical Pharmacology (12.8)].
The recommended cyclosporine therapeutic ranges when administered with Zortress are 100 to 200 ng/mL through Month 1 posttransplant, 75 to 150 ng/mL at Months 2 and 3 posttransplant, 50 to 100 ng/mL at Month 4 posttransplant, and 25 to 50 ng/mL from Month 6 through Month 12 posttransplant. The median trough concentrations observed in the clinical trial ranged between 161 to 185 ng/mL through Month 1 posttransplant and between 111 to 140 ng/mL at Months 2 and 3 posttransplant. The median trough concentration was 99 ng/mL at Month 4 posttransplant and ranged between 46 to 75 ng/mL from Months 6 through Month 12 posttransplant [see Clinical Pharmacology (12.8), Clinical Studies (14.1)].
Cyclosporine, USP Modified is to be administered as oral capsules twice daily unless cyclosporine oral solution or intravenous administration of cyclosporine cannot be avoided. Cyclosporine, USP Modified should be initiated as soon as possible, and no later than 48 hours after reperfusion of the graft and dose adjusted to target concentrations from Day 5 onwards.
If impairment of renal function is progressive, the treatment regimen should be adjusted. In renal transplant patients, the cyclosporine dose should be based on cyclosporine whole blood trough concentrations [see Clinical Pharmacology (12.8)].
In renal transplantation, there are limited data regarding dosing Zortress with reduced cyclosporine trough concentrations of 25 to 50 ng/mL after 12 months. Zortress has not been evaluated in clinical trials with other formulations of cyclosporine. Prior to dose reduction of cyclosporine, it should be ascertained that steady state everolimus whole blood trough concentration is at least 3 ng/mL. There is an interaction of cyclosporine on everolimus, and consequently, everolimus concentrations may decrease if cyclosporine exposure is reduced [see Drug Interactions (7.2)].
2.5 Therapeutic Drug Monitoring (TDM) - Tacrolimus in Liver Transplant Patients
Both tacrolimus doses and the target range for whole blood trough concentrations should be reduced, when given in a regimen with Zortress, in order to minimize the potential risk of nephrotoxicity [see Warnings and Precautions (5.6), Clinical Pharmacology (12.9)].
The recommended tacrolimus therapeutic range when administered with Zortress are whole blood trough (C-0h) concentrations of 3 to 5 ng/mL by three weeks after the first dose of Zortress (approximately Month 2) and through Month 12 posttransplant.
The median tacrolimus trough concentrations observed in the clinical trial ranged between 8.6 to 9.5 ng/mL at Weeks 2 and 4 posttransplant (prior to initiation of everolimus). The median tacrolimus trough concentrations ranged between 7 to 8.1 ng/mL at Weeks 5 and 6 posttransplant, between 5.2 to 5.6 ng/mL at Months 2 and 3 posttransplant, and between 4.3 to 4.9 ng/mL between Months 4 and 12 posttransplant [see Clinical Pharmacology (12.9), Clinical Studies (14.2)].
Tacrolimus is to be administered as oral capsules twice daily unless intravenous administration of tacrolimus cannot be avoided.
In liver transplant patients, the tacrolimus dose should be based on tacrolimus whole blood trough concentrations [see Clinical Pharmacology (12.9)].
In liver transplantation, there are limited data regarding dosing Zortress with reduced tacrolimus trough concentrations of 3 to 5 ng/mL after 12 months. Prior to dose reduction of tacrolimus it should be ascertained that the steady-state everolimus whole blood trough concentration is at least 3 ng/mL. Unlike the interaction between cyclosporine and everolimus, tacrolimus does not affect everolimus trough concentrations, and consequently, everolimus concentrations do not decrease if the tacrolimus exposure is reduced.
2.6 Administration
Zortress tablets should be swallowed whole with a glass of water and not crushed before use.
Administer Zortress consistently approximately 12 hours apart with or without food to minimize variability in absorption and at the same time as cyclosporine or tacrolimus [see Clinical Pharmacology (12.3)].
2.7 Hepatic Impairment
Whole blood trough concentrations of everolimus should be closely monitored in patients with impaired hepatic function. For patients with mild hepatic impairment (Child-Pugh Class A), the initial daily dose should be reduced by approximately one-third of the normally recommended daily dose. For patients with moderate or severe hepatic impairment (Child-Pugh B or C), the initial daily dose should be reduced to approximately one-half of the normally recommended daily dose. Further dose adjustment and/or dose titration should be made if a patient’s whole blood trough concentration of everolimus, as measured by an LC/MS/MS assay, is not within the target trough concentration range of 3 to 8 ng/mL [see Clinical Pharmacology (12.6)].
3. Dosage Forms and Strengths
Zortress is available as 0.25 mg, 0.5 mg, 0.75 mg, and 1 mg tablets.
| Dosage Strength | 0.25 mg | 0.5 mg | 0.75 mg | 1 mg |
| Appearance | White to yellowish, marbled, round, flat tablets with bevelled edge | |||
| Imprint | “C” on one side and “NVR” on the other | “CH” on one side and “NVR” on the other | “CL” on one side and “NVR” on the other | “CU” on one side and “NVR” on the other |
5. Warnings and Precautions
5.1 Management of Immunosuppression
Only physicians experienced in management of systemic immunosuppressant therapy in transplantation should prescribe Zortress. Patients receiving the drug should be managed in facilities equipped and staffed with adequate laboratory and supportive medical resources. The physician responsible for the maintenance therapy should have complete information requisite for the follow-up of the patient. In limited data with the complete elimination of calcineurin inhibition (CNI), there was an increased risk of acute rejection.
5.2 Lymphomas and Other Malignancies
Patients receiving immunosuppressants, including Zortress, are at increased risk of developing lymphomas and other malignancies, particularly of the skin. The risk appears to be related to the intensity and duration of immunosuppression rather than to the use of any specific agent.
As usual for patients with increased risk for skin cancer, exposure to sunlight and ultraviolet light should be limited by wearing protective clothing and using a sunscreen with a high protection factor.
5.3 Serious Infections
Patients receiving immunosuppressants, including Zortress, are at increased risk of developing bacterial, viral, fungal, and protozoal infections, including opportunistic infections [see Warnings and Precautions (5.13), Adverse Reactions (6.1, 6.2)]. These infections may lead to serious, including fatal, outcomes. Because of the danger of over-immunosuppression, which can cause increased susceptibility to infection, combination immunosuppressant therapy should be used with caution.
Antimicrobial prophylaxis for Pneumocystis jiroveci (carinii) pneumonia and prophylaxis for cytomegalovirus (CMV) is recommended in transplant recipients.
5.4 Kidney Graft Thrombosis
An increased risk of kidney arterial and venous thrombosis, resulting in graft loss, has been reported, usually within the first 30 days posttransplantation [see Boxed Warning].
5.5 Hepatic Artery Thrombosis
Mammalian target of rapamycin (mTOR) inhibitors are associated with an increase in hepatic artery thrombosis (HAT). Reported cases mostly have occurred within the first 30 days posttransplant and most also lead to graft loss or death. Therefore, Zortress should not be administered earlier than 30 days after liver transplant.
5.6 Zortress and Calcineurin Inhibitor-Induced Nephrotoxicity
In kidney transplant recipients, Zortress with standard dose cyclosporine increases the risk of nephrotoxicity resulting in a lower glomerular filtration rate. Reduced doses of cyclosporine are required for use in combination with Zortress in order to reduce renal dysfunction [see Boxed Warning, Indications and Usage (1.1), Clinical Pharmacology (12.8)].
In liver transplant recipients, Zortress has not been studied with standard dose tacrolimus. Reduced doses of tacrolimus should be used in combination with Zortress in order to minimize the potential risk of nephrotoxicity [see Indications and Usage (1.2), Clinical Pharmacology (12.9)].
Renal function should be monitored during the administration of Zortress. Consider switching to other immunosuppressive therapies if renal function does not improve after dose adjustments or if the dysfunction is thought to be drug related. Caution should be exercised when using other drugs which are known to impair renal function.
5.7 Heart Transplantation
In a clinical trial of de novo heart transplant patients, Zortress in an immunosuppressive regimen with or without induction therapy, resulted in an increased mortality often associated with serious infections within the first three months posttransplantation compared to the control regimen. Use of Zortress in heart transplantation is not recommended.
5.8 Angioedema
Zortress has been associated with the development of angioedema. The concomitant use of Zortress with other drugs known to cause angioedema, such as angiotensin converting enzyme (ACE) inhibitors may increase the risk of developing angioedema.
5.9 Wound Healing and Fluid Accumulation
Zortress increases the risk of delayed wound healing and increases the occurrence of wound-related complications like wound dehiscence, wound infection, incisional hernia, lymphocele and seroma. These wound-related complications may require more surgical intervention. Generalized fluid accumulation, including peripheral edema (e.g., lymphoedema) and other types of localized fluid collection, such as pericardial and pleural effusions and ascites have also been reported.
5.10 Interstitial Lung Disease (ILD)/Non-Infectious Pneumonitis
A diagnosis of interstitial lung disease (ILD) should be considered in patients presenting with symptoms consistent with infectious pneumonia but not responding to antibiotic therapy and in whom infectious, neoplastic and other non-drug causes have been ruled out through appropriate investigations. Cases of ILD, implying lung intraparenchymal inflammation (pneumonitis) and/or fibrosis of non-infectious etiology, some reported with pulmonary hypertension [including pulmonary arterial hypertension (PAH)] as a secondary event, have occurred in patients receiving rapamycins and their derivatives, including Zortress. Most cases generally resolve on drug interruption with or without glucocorticoid therapy. However, fatal cases have also occurred.
5.11 Hyperlipidemia
Increased serum cholesterol and triglycerides, requiring the need for anti-lipid therapy, have been reported to occur following initiation of Zortress and the risk of hyperlipidemia is increased with higher everolimus whole blood trough concentrations [see Adverse Reactions (6.2)]. Use of anti-lipid therapy may not normalize lipid levels in patients receiving Zortress.
Any patient who is administered Zortress should be monitored for hyperlipidemia. If detected, interventions, such as diet, exercise, and lipid-lowering agents should be initiated as outlined by the National Cholesterol Education Program guidelines. The risk/benefit should be considered in patients with established hyperlipidemia before initiating an immunosuppressive regimen containing Zortress. Similarly, the risk/benefit of continued Zortress therapy should be reevaluated in patients with severe refractory hyperlipidemia. Zortress has not been studied in patients with baseline cholesterol levels greater than 350 mg/dL.
Due to an interaction with cyclosporine, clinical trials of Zortress and cyclosporine in kidney transplant patients strongly discouraged patients from receiving the HMG-CoA reductase inhibitors simvastatin and lovastatin. During Zortress therapy with cyclosporine, patients administered an HMG-CoA reductase inhibitor and/or fibrate should be monitored for the possible development of rhabdomyolysis and other adverse effects, as described in the respective labeling for these agents [see Drug Interactions (7.7)].
5.12 Proteinuria
The use of Zortress in transplant patients has been associated with increased proteinuria. The risk of proteinuria increased with higher everolimus whole blood trough concentrations. Patients receiving Zortress should be monitored for proteinuria [see Adverse Reactions (6.2)].
5.13 Polyoma Virus Infections
Patients receiving immunosuppressants, including Zortress, are at increased risk for opportunistic infections; including polyoma virus infections. Polyoma virus infections in transplant patients may have serious, and sometimes fatal, outcomes. These include polyoma virus-associated nephropathy (PVAN), mostly due to BK virus infection, and JC virus associated progressive multiple leukoencephalopathy (PML). PVAN has been observed in patients receiving immunosuppressants, including Zortress. PVAN is associated with serious outcomes; including deteriorating renal function and kidney graft loss [see Adverse Reactions (6.2)]. Patient monitoring may help detect patients at risk for PVAN. Reductions in immunosuppression should be considered for patients who develop evidence of PVAN or PML. Physicians should also consider the risk that reduced immunosuppression represents to the functioning allograft.
5.14 Interaction with Strong Inhibitors and Inducers of CYP3A4
Coadministration of Zortress with strong CYP3A4-inhibitors (e.g., ketoconazole, itraconazole, voriconazole, clarithromycin, telithromycin, ritonavir, boceprevir, telaprevir) and strong CYP3A4 inducers (e.g., rifampin, rifabutin) is not recommended without close monitoring of everolimus whole blood trough concentrations [see Drug Interactions (7)].
5.15 Thrombotic Microangiopathy/Thrombotic Thrombocytopenic Purpura/Hemolytic Uremic Syndrome
The concomitant use of Zortress with cyclosporine may increase the risk of thrombotic microangiopathy (TMA)/thrombotic thrombocytopenic purpura (TTP)/hemolytic uremic syndrome (HUS). Monitor hematologic parameters [see Adverse Reactions (6.2)].
5.16 New Onset Diabetes after Transplant
Zortress has been shown to increase the risk of new onset diabetes mellitus after transplant. Blood glucose concentrations should be monitored closely in patients using Zortress.
5.17 Embryo-Fetal Toxicity
Based on animal studies and the mechanism of action [see Clinical Pharmacology (12.1)], Zortress may cause fetal harm when administered to a pregnant woman. In animal studies, everolimus caused embryo-fetal toxicity when administered during the period of organogenesis at maternal exposures that were equal to or less than human exposures at the recommended lowest starting dose. Advise pregnant women of the potential risk to a fetus. Advise female patients of reproductive potential to avoid becoming pregnant and to use effective contraception while using Zortress and for 8 weeks after ending treatment [see Use in Specific Populations (8.1, 8.3)].
5.18 Male Infertility
Azospermia or oligospermia may be observed [see Adverse Reactions (6.2), Nonclinical Toxicology (13.1)]. Zortress is an anti-proliferative drug and affects rapidly dividing cells like the germ cells.
6. Adverse Reactions/Side Effects
6.1 Serious and Otherwise Important Adverse Reactions
The following adverse reactions are discussed in greater detail in other sections of the label.
- Hypersensitivity Reactions [see Contraindications (4.1)]
- Lymphomas and Other Malignancies [see Boxed Warning, Warnings and Precautions (5.2)]
- Serious Infections [see Warnings and Precautions (5.3)]
- Kidney Graft Thrombosis [see Warnings and Precautions (5.4)]
- Hepatic Artery Thrombosis [see Warnings and Precautions (5.5)]
- Zortress and Calcineurin Inhibitor-Induced Nephrotoxicity [see Warnings and Precautions (5.6)]
- Heart Transplantation [see Warnings and Precautions (5.7)]
- Angioedema [see Warnings and Precautions (5.8)]
- Wound Healing and Fluid Accumulation [see Warnings and Precautions (5.9)]
- Interstitial Lung Disease/Non-Infectious Pneumonitis [see Warnings and Precautions (5.10)]
- Hyperlipidemia [see Warnings and Precautions (5.11)]
- Proteinuria [see Warnings and Precautions (5.12)]
- Polyoma Virus Infections [see Warnings and Precautions (5.13)]
- Thrombotic Microangiopathy/Thrombotic Thrombocytopenic Purpura/Hemolytic Uremic Syndrome (TMA/TTP/HUS) [see Warnings and Precautions (5.15)]
- New Onset Diabetes After Transplant [see Warnings and Precautions (5.16)]
- Male Infertility [see Warnings and Precautions (5.18)]
7. Drug Interactions
7.1 Interactions with Strong Inhibitors or Inducers of CYP3A4 and P-glycoprotein
Everolimus is mainly metabolized by CYP3A4 in the liver and to some extent in the intestinal wall and is a substrate for the multidrug efflux pump, P-glycoprotein (P-gp). Therefore, absorption and subsequent elimination of systemically absorbed everolimus may be influenced by medicinal products that affect CYP3A4 and/or P-gp. Concurrent treatment with strong inhibitors (e.g., ketoconazole, itraconazole, voriconazole, clarithromycin, telithromycin, ritonavir, boceprevir, telaprevir) and inducers (e.g., rifampin, rifabutin) of CYP3A4 is not recommended. Inhibitors of P-gp (e.g., digoxin, cyclosporine) may decrease the efflux of everolimus from intestinal cells and increase everolimus blood concentrations. In vitro, everolimus was a competitive inhibitor of CYP3A4 and of CYP2D6, potentially increasing the concentrations of medicinal products eliminated by these enzymes. Thus, caution should be exercised when coadministering Zortress with CYP3A4 and CYP2D6 substrates with a narrow therapeutic index [see Dosage and Administration (2.3)].
All in vivo interaction studies were conducted without concomitant cyclosporine. Pharmacokinetic interactions between Zortress and concomitantly administered drugs are discussed below. Drug interaction studies have not been conducted with drugs other than those described below.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on animal studies and the mechanism of action [see Clinical Pharmacology (12.1)], Zortress can cause fetal harm when administered to a pregnant woman. There are limited case reports of Zortress use in pregnant women; however, these reports are insufficient to inform a drug associated risk of adverse developmental outcomes. Reproductive studies in animals have demonstrated that everolimus was maternally toxic in rabbits and caused embryo-fetal toxicities in rats and rabbits, at exposures near or below those achieved in human transplant patients. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, in the U.S. general population, the estimated background risk of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
Everolimus crossed the placenta and was toxic to the conceptus.
Everolimus administered daily to pregnant rats by oral gavage at 0.1 mg/kg (approximately one tenth the exposure in humans administered the lowest starting dose of 0.75 mg twice daily), from before mating through organogenesis, resulted in increased preimplantation loss and embryonic resorptions. These effects occurred in the absence of maternal toxicities.
Everolimus administered daily by oral gavage to pregnant rabbits during organogenesis resulted in abortions, maternal toxicity and lethality, and increased fetal resorptions. At these doses, exposure to everolimus (AUC) was approximately one-tenth, one-half, and one and one-half fold the exposures in humans administered the starting clinical dose, respectively.
In a pre- and post-natal development study in rats, animals were dosed from implantation through lactation. At a dose of 0.1 mg/kg (0.6 mg/m2), there were no adverse effects on delivery and lactation or signs of maternal toxicity; however, there were reductions in body weight (up to 9% reduction) and in survival of offspring (~5%). There were no drug-related effects on the developmental parameters (morphological development, motor activity, learning, or fertility assessment) in the offspring.
8.2 Lactation
Risk Summary
There is no data regarding the presence of Zortress in human milk, the effects on breastfed infants, or the effects on milk production. Everolimus and/or its metabolites are readily transferred into milk of lactating rats at a concentration 3.5 times higher than in maternal rat serum. In pre-post-natal and juvenile studies in rats, exposure to everolimus during the postnatal period caused developmental toxicity [see Use in Specific Populations (8.1), Nonclinical Toxicology (13.2)]. Advise lactating women not to breastfeed because of the potential for serious adverse reactions in infants exposed to everolimus.
8.3 Females and Males of Reproductive Potential
Contraception
Females should not be pregnant or become pregnant while receiving Zortress. Advise females of reproductive potential that animal studies have been performed showing Zortress to be harmful to the mother and developing fetus [see Use in Specific Populations (8.1)]. Females of reproductive potential are recommended to use highly effective contraception methods while receiving Zortress and up to 8 weeks after treatment has been stopped.
Infertility
Females
Amenorrhea occurred in female patients taking Zortress [see Adverse Reactions (6.2)]. Zortress may cause pre-implantation loss in females based on animal data [see Nonclinical Toxicology (13.1)].
Female fertility may be compromised by treatment with Zortress.
Males
Zortress treatment may impair fertility in males based on human [see Warnings and Precautions (5.18), Adverse Reactions (6.2, 6.3)] and animal findings [see Nonclinical Toxicology (13.1)].
![Zortress structural formula of everolimus is (1R, 9S, 12S, 15R, 16E, 18R, 19R, 21R, 23S, 24E, 26E, 28E, 30S, 32S, 35R)-1, 18-dihydroxy-12 -{(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl}-19,30-dimethoxy-15, 17, 21, 23, 29, 35-hexamethyl-11, 36-dioxa-4-aza-tricyclo[30.3.1.04,9] hexatriaconta-16,24,26,28-tetraene-2, 3,10,14,20-pentaone.](https://cdn.themeditary.com/images/2023/09/06/zortress-01.webp)
12. Zortress - Clinical Pharmacology
12.3 Pharmacokinetics
Everolimus pharmacokinetics have been characterized after oral administration of single and multiple doses to adult kidney transplant patients, hepatically-impaired patients, and healthy subjects.
Absorption
After oral dosing, peak everolimus concentrations occur 1 to 2 hours post dose. Over the dose range of 0.5 mg to 2 mg twice daily, everolimus Cmax and AUC are dose proportional in transplant patients at steady-state.
Food Effect
In 24 healthy subjects, a high-fat breakfast (44.5 g fat) reduced everolimus Cmax by 60%, delayed Tmax by a median 1.3 hours, and reduced AUC by 16% compared with a fasting administration. To minimize variability, everolimus should be taken consistently with or without food [see Dosage and Administration (2.6)].
Distribution
The blood-to-plasma ratio of everolimus is concentration dependent ranging from 17% to 73% over the range of 5 ng/mL to 5000 ng/mL. Plasma protein binding is approximately 74% in healthy subjects and in patients with moderate hepatic impairment. The apparent distribution volume associated with the terminal phase (Vz/F) from a single-dose pharmacokinetic study in maintenance kidney transplant patients is 342 to 107 L (range 128 to 589 L).
Elimination
Metabolism
Everolimus is a substrate of CYP3A4 and P-gp. Following oral administration, everolimus is the main circulating component in human blood. Six main metabolites of everolimus have been detected in human blood, including 3 monohydroxylated metabolites, 2 hydrolytic ring-opened products, and a phosphatidylcholine conjugate of everolimus. These metabolites were also identified in animal species used in toxicity studies and showed approximately 100 times less activity than everolimus itself.
Excretion
After a single dose of radiolabeled everolimus was given to transplant patients receiving cyclosporine, the majority (80%) of radioactivity was recovered from the feces and only a minor amount (5%) was excreted in urine. Parent drug was not detected in urine and feces.
Pharmacokinetics in Kidney Transplant Patients
Steady-state is reached by Day 4 with an accumulation in blood concentrations of 2- to 3-fold compared with the exposure after the first dose. Table 4 below provides a summary of the steady-state pharmacokinetic parameters.
| 1Population pharmacokinetic analysis. | |||||
| Cmax | Tmax | AUC | CL/F1 | Vc/F1 | Half-life (T1/2) |
| 11.1 + 4.6 ng/mL | 1-2 h | 75 + 31 ng•h/mL | 8.8 L/h | 110 L | 30 ± 11h |
The half-life estimates from 12 maintenance renal transplant patients who received single doses of everolimus capsules at 0.75 mg or 2.5 mg with their maintenance cyclosporine regimen indicate that the pharmacokinetics of everolimus are linear over the clinically-relevant dose range. Results indicate the half-life of everolimus in maintenance renal transplant patients receiving single doses of 0.75 mg or 2.5 mg Zortress during steady-state cyclosporine treatment was 30 ± 11 hours (range 19 to 53 hours).
12.6 Specific Populations
Hepatic Impairment
Relative to the AUC of everolimus in subjects with normal hepatic function, the average AUC in 6 patients with mild hepatic impairment (Child-Pugh Class A) was 1.6-fold higher following administration of a 10 mg single-dose. In 2 independently studied groups of 8 and 9 patients with moderate hepatic impairment (Child-Pugh Class B) the average AUC was 2.1-fold and 3.3-fold higher following administration of a 2 mg or a 10 mg single-dose, respectively; and in 6 patients with severe hepatic impairment (Child-Pugh Class C) the average AUC was 3.6-fold higher following administration of a 10 mg single-dose. For patients with mild hepatic impairment (Child-Pugh Class A), the dose should be reduced by approximately one-third of the normally recommended daily dose. For patients with moderate or severe hepatic impairment (Child-Pugh B or C), the initial daily dose should be reduced to approximately one-half of the normally recommended daily dose. Further dose adjustment and/or dose titration should be made if a patient’s whole blood trough concentration of everolimus, as measured by an LC/MS/MS assay, is not within the target trough concentration range of 3 to 8 ng/mL [see Dosage and Administration (2.7)].
Renal Impairment
No pharmacokinetic studies in patients with renal impairment were conducted. Posttransplant renal function (creatinine clearance range 11 to 107 mL/min) did not affect the pharmacokinetics of everolimus, therefore, no dosage adjustments are needed in patients with renal impairment.
Geriatrics
A limited reduction in everolimus oral CL/F of 0.33% per year was estimated in adults (age range studied was 16 to 70 years). There is no evidence to suggest that elderly patients will require a different dosage recommendation from younger adult patients.
Race
Based on analysis of population pharmacokinetics, oral clearance (CL/F) is, on average, 20% higher in black transplant patients.
12.7 Everolimus Whole Blood Concentrations Observed in Kidney and in Liver Transplant Patients
Everolimus in Kidney Transplantation
Based on exposure-efficacy and exposure-safety analyses of clinical trials and using an LC/MS/MS assay method, kidney transplant patients achieving everolimus whole blood trough concentrations greater than or equal to 3 ng/mL have been found to have a lower incidence of treated biopsy-proven acute rejection compared with patients whose trough concentrations were below 3 ng/mL. Patients who attained everolimus trough concentrations within the range of 6 to 12 ng/mL had similar efficacy and more adverse reactions than patients who attained lower trough concentrations between 3 to 8 ng/mL [see Dosage and Administration (2.3)].
In the kidney clinical trial [see Clinical Studies (14.1)], everolimus whole blood trough concentrations were measured at Days 3, 7, and 14 and Months 1, 2, 3, 4, 6, 7, 9, and 12. The proportion of patients receiving 0.75 mg twice daily Zortress treatment regimen who had everolimus whole blood trough concentrations within the protocol specified target range of 3 to 8 ng/mL at Days 3, 7, and 14 were 55%, 71% and 69%, respectively. Approximately 80% of patients had everolimus whole blood trough concentrations within the 3 to 8 ng/mL target range by Month 1 and remained stable within range through Month 12 posttransplant. The median everolimus trough concentration for the 0.75 mg twice daily treatment group was between 3 and 8 ng/mL throughout the study duration.
Everolimus in Liver Transplantation
In the liver clinical trial [see Clinical Studies (14.2)], Zortress dosing was initiated after 30 days following transplantation. Whole blood trough everolimus concentrations were measured within 5 days after first dose, followed by weekly intervals for 3 to 4 weeks, and then monthly thereafter. Approximately 49%, 37%, and 18% of patients, respectively, were below 3 ng/mL at 1, 2, and 4 weeks after initiation of Zortress dosing. The majority of patients (approximately 70% to 80%) had everolimus trough blood concentrations within the target range of 3 to 8 ng/mL from Month 2 through Month 24 posttransplant.
12.8 Cyclosporine Concentrations Observed in Kidney Transplant Patients
In the kidney transplant clinical trial [see Clinical Studies (14.1)], the target cyclosporine whole blood trough concentration for the Zortress treatment arm of 0.75 mg twice daily were 100 to 200 ng/mL through Month 1 posttransplant, 75 to 150 ng/mL at Months 2 and 3 posttransplant, 50 to 100 ng/mL at Month 4 posttransplant, and 25 to 50 ng/mL from Month 6 through Month 12 posttransplant. Table 5 below provides a summary of the observed cyclosporine whole blood trough concentrations during the study.
| Treatment Group | Visit | N | Target
(ng/mL) | Median | 10th Percentile | 90th Percentile |
| Zortress 0.75 mg twice daily | Day 3 | 242 | 100-200 | 172 | 46 | 388 |
| Day 7 | 265 | 100-200 | 185 | 75 | 337 | |
| Day 14 | 243 | 100-200 | 182 | 97 | 309 | |
| Month 1 | 245 | 100-200 | 161 | 85 | 274 | |
| Month 2 | 232 | 75-150 | 140 | 84 | 213 | |
| Month 3 | 220 | 75-150 | 111 | 68 | 187 | |
| Month 4 | 208 | 50-100 | 99 | 56 | 156 | |
| Month 6 | 200 | 25-50 | 75 | 43 | 142 | |
| Month 7 | 199 | 25-50 | 59 | 36 | 117 | |
| Month 9 | 194 | 25-50 | 49 | 28 | 91 | |
| Month 12 | 186 | 25-50 | 46 | 25 | 100 |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Everolimus was not carcinogenic in mice or rats when administered daily by oral gavage for 2 years at doses up to 0.9 mg/kg, the highest dose tested. In these studies, AUCs in mice were higher (at least 20 times) than those in humans receiving 0.75 mg twice daily, and AUCs in rats were in the same range as those in humans receiving 0.75 mg twice daily.
Everolimus was not mutagenic in the bacterial reverse mutation, the mouse lymphoma thymidine kinase assay, or the chromosome aberration assay using V79 Chinese hamster cells, or in vivo following two daily doses of 500 mg/kg in the mouse micronucleus assay.
In a 13-week male fertility oral gavage study in rats, testicular morphology was affected at 0.5 mg/kg and above, and sperm motility, sperm head count and plasma testosterone concentrations were diminished at 5 mg/kg which caused a decrease in male fertility. There was evidence of reversibility of these findings in animals examined after 13 weeks post-dosing. The 0.5 mg/kg dose in male rats resulted in AUCs in the range of clinical exposures, and the 5 mg/kg dose resulted in AUCs approximately 5 times the AUCs in humans receiving 0.75 mg twice daily.
Oral doses of everolimus in female rats greater or equal to 0.1 mg/kg (approximately 0.13-fold the estimated AUC 0-24h in patients receiving the starting dose 0.75 mg twice daily) resulted in increased incidence of pre-implantation loss.
![Figure 1. Mean and 95% CI of eGFR (MDRD 4) [mL/min/1.73 m2] by Visit Window and Treatment After Liver Transplantation (ITT population 24 Month Analysis)*](https://cdn.themeditary.com/images/2023/09/06/zortress-05.webp)

| ZORTRESS
everolimus tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ZORTRESS
everolimus tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ZORTRESS
everolimus tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ZORTRESS
everolimus tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
