Drug Class: Viral vaccines
Highlights of Prescribing Information
AFLURIA QUADRIVALENT, Influenza Vaccine
Suspension for Intramuscular Injection
2023-2024 Formula
Initial U.S. Approval (AFLURIA QUADRIVALENT): 2016
Recent Major Changes
| Dosage and Administration (2) | 08/2020 |
Indications and Usage for Afluria Quadrivalent
- AFLURIA QUADRIVALENT is an inactivated influenza vaccine indicated for active immunization against influenza disease caused by influenza A subtype viruses and type B viruses contained in the vaccine. (1)
- AFLURIA QUADRIVALENT is approved for use in persons 6 months of age and older. (1)
Afluria Quadrivalent Dosage and Administration
For intramuscular injection only, by needle and syringe (6 months and older) or by PharmaJet®Stratis® Needle-Free Injection System (18 through 64 years). (2)
|
a1 or 2 doses depends on vaccination history as per Advisory Committee on Immunization Practices annual recommendations on prevention and control of influenza with vaccines. (2) |
||
| Age | Dose | Schedule |
| 6 months through 35 months | One or two dosesa, 0.25 mL each | If 2 doses, administer at least 1 month apart |
| 36 months through 8 years | One or two dosesa, 0.5 mL each | If 2 doses, administer at least 1 month apart |
| 9 years and older | One dose, 0.5 mL | Not Applicable |
Dosage Forms and Strengths
AFLURIA QUADRIVALENT is a suspension for injection supplied in two presentations:
- 0.5 mL pre-filled syringe (single dose) (3, 11)
- 5 mL multi-dose vial (0.25 mL or 0.5 mL) (3, 11)
Contraindications
- Severe allergic reaction (e.g., anaphylaxis) to any component of the vaccine including egg protein, or to a previous dose of any influenza vaccine. (4, 11)
Warnings and Precautions
- If Guillain-Barré Syndrome (GBS) has occurred within 6 weeks of previous influenza vaccination, the decision to give AFLURIA QUADRIVALENT should be based on careful consideration of the potential benefits and risks. (5.1)
- Appropriate medical treatment and supervision must be available to manage possible anaphylactic reactions following administration of the vaccine. (5.2)
Adverse Reactions/Side Effects
AFLURIA QUADRIVALENT administered by needle and syringe:
- In adults 18 through 64 years, the most commonly reported injection-site adverse reaction was pain (≥ 40%). The most common systemic adverse events were myalgia and headache (≥ 20%). (6.1)
- In adults 65 years of age and older, the most commonly reported injection-site adverse reaction was pain (≥ 20%). The most common systemic adverse event was myalgia (≥ 10%). (6.1)
- In children 5 through 8 years, the most commonly reported injection-site adverse reactions were pain (≥ 50%), redness and swelling (≥ 10%). The most common systemic adverse event was headache (≥ 10%). (6.1)
- In children 9 through 17 years, the most commonly reported injection-site adverse reactions were pain (≥ 50%), redness and swelling (≥ 10%). The most common systemic adverse events were headache, myalgia, and malaise and fatigue (≥ 10%). (6.1)
- In children 6 months through 35 months of age, the most commonly reported injection-site reactions were pain and redness (≥ 20%).The most common systemic adverse events were irritability (≥ 30%), diarrhea and loss of appetite (≥ 20%). (6.1)
- In children 36 through 59 months of age, the most commonly reported injection site reactions were pain (≥ 30%) and redness (≥ 20%). The most commonly reported systemic adverse events were malaise and fatigue, and diarrhea (≥ 10%). (6.1)
AFLURIA (trivalent formulation) administered by the PharmaJet Stratis Needle-Free Injection System:
- In adults 18 through 64 years of age, the most commonly reported injection-site adverse reactions were tenderness (≥ 80%), swelling, pain, redness (≥ 60%), itching (≥ 20%) and bruising (≥ 10%). The most common systemic adverse events were myalgia, malaise (≥ 30%), and headache (≥ 20%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Seqirus USA Inc. at 1-855-358-8966 or VAERS at 1-800-822-7967 or www.vaers.hhs.gov.
Use In Specific Populations
- The safety and effectiveness of AFLURIA QUADRIVALENT in persons less than 6 months of age have not been established. (8.4)
- Antibody responses were lower in geriatric subjects than in younger adults. (8.5)
- Pregnancy: There is a pregnancy exposure registry that monitors outcomes in women exposed to AFLURIA QUADRIVALENT during pregnancy. Enroll in the pregnancy registry by calling 1-855-358-8966 or sending an email to [email protected]. (8.1).
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Afluria Quadrivalent
AFLURIA® QUADRIVALENT is an inactivated influenza vaccine indicated for active immunization against influenza disease caused by influenza A subtype viruses and type B viruses contained in the vaccine.
AFLURIA QUADRIVALENT is approved for use in persons 6 months of age and older.
2. Afluria Quadrivalent Dosage and Administration
For intramuscular (IM) use only.
- By needle and syringe (6 months of age and older)
- By PharmaJet® Stratis® Needle-Free Injection System (18 through 64 years of age)
The dose and schedule for AFLURIA QUADRIVALENT are presented in Table 1.
|
a 1 or 2 doses depends on vaccination history as per Advisory Committee on Immunization Practices annual recommendations on prevention and control of influenza with vaccines. |
||
| Age | Dose | Schedule |
| 6 months through 35 months | One or two doses a, 0.25 mL each | If 2 doses, administer at least 1 month apart |
| 36 months through 8 years | One or two doses a, 0.5 mL each | If 2 doses, administer at least 1 month apart |
| 9 years and older | One dose, 0.5mL | Not Applicable |
Immediately before use, shake thoroughly and inspect visually. Parenteral drug products should be inspected visually for foreign particulate matter and discoloration prior to administration, whenever suspension and container permit. If either of these conditions exists, the vaccine should not be administered.
When using the single-dose pre-filled syringe, shake the syringe thoroughly and administer the dose immediately.
When using the multi-dose vial, shake the vial thoroughly before withdrawing each dose, and administer the dose immediately. The number of needle punctures should not exceed 20 per multi-dose vial.
- Needle and Syringe: Draw up the exact dose using a separate sterile needle and syringe for each individual patient. It is recommended that small syringes (0.5 mL or 1 mL) be used to minimize any product loss.
- PharmaJet Stratis Needle-Free Injection System: For instructions on withdrawal of a 0.5 mL dose and use of the PharmaJet Stratis Needle-Free Injection System, refer to the Instructions For Use for the PharmaJet Stratis Needle-Free Injection System.
The preferred sites for intramuscular injection are the anterolateral aspect of the thigh in infants 6 months through 11 months of age, the anterolateral aspect of the thigh (or the deltoid muscle of the upper arm if muscle mass is adequate) in persons 12 months through 35 months of age, or the deltoid muscle of the upper arm in persons ≥ 36 months of age.
3. Dosage Forms and Strengths
AFLURIA QUADRIVALENT is a sterile suspension for intramuscular injection (see Description [11]).
AFLURIA QUADRIVALENT is supplied in two presentations:
- 0.5 mL pre-filled syringe (single dose, for persons 36 months of age and older)
- 5 mL multi-dose vial ( for persons 6 months of age and older)
4. Contraindications
AFLURIA QUADRIVALENT is contraindicated in individuals with known severe allergic reactions (e.g., anaphylaxis) to any component of the vaccine including egg protein, or to a previous dose of any influenza vaccine (see Description [11]).
5. Warnings and Precautions
5.1 Guillain-Barré Syndrome
If Guillain-Barré Syndrome (GBS) has occurred within 6 weeks of previous influenza vaccination, the decision to give AFLURIA QUADRIVALENT should be based on careful consideration of the potential benefits and risks.
The 1976 swine influenza vaccine was associated with an increased frequency of GBS. Evidence for a causal relation of GBS with subsequent vaccines prepared from other influenza viruses is unclear. If influenza vaccine does pose a risk, it is probably slightly more than one additional case per 1 million persons vaccinated.
5.2 Preventing and Managing Allergic Reactions
Appropriate medical treatment and supervision must be available to manage possible anaphylactic reactions following administration of the vaccine.
6. Adverse Reactions/Side Effects
In adults 18 through 64 years of age, the most commonly reported injection-site adverse reaction observed in clinical studies with AFLURIA QUADRIVALENT administered by needle and syringe was pain (≥ 40%). The most common systemic adverse events observed were myalgia and headache (≥ 20%).
In adults 65 years of age and older, the most commonly reported injection-site adverse reaction observed in clinical studies with AFLURIA QUADRIVALENT administered by needle and syringe was pain (≥ 20%). The most common systemic adverse event observed was myalgia (≥ 10%).
The safety experience with AFLURIA (trivalent formulation) is relevant to AFLURIA QUADRIVALENT because both vaccines are manufactured using the same process and have overlapping compositions (see Description [11]).
In adults 18 through 64 years of age, the most commonly reported injection-site adverse reactions observed in a clinical study with AFLURIA (trivalent formulation) using the PharmaJet Stratis Needle-Free Injection System were tenderness (≥ 80%), swelling, pain, redness (≥ 60%), itching (≥ 20%) and bruising (≥ 10%). The most common systemic adverse events were myalgia, malaise (≥ 30%) and headache (≥ 20%).
In children 5 through 8 years, the most commonly reported injection-site adverse reactions when AFLURIA QUADRIVALENT was administered by needle and syringe were pain (≥ 50%) and redness and swelling (≥ 10%). The most common systemic adverse event was headache (≥ 10%).
In children 9 through 17 years, the most commonly reported injection-site adverse reactions when AFLURIA QUADRIVALENT was administered by needle and syringe were pain (≥ 50%) and redness and swelling (≥ 10%). The most common systemic adverse events were headache, myalgia, and malaise and fatigue (≥ 10%).
In children 6 months through 35 months of age, the most frequently reported injection site reactions in the clinical study with AFLURIA QUADRIVALENT administered by needle and syringe were pain and redness (≥ 20%). The most common systemic adverse events were irritability (≥ 30%), diarrhea and loss of appetite (≥ 20%).
In children 36 through 59 months of age, the most commonly reported injection site reactions were pain (≥ 30%) and redness (≥ 20%). The most commonly reported systemic adverse events were malaise and fatigue, and diarrhea (≥ 10%).
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a vaccine cannot be directly compared to rates in the clinical studies of another vaccine and may not reflect the rates observed in clinical practice.
Adults
Clinical safety data for AFLURIA QUADRIVALENT in adults have been collected in one clinical trial, Study 1, a randomized, double-blind, active-controlled trial conducted in the U.S. in 3449 subjects ages 18 years and older. Subjects in the safety population received one dose of either AFLURIA QUADRIVALENT (N=1721) or one of two formulations of comparator trivalent influenza vaccine (AFLURIA, TIV-1 N=864 or TIV-2 N=864) each containing an influenza type B virus that corresponded to one of the two B viruses in AFLURIA QUADRIVALENT (a type B virus of the Yamagata lineage or a type B virus of the Victoria lineage), respectively. The mean age of the population was 58 years, 57% were female, and racial groups consisted of 82% White, 16% Black, and 2% other; 5% of subjects were Hispanic/Latino. The age sub-groups were 18 through 64 years and 65 years and older with mean ages of 43 years and 73 years, respectively. In this study, AFLURIA QUADRIVALENT and comparator trivalent influenza vaccines were administered by needle and syringe (see Clinical Studies [14]).
Local (injection-site) adverse reactions and systemic adverse events were solicited for 7 days post-vaccination (Table 2). Injection site cellulitis, cellulitis-like reactions (defined as concurrent Grade 3 pain, redness, and swelling/lump), and Grade 3 swelling/lump were monitored for 28 days post-vaccination. Unsolicited adverse events were collected for 28 days post-vaccination. Serious adverse events (SAEs), including deaths, were collected for 180 days post-vaccination.
|
Abbreviations: Gr 3, Grade 3. |
||||||||||||
|
a NCT02214225 |
||||||||||||
|
b Proportion of subjects reporting each solicited local adverse reaction or systemic adverse event by study vaccine group based on the number of subjects contributing any follow up safety information for at least one data value of an individual sign/symptom. |
||||||||||||
|
c N = number of subjects in the Safety Population for each study vaccine group. |
||||||||||||
|
d Local adverse reactions: Grade 3 pain is that which prevents daily activity; Swelling/Lump and redness: any = ≥ 20mm diameter, Grade 3 = ≥ 100mm diameter. |
||||||||||||
|
e Systemic adverse events: Fever: any = ≥ 100.4°F (Oral), Grade 3 = ≥ 102.2°F (Oral); Grade 3 for all other adverse events is that which prevents daily activity. |
||||||||||||
| Percentage (%) b of Subjects in each Age Cohort Reporting an Event | ||||||||||||
| Subjects 18 through 64 years | Subjects ≥ 65 years | |||||||||||
| AFLURIA Quadrivalent
N=854 c | TIV-1
N=428 c | TIV-2
N=430 c | AFLURIA Quadrivalent
N=867 c | TIV-1
N=436 c | TIV-2
N=434 c |
|||||||
| Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | |
| Local Adverse Reactions d | ||||||||||||
| Pain | 47.9 | 0.7 | 43.7 | 1.4 | 50.7 | 1.2 | 24.6 | 0.1 | 22.7 | 0 | 21.0 | 0.2 |
| Swelling/Lump | 3.7 | 0.1 | 2.3 | 0 | 3.5 | 0.2 | 3.2 | 0.5 | 1.8 | 0 | 1.6 | 0 |
| Redness | 2.9 | 0 | 2.8 | 0 | 2.8 | 0 | 4.2 | 0.3 | 2.1 | 0 | 2.5 | 0.2 |
| Systemic Adverse Events e | ||||||||||||
| Myalgia (muscle ache) | 25.5 | 1.9 | 23.4 | 1.4 | 24.2 | 1.2 | 12.7 | 0.3 | 14.0 | 0.7 | 12.2 | 0.5 |
| Headache | 21.7 | 1.7 | 15.2 | 0.9 | 19.1 | 1.2 | 8.4 | 0 | 7.1 | 0.2 | 7.8 | 0.7 |
| Malaise | 8.9 | 0.7 | 9.1 | 0 | 9.3 | 0.7 | 4.4 | 0.5 | 5.0 | 0.2 | 5.1 | 0.2 |
| Nausea | 6.9 | 0.6 | 7.7 | 0.5 | 6.3 | 1.2 | 1.6 | 0 | 1.8 | 0 | 2.1 | 0.2 |
| Chills | 4.8 | 0.6 | 4.4 | 0.2 | 4.7 | 0.5 | 2.0 | 0 | 2.1 | 0.5 | 1.4 | 0.2 |
| Vomiting | 1.5 | 0.4 | 0.9 | 0 | 2.3 | 0.7 | 0.5 | 0.1 | 0 | 0 | 0.7 | 0.2 |
| Fever | 1.1 | 0.4 | 0.9 | 0 | 0.5 | 0 | 0.2 | 0 | 0.9 | 0 | 0.5 | 0.2 |
In the 28 days following vaccination, no subject experienced cellulitis or a cellulitis-like reaction. All Grade 3 swelling/lump reactions began within 7 days of vaccination and are included in Table 2.
In the 28 days following vaccination, 20.5%, 20.1%, and 20.7% of adults 18 through 64 years and 20.3%, 24.1%, and 20.0% of adults ≥ 65 years who received AFLURIA QUADRIVALENT, TIV-1, and TIV-2, respectively, reported unsolicited adverse events. Rates of individual events were similar between treatment groups, and most events were mild to moderate in severity.
In the 180 days following vaccination, 2.3%, 1.6%, and 1.5% of all subjects who received AFLURIA QUADRIVALENT, TIV-1, and TIV-2, respectively, experienced SAEs, including six deaths, five in the AFLURIA QUADRIVALENT group and one in the TIV-2 group. The majority of SAEs occurred after Study Day 28 and in subjects ≥ 65 years of age who had co-morbid illnesses. No SAEs or deaths appeared related to the study vaccines.
Safety information has also been collected in a clinical study of AFLURIA (trivalent formulation) administered using the PharmaJet Stratis Needle-Free Injection System (Study 2). Study 2 included 1,247 subjects for safety analysis, ages 18 through 64 years, randomized to receive AFLURIA by either the PharmaJet Stratis Needle-Free Injection System (624 subjects) or needle and syringe (623 subjects). No deaths or vaccine-related serious adverse events were reported in Study 2. Local (injection-site) adverse reactions and systemic adverse events were solicited for 7 days post-vaccination (Table 3).
|
a NCT01688921 |
||||
|
b Proportion of subjects reporting each local adverse reaction or systemic adverse event by treatment group based on the number of subjects contributing at least one data value for an individual sign/symptom (individual event denominators). |
||||
|
c N = number of subjects in the Safety Population for each treatment group. Denominators for the PharmaJet Stratis Needle-Free Injection System group were: N=540 for itching and N=605-616 for all other parameters. Denominators for the needle and syringe group were: N=527 for itching and N=599-606 for all other parameters. |
||||
|
d Local adverse reactions: Grade 3 is pain, tenderness or itching that prevents daily activity; Swelling, redness or bruising: any = ≥ 25mm diameter, Grade 3 = > 100mm diameter. |
||||
|
e Systemic adverse events: Fever: any = ≥ 100.4°F (Oral), Grade 3 = ≥ 102.2°F (Oral); Grade 3 for all other adverse events is that which prevents daily activity. |
||||
|
f A total of 155 subjects (approximately randomly distributed between PharmaJet Stratis Needle-Free Injection System and needle and syringe groups) received Diary Cards without itching listed as a solicited symptom. |
||||
| Percentage b of Subjects Reporting Event | ||||
| Subjects 18 through 64 years | ||||
| AFLURIA (trivalent formulation) | ||||
| PharmaJet Stratis Needle-Free Injection System
N=540-616 c | Needle and Syringe
N=599-606 c |
|||
| Any | Grade 3 | Any | Grade 3 | |
| Local Adverse Reactions d | ||||
| Tenderness | 89.4 | 2.1 | 77.9 | 1.0 |
| Swelling | 64.8 | 1.7 | 19.7 | 0.2 |
| Pain | 64.4 | 0.8 | 49.3 | 0.7 |
| Redness | 60.1 | 1.3 | 19.2 | 0.3 |
| Itching f | 28.0 | 0.0 | 9.5 | 0.2 |
| Bruising | 17.6 | 0.2 | 5.3 | 0.0 |
| Systemic Adverse Events e | ||||
| Myalgia | 36.4 | 0.8 | 35.5 | 1.0 |
| Malaise | 31.2 | 0.7 | 28.4 | 0.5 |
| Headache | 24.7 | 1.3 | 22.1 | 1.3 |
| Chills | 7.0 | 0.2 | 7.2 | 0.2 |
| Nausea | 6.6 | 0.2 | 6.5 | 0.0 |
| Vomiting | 1.3 | 0.0 | 1.8 | 0.2 |
| Fever | 0.3 | 0.0 | 0.3 | 0.0 |
In adults 18 through 64 years who received AFLURIA (trivalent formulation) administered by PharmaJet Stratis Needle-Free Injection System, commonly reported unsolicited adverse events were headache (4.2%), injection site hematoma (1.8%), injection site erythema (1.1%), myalgia (1.0%) and nausea (1.0%).
Children 5 Years Through 17 Years of Age
Clinical safety data for AFLURIA QUADRIVALENT in older children and adolescents have been collected in one clinical trial, Study 3, a randomized, observer-blinded, comparator-controlled trial conducted in the U.S. in 2278 subjects aged 5 through 17 years. Subjects were stratified into one of two age cohorts of 5 through 8 years or 9 through 17 years (51.2% and 48.8% of the study population, respectively). The mean age of the population was 9.5 years, 52.1% were male, and racial groups consisted of 73.3% White, 20.7% Black, 0.8% Asian, 0.3% American Indian/Native American, and 0.7% Native Hawaiian/Pacific Islander; 23.8% of subjects were Hispanic/Latino. The mean ages of subjects 5 through 8 years and 9 through 17 years were 6.7 years and 12.5 years, respectively. Subjects in the safety population (N=2252) received either AFLURIA QUADRIVALENT (N=1692) or a U.S.-licensed comparator quadrivalent influenza vaccine (N=560). Study subjects were scheduled to receive either a single vaccination or two vaccinations 28 days apart based on their previous vaccination history. In this study, AFLURIA QUADRIVALENT and comparator vaccine were administered by needle and syringe (see Clinical Studies [14]).
Local (injection site) adverse reactions and systemic adverse events were solicited for 7 days post-vaccination. Cellulitis-like reactions (defined as concurrent Grade 3 pain, redness, and swelling/lump) at the injection site were monitored for 28 days post-vaccination. Subjects were instructed to report and return to clinic within 24 hours in the event of a cellulitis-like reaction. Unsolicited adverse events were collected for 28 days post-vaccination. All solicited local adverse reactions and systemic adverse events following any vaccination (first or second dose) are presented in Table 4.
|
Abbreviations: Gr 3, Grade 3 (severe); Comparator, Comparator quadrivalent influenza vaccine [Fluarix® Quadrivalent (GlaxoSmithKline Biologicals)] |
||||||||
|
a NCT02545543 |
||||||||
|
b Percent (%) is derived from the number of subjects that reported the event divided by the number of subjects in the Solicited Safety Population with non-missing data for each age cohort, treatment group, and each solicited parameter. |
||||||||
|
c N = number of subjects in the Solicited Safety Population (subjects who were vaccinated and provided any solicited safety data) for each study vaccine group. |
||||||||
|
d Local adverse reactions: Grade 3 pain is that which prevents daily activity; swelling/lump and redness: any = > 0mm diameter, Grade 3 = > 30mm diameter. |
||||||||
|
e Systemic adverse events: Fever: any = ≥ 100.4°F (Oral), Grade 3 = ≥ 102.2°F (Oral); Grade 3 for all other adverse events is that which prevents daily activity or requires significant medical intervention. |
||||||||
| Percentage (%) b of Subjects in each Age Cohort Reporting an Event | ||||||||
| Subjects 5 through 8 years | Subjects 9 through 17 years | |||||||
| AFLURIA Quadrivalent
N=828-829 c | Comparator
N=273-274 c | AFLURIA Quadrivalent
N=790-792 c | Comparator
N=261 c |
|||||
| Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | |
| Local Adverse Reactions d | ||||||||
| Pain | 51.3 | 0.8 | 49.6 | 0.7 | 51.5 | 0.3 | 45.2 | 0.4 |
| Redness | 19.4 | 3.5 | 18.6 | 1.8 | 14.8 | 1.9 | 16.1 | 1.9 |
| Swelling/Lump | 15.3 | 3.4 | 12.4 | 2.2 | 12.2 | 2.0 | 10.7 | 1.9 |
| Systemic Adverse Events e | ||||||||
| Headache | 12.3 | 0.1 | 10.6 | 0.4 | 18.8 | 0.4 | 14.6 | 0.4 |
| Myalgia | 9.8 | 0.1 | 11.3 | 0.4 | 16.7 | 0.3 | 11.1 | 0.4 |
| Malaise and Fatigue | 8.8 | 0.4 | 5.8 | 0 | 10.0 | 0.4 | 7.7 | 0 |
| Nausea | 7.1 | 0.1 | 8.4 | 0 | 7.7 | 0 | 8.0 | 0 |
| Diarrhea | 5.2 | 0 | 3.6 | 0 | 5.4 | 0 | 4.2 | 0 |
| Fever | 4.5 | 1.2 | 3.6 | 0.7 | 2.1 | 0.5 | 0.8 | 0 |
| Vomiting | 2.4 | 0.2 | 4.4 | 0 | 1.8 | 0 | 2.3 | 0 |
In subjects 5 through 8 years of age, all solicited local adverse reactions and systemic adverse events were reported at lower frequencies after the second vaccination than after the first vaccination with AFLURIA QUADRIVALENT with the exception of vomiting (which occurred at the same rate of 2.2% after each vaccination).
One subject, 8 years of age, experienced a cellulitis-like reaction at the injection site after vaccination with AFLURIA QUADRIVALENT.
The most commonly reported unsolicited adverse events in the 28 days following the first or second dose of AFLURIA QUADRIVALENT in subjects 5 through 8 years of age were cough (2.4%), pyrexia (1.8%), rhinorrhea (1.2%), and headache (1.0%), and were similar to the comparator.
For subjects ages 9 through 17 years who received AFLURIA QUADRIVALENT, the most commonly reported unsolicited adverse events in the 28 days following vaccination were oropharyngeal pain (1.6%), cough (1.3%), and upper respiratory tract infection (1.0%), and were similar to the comparator.
No deaths were reported in Study 3. In the 180 days following vaccinations, AFLURIA QUADRIVALENT and comparator vaccine recipients experienced similar rates of serious adverse events (SAEs). None of the SAEs appeared related to the study vaccines except for one case of influenza B infection (considered a vaccine failure) in an AFLURIA QUADRIVALENT recipient.
Children 6 Months Through 59 Months of Age
Clinical safety data for AFLURIA QUADRIVALENT in infants and young children have been collected in one clinical trial, Study 4, a randomized, observer-blind, comparator-controlled trial conducted in the U.S. in 2247 subjects aged 6 through 59 months. Subjects were stratified into one of two age cohorts of 6 through 35 months or 36 through 59 months (41.6% and 58.4% of the study population, respectively). The mean age of the population was 36.6 months, 51.6% were male, and racial groups consisted of 71.0% White, 21.5% Black, 1.1% Asian, 0.7% Native Hawaiian/Pacific Islander, and 0.3% American Indian/Native American; 26.4% of subjects were Hispanic/Latino. The mean ages of subjects 6 through 35 months and 36 through 59 months were 21.7 months and 47.1 months, respectively. Subjects in the safety population (N=2232) received either AFLURIA QUADRIVALENT (N=1673) or a U.S.-licensed comparator quadrivalent influenza vaccine (N=559). Study subjects were scheduled to receive either a single vaccination or two vaccinations 28 days apart based on their previous vaccination history. In this study, AFLURIA QUADRIVALENT and comparator vaccine were administered by needle and syringe (see Clinical Studies [14]).
Local (injection site) adverse reactions and systemic adverse events were solicited for 7 days post-vaccination. Cellulitis-like reactions (defined as concurrent Grade 3 pain, redness, and swelling/lump) at the injection site were monitored for 28 days post-vaccination. Subjects were instructed to report and return to clinic within 24 hours in the event of a cellulitis-like reaction. Unsolicited adverse events were collected for 28 days post-vaccination, and SAEs for 6 months following the last vaccination. All solicited local adverse reactions and systemic adverse events following any vaccination (first or second dose) are presented in Table 5.
|
Abbreviations: Gr 3, Grade 3 (severe); Comparator, Comparator quadrivalent influenza vaccine [Fluzone® Quadrivalent (Sanofi Pasteur)] |
||||||||
|
a NCT02914275 |
||||||||
|
b Percent (%) is derived from the number of subjects that reported the event divided by the number of subjects in the Solicited Safety Population with non-missing data for each age cohort, treatment group, and each solicited parameter. |
||||||||
|
c N = number of subjects in the Solicited Safety Population (subjects who were vaccinated and provided any solicited safety data) for each study vaccine group. |
||||||||
|
d Local adverse reactions: Grade 3 pain is that which prevents daily activity (36 through 59 month subjects); or cried when limb was moved or spontaneously painful (6 through 35 month subjects); Swelling/Lump and redness: any = ≥ 0mm diameter, Grade 3 = ≥ 30mm diameter. |
||||||||
|
e Systemic adverse events: Fever: any = ≥ 99.5°F (Axillary), Grade 3 = ≥ 101.3°F (Axillary); Grade 3 for all other adverse events is that which prevents daily activity; Irritability, Loss of Appetite, Malaise and Fatigue, Myalgia and Headache are age specific systemic adverse events, where “-” denotes event was not applicable to that age cohort. |
||||||||
|
f Prophylactic antipyretics (acetaminophen or ibuprophen-containing medications) were not permitted. Antipyretics used to treat fever were permitted and rates of use were as follows: 6 through 35 months (Afluria QIV 5.9%, Comparator QIV 9.0%); 36 through 59 months (Afluria QIV 3.7%, Comparator QIV 2.5%). |
||||||||
| Percentage (%) b of Subjects in each Age Cohort Reporting an Event | ||||||||
| 6 through 35 months | 36 through 59 months | |||||||
| AFLURIA Quadrivalent
N=668-669 c | Comparator
N=226-227 c | AFLURIA Quadrivalent
N=947-949 c | Comparator
N=317-318 c |
|||||
| Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | Any | Gr 3 | |
| Local Adverse Reactions d | ||||||||
| Pain | 20.8 | 0.1 | 25.6 | 0.4 | 35.5 | 0 | 31.4 | 0.6 |
| Redness | 20.8 | 0.6 | 17.6 | 1.8 | 22.4 | 2.3 | 20.8 | 5.3 |
| Swelling/Lump | 6.1 | 0.4 | 6.2 | 0.9 | 10.1 | 1.7 | 12.9 | 2.5 |
| Systemic Adverse Events e | ||||||||
| Irritability | 32.9 | 0.7 | 28.2 | 0.4 | - | - | - | - |
| Diarrhea | 24.2 | 0.1 | 25.6 | 0.4 | 12.1 | 0.1 | 8.8 | 0.6 |
| Loss of Appetite | 20.0 | 0.3 | 19.4 | 0.4 | - | - | - | - |
| Malaise and Fatigue | - | - | - | - | 14.3 | 0.5 | 13.2 | 0.3 |
| Myalgia | - | - | - | - | 9.9 | 0.1 | 9.4 | 0 |
| Nausea and/or vomiting | 9.4 | 0.7 | 11.0 | 0 | 9.2 | 0.4 | 6.6 | 0.3 |
| Headache | - | - | - | - | 6.2 | 0.4 | 5.0 | 0 |
| Fever f | 7.2 | 2.5 | 11.9 | 2.6 | 4.8 | 1.2 | 6.0 | 0.9 |
In subjects 6 through 35 months of age, all solicited local adverse reactions and systemic adverse events were reported at lower frequencies after the second vaccination than after the first vaccination with AFLURIA QUADRIVALENT.
In subjects 36 through 59 months of age, all solicited local adverse reactions and systemic adverse events were reported at lower frequencies after the second vaccination than after the first vaccination with AFLURIA QUADRIVALENT.
The most commonly reported unsolicited adverse events in the 28 days following the first or second dose of AFLURIA QUADRIVALENT in subjects 6 through 35 months of age were rhinorrhea (11.2%), cough (10.4%), pyrexia (6.3%), upper respiratory tract infection (4.8%), diarrhea (3.7%), otitis media (2.4%), vomiting (2.4%), nasal congestion (2.4%), nasopharyngitis (1.9%), irritability (1.7%), ear infection (1.6%), croup infectious (1.4%), teething (1.3%), rash (1.2%), influenza like illness (1.0%) and fatigue (1.0%), and were similar to comparator.
The most commonly reported unsolicited adverse events in the 28 days following the first or second dose of AFLURIA QUADRIVALENT in subjects 36 through 59 months of age were cough (7.7%), rhinorrhea (4.9%), pyrexia (3.7%), upper respiratory tract infection (2.5%), vomiting (2.1%), nasal congestion (1.6%), nasopharyngitis (1.7%), ororpharyngeal pain (1.2%), diarrhea (1.1%) and fatigue (1.1%), and were similar to the comparator.
No deaths were reported in Study 4. In the 180 days following vaccinations, AFLURIA QUADRIVALENT and comparator vaccine recipients experienced similar rates of serious adverse events (SAEs), none of which were related to study vaccines. No vaccine-related febrile seizures occurred in Study 4. Unrelated SAEs of febrile seizures occurred in two AFLURIA QUADRIVALENT recipients (6 through 35 months age group) at 43 and 104 days post-vaccinations.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of AFLURIA or AFLURIA QUADRIVALENT. Because post-marketing reporting of adverse events is voluntary and from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to vaccine exposure. The adverse reactions have been included in this section based on strength of evidence for a causal relationship to AFLURIA or AFLURIA QUADRIVALENT, seriousness or frequency of reporting.
Blood and lymphatic system disorders
Thrombocytopenia
Immune system disorders
Allergic or immediate hypersensitivity reactions including anaphylactic shock and serum sickness
Nervous system disorders
Neuralgia, paresthesia, convulsions (including febrile seizures), dizziness, encephalomyelitis, encephalopathy, neuritis or neuropathy, transverse myelitis, GBS, syncope and presyncope
Vascular disorders
Vasculitis which may be associated with renal involvement
Musculoskeletal and Connective Tissue Disorders
Musculoskeletal pain and pain in the extremity
Skin and subcutaneous tissue disorders
Pruritus, urticaria, and rash
General disorders and administration site conditions
Cellulitis and large injection site swelling
Influenza-like illness, injected limb mobility decreased, pyrexia, injection site erythema and injection site reaction
7. Drug Interactions
No interaction studies have been performed on interaction between influenza vaccines in general and other vaccines or medications.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to AFLURIA QUADRIVALENT during pregnancy. Women who are vaccinated with AFLURIA QUADRIVALENT during pregnancy are encouraged to enroll in the registry by calling 1-855-358-8966 or sending an email to Seqirus at [email protected].
Risk summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. Data for AFLURIA (trivalent formulation) administered to pregnant women are relevant to AFLURIA QUADRIVALENT because both vaccines are manufactured using the same process and have overlapping compositions (see Description [11]). There are limited data for AFLURIA QUADRIVALENT administered to pregnant women, and available data for AFLURIA (trivalent formulation) administered to pregnant women are insufficient to inform vaccine-associated risks in pregnancy.
There were no developmental toxicity studies of AFLURIA QUADRIVALENT performed in animals. A developmental toxicity study of AFLURIA (trivalent formulation) has been performed in female rats administered a single human dose [0.5 mL (divided)] of AFLURIA (trivalent formulation) prior to mating and during gestation. This study revealed no evidence of harm to the fetus due to AFLURIA (trivalent formulation) (see 8.1 Data).
Data
Animal Data
In a developmental toxicity study, female rats were administered a single human dose [0.5 mL (divided)] of AFLURIA (trivalent formulation) by intramuscular injection 21 days and 7 days prior to mating, and on gestation day 6. Some rats were administered an additional dose on gestation day 20. No vaccine-related fetal malformations or variations and no adverse effects on pre-weaning development were observed in the study.
8.2 Lactation
Risk Summary
It is not known whether AFLURIA QUADRIVALENT is excreted in human milk. Data are not available to assess the effects of AFLURIA QUADRIVALENT on the breastfed infant or on milk production/excretion.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for AFLURIA QUADRIVALENT and any potential adverse effects on the breastfed child from AFLURIA QUADRIVALENT or from the underlying maternal condition. For preventive vaccines, the underlying maternal condition is susceptibility to disease prevented by the vaccine.
8.4 Pediatric Use
The safety and effectiveness of AFLURIA QUADRIVALENT in persons less than 6 months of age have not been established.
The PharmaJet Stratis Needle-Free Injection System is not approved as a method of administering AFLURIA QUADRIVALENT to children and adolescents less than 18 years of age due to lack of adequate data supporting safety and effectiveness in this population.
8.5 Geriatric Use
In clinical studies, AFLURIA QUADRIVALENT has been administered to, and safety information collected for, 867 subjects aged 65 years and older (see Adverse Reactions [6]). The 65 years and older age group included 539 subjects 65 through 74 years and 328 subjects 75 years and older. After administration of AFLURIA QUADRIVALENT, hemagglutination-inhibiting antibody responses were non-inferior to comparator trivalent influenza (TIV-1 and TIV-2) in persons 65 years of age and older, but were lower than younger adult subjects (see Clinical Studies [14]).
The PharmaJet Stratis Needle-Free Injection System is not approved as a method of administering AFLURIA QUADRIVALENT to adults 65 years of age and older due to lack of adequate data supporting safety and effectiveness in this population.
11. Afluria Quadrivalent Description
AFLURIA QUADRIVALENT, Influenza Vaccine for intramuscular injection, is a sterile, clear, colorless to slightly opalescent suspension with some sediment that resuspends upon shaking to form a homogeneous suspension. AFLURIA QUADRIVALENT is prepared from influenza virus propagated in the allantoic fluid of embryonated chicken eggs. Following harvest, the virus is purified in a sucrose density gradient using continuous flow zonal centrifugation. The purified virus is inactivated with beta-propiolactone, and the virus particles are disrupted using sodium taurodeoxycholate to produce a “split virion”. The disrupted virus is further purified and suspended in a phosphate buffered isotonic solution.
AFLURIA QUADRIVALENT is standardized according to USPHS requirements for the 2023-2024 influenza season and is formulated to contain 60 mcg hemagglutinin (HA) per 0.5 mL dose in the recommended ratio of 15 mcg HA for each of the four influenza strains recommended for the 2023-2024 Northern Hemisphere influenza season:
A/Victoria/4897/2022 IVR-238 (an A/Victoria/4897/2022 (H1N1)pdm09-like virus), A/Darwin/6/2021 IVR-227 (an A/Darwin/9/2021 (H3N2)-like virus, B/Austria/1359417/2021 BVR-26 (a B/Austria/1359417/2021-like virus) and B/Phuket/3073/2013 BVR-1B (a B/Phuket/3073/2013-like virus). A 0.25 mL dose contains 7.5 mcg HA of each of the same four influenza strains.
Thimerosal, a mercury derivative, is not used in the manufacturing process for the single dose presentation. This presentation does not contain preservative. The multi-dose presentation contains thimerosal added as a preservative; each 0.5 mL dose contains 24.5 mcg of mercury and each 0.25 mL dose contains 12.25 mcg of mercury.
A single 0.5 mL dose of AFLURIA QUADRIVALENT contains sodium chloride (4.1 mg), monobasic sodium phosphate (80 mcg), dibasic sodium phosphate (300 mcg), monobasic potassium phosphate (20 mcg), potassium chloride (20 mcg), and calcium chloride (0.5 mcg). From the manufacturing process, each 0.5 mL dose may also contain residual amounts of sodium taurodeoxycholate (≤ 10 ppm), ovalbumin (< 1 mcg), sucrose (< 10 mcg), neomycin sulfate (≤ 81.8 nanograms [ng]), polymyxin B (≤ 14 ng), beta-propiolactone (≤ 1.5 ng) and hydrocortisone (≤ 0.56 ng). A single 0.25 mL dose of AFLURIA QUADRIVALENT contains half of these quantities.
The rubber tip cap and plunger used for the preservative-free, single-dose syringes and the rubber stoppers used for the multi-dose vial are not made with natural rubber latex.
12. Afluria Quadrivalent - Clinical Pharmacology
12.1 Mechanism of Action
Influenza illness and its complications follow infection with influenza viruses. Global surveillance of influenza identifies yearly antigenic variants. For example, since 1977 antigenic variants of influenza A (H1N1 and H3N2) and influenza B viruses have been in global circulation. Since 2001, two distinct lineages of influenza B (Victoria and Yamagata lineages) have co-circulated worldwide. Specific levels of hemagglutination inhibition (HI) antibody titers post-vaccination with inactivated influenza vaccine have not been correlated with protection from influenza virus. In some human studies, antibody titers of 1:40 or greater have been associated with protection from influenza illness in up to 50% of subjects.2,3
Antibody against one influenza virus type or subtype confers limited or no protection against another. Furthermore, antibody to one antigenic variant of influenza virus might not protect against a new antigenic variant of the same type or subtype. Frequent development of antigenic variants through antigenic drift is the virologic basis for seasonal epidemics and the reason for the usual change to one or more new strains in each year's influenza vaccine. Therefore, inactivated influenza vaccines are standardized to contain the HA of four strains (i.e., typically two type A and two type B) representing the influenza viruses likely to be circulating in the U.S. during the upcoming winter.
Annual revaccination with the current vaccine is recommended because immunity declines during the year after vaccination and circulating strains of influenza virus change from year to year.1
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
AFLURIA QUADRIVALENT has not been evaluated for carcinogenic or mutagenic potential, or male infertility in animals. A developmental toxicity study conducted in rats vaccinated with AFLURIA (trivalent formulation) revealed no impact on female fertility (see Pregnancy [8.1]).
14. Clinical Studies
14.1 Efficacy Against Laboratory-Confirmed Influenza
The efficacy of AFLURIA (trivalent formulation) is relevant to AFLURIA QUADRIVALENT because both vaccines are manufactured using the same process and have overlapping compositions (see Description [11]).
The efficacy of AFLURIA (trivalent formulation) was demonstrated in Study 5, a randomized, observer-blind, placebo-controlled study conducted in 15,044 subjects. Healthy subjects 18 through 64 years of age were randomized in a 2:1 ratio to receive a single dose of AFLURIA (trivalent formulation) (enrolled subjects: 10,033; evaluable subjects: 9,889) or placebo (enrolled subjects: 5,011; evaluable subjects: 4,960). The mean age of all randomized subjects was 35.5 years. 54.4% were female and 90.2% were White. Laboratory-confirmed influenza was assessed by active and passive surveillance of influenza-like illness (ILI) beginning 2 weeks post-vaccination until the end of the influenza season, approximately 6 months post-vaccination. ILI was defined as at least one respiratory symptom (e.g., cough, sore throat, nasal congestion) and at least one systemic symptom (e.g., oral temperature of 100.0ºF or higher, feverishness, chills, body aches). Nasal and throat swabs were collected from subjects who presented with an ILI for laboratory confirmation by viral culture and real-time reverse transcription polymerase chain reaction. Influenza virus strain was further characterized using gene sequencing and pyrosequencing.
Attack rates and vaccine efficacy, defined as the relative reduction in the influenza infection rate for AFLURIA (trivalent formulation) compared to placebo, were calculated using the Per Protocol Population. Vaccine efficacy against laboratory-confirmed influenza infection due to influenza A or B virus strains contained in the vaccine was 60% with a lower limit of the 95% CI of 41% (Table 6).
|
Abbreviations: CI, confidence interval. |
|||||
|
a NCT00562484 |
|||||
|
b The Per Protocol Population was identical to the Evaluable Population in this study. |
|||||
|
c Vaccine efficacy = 1 minus the ratio of AFLURIA (trivalent formulation) /placebo infection rates. The objective of the study was to demonstrate that the lower limit of the CI for vaccine efficacy was greater than 40%. |
|||||
| Subjects b | Laboratory-Confirmed Influenza Cases | Influenza Infection Rate | Vaccine Efficacy c | ||
| N | N | n/N % | % | Lower Limit of the 95% CI | |
| Vaccine-matched Strains | |||||
| AFLURIA | 9889 | 58 | 0.59 | 60 | 41 |
| Placebo | 4960 | 73 | 1.47 | ||
| Any Influenza Virus Strain | |||||
| AFLURIA | 9889 | 222 | 2.24 | 42 | 28 |
| Placebo | 4960 | 192 | 3.87 | ||
14.2 Immunogenicity of AFLURIA QUADRIVALENT in Adults and Older Adults Administered by Needle and Syringe
Study 1 was a randomized, double-blind, active-controlled trial conducted in the U.S. in adults aged 18 years of age and older. Subjects received one dose of either AFLURIA QUADRIVALENT (N=1691) or one of two formulations of comparator trivalent influenza vaccine (AFLURIA, TIV-1 N=854 or TIV-2 N=850) each containing an influenza type B virus that corresponded to one of the two B viruses in AFLURIA QUADRIVALENT (a type B virus of the Yamagata lineage or a type B virus of the Victoria lineage, respectively).
Post-vaccination immunogenicity was evaluated on sera obtained 21 days after administration of a single dose of AFLURIA QUADRIVALENT or TIV comparator. The co-primary endpoints were HI Geometric Mean Titer (GMT) ratios (adjusted for baseline HI titers) and the difference in seroconversion rates for each vaccine strain, 21 days after vaccination. Pre-specified non-inferiority criteria required that the upper bound of the 2-sided 95% CI of the GMT ratio (TIV/AFLURIA QUADRIVALENT) did not exceed 1.5 and the upper bound of the 2-sided 95% CI of the seroconversion rate difference (TIV minus AFLURIA QUADRIVALENT) did not exceed 10.0% for each strain.
Serum HI antibody responses to AFLURIA QUADRIVALENT were non-inferior to both TIVs for all influenza strains for subjects 18 years of age and older. Additionally, non-inferiority was demonstrated for both endpoints in both age sub-groups, adults aged 18 through 64 years and 65 years and older, for all strains (Table 7). Superiority of the immune response to each of the influenza B strains contained in AFLURIA QUADRIVALENT was shown relative to the antibody response after vaccination with TIV formulations not containing that B lineage strain for subjects 18 years of age and older. Superiority against the alternate B strain was also demonstrated for each of the influenza B strains in both age sub-groups; 18 through 64 years and 65 years and older. Post-hoc analyses of immunogenicity endpoints by gender did not demonstrate meaningful differences between males and females. The study population was not sufficiently diverse to assess differences between races or ethnicities.
| Post-vaccination GMT | GMT Ratio b | Seroconversion % c | Difference | Met both pre-defined non-inferiority criteria? d | |||
|---|---|---|---|---|---|---|---|
| Strain | AFLURIA Quadrivalent | Pooled TIV
or TIV-1 (B Yamagata) or TIV-2 (B Victoria) | Pooled TIV or TIV-1 or TIV-2 over AFLURIA Quadrivalent (95% CI) | AFLURIA Quadrivalent N=1691 | Pooled TIV
or TIV-1 (B Yamagata) or TIV-2 (B Victoria) | Pooled TIV or TIV-1 or TIV-2 minus AFLURIA Quadrivalent (95% CI) |
|
|
Abbreviations: CI, confidence interval; GMT, geometric mean titer. |
|||||||
|
a NCT02214225 |
|||||||
|
b GMT ratio was computed after fitting a multi-variable model on the post-vaccination titers including sex, vaccination history, pre-vaccination HI titers and other factors. |
|||||||
|
c Seroconversion rate is defined as a 4-fold increase in post-vaccination HI antibody titer from pre-vaccination titer ≥ 1:10 or an increase in titer from < 1:10 to ≥ 1:40. |
|||||||
|
d Non-inferiority (NI) criterion for the GMT ratio: upper bound of 2-sided 95% CI on the GMT ratio of Pooled TIV or TIV-1 (B Yamagata) or TIV-2 (B Victoria)/AFLURIA Quadrivalent should not exceed 1.5. NI criterion for the SCR difference: upper bound of 2-sided 95% CI on the difference between SCR Pooled TIV or TIV-1 (B Yamagata) or TIV-2 (B Victoria) minus AFLURIA Quadrivalent should not exceed 10%. |
|||||||
|
e Pooled TIV/AFLURIA Quadrivalent |
|||||||
|
f TIV-1 (B Yamagata)/AFLURIA Quadrivalent |
|||||||
|
g TIV-2 (B Victoria)/AFLURIA Quadrivalent |
|||||||
|
h Pooled TIV – AFLURIA Quadrivalent |
|||||||
|
i TIV-1 (B Yamagata) - AFLURIA Quadrivalent |
|||||||
|
j TIV-2 (B Victoria) - AFLURIA Quadrivalent |
|||||||
| 18 through 64 years | AFLURIA Quadrivalent N=835, Pooled TIV N=845, TIV-1 N=424, TIV-2 N=421 | ||||||
| A(H1N1) | 432.7 | 402.8 | 0.93 e
(0.85, 1.02) | 51.3 | 49.1 | -2.1 h
(-6.9, 2.7) | Yes |
| A(H3N2) | 569.1 | 515.1 | 0.91 e
(0.83, 0.99) | 56.3 | 51.7 | -4.6 h
(-9.4, 0.2) | Yes |
| B/Massachusetts/ 2/2012 (B Yamagata) | 92.3 | 79.3 | 0.86 f
(0.76, 0.97) | 45.7 | 41.3 | -4.5 i
(-10.3, 1.4) | Yes |
| B/Brisbane/ 60/2008 (B Victoria) | 110.7 | 95.2 | 0.86 g
(0.76, 0.98) | 57.6 | 53.0 | -4.6 j
(-10.5, 1.2) | Yes |
| ≥ 65 years | AFLURIA Quadrivalent N=856, Pooled TIV N=859, TIV-1 N=430, TIV-2 N=429 | ||||||
| A(H1N1) | 211.4 | 199.8 | 0.95 e
(0.88, 1.02) | 26.6 | 26.4 | -0.2 h
(-5.0, 4.5) | Yes |
| A(H3N2) | 419.5 | 400.0 | 0.95 e
(0.89, 1.02) | 25.9 | 27.0 | 1.1 h
(-3.7, 5.8) | Yes |
| B/Massachusetts/ 2/2012 (B Yamagata) | 43.3 | 39.1 | 0.90 f
(0.84, 0.97) | 16.6 | 14.4 | -2.2 i
(-8.0, 3.6) | Yes |
| B/Brisbane/ 60/2008 (B Victoria) | 66.1 | 68.4 | 1.03 g
(0.94, 1.14) | 23.5 | 24.7 | 1.2 j
(-4.6, 7.0) | Yes |
14.3 Immunogenicity of AFLURIA (trivalent formulation) Administered by PharmaJet Stratis Needle-Free Injection System
Study 2 was a randomized, comparator-controlled, non-inferiority study that enrolled 1,250 subjects 18 through 64 years of age. This study compared the immune response following administration of AFLURIA (trivalent formulation) when delivered intramuscularly using either the PharmaJet Stratis Needle-Free Injection System or needle and syringe. Immunogenicity assessments were performed prior to vaccination and at 28 days after vaccination in the immunogenicity population (1130 subjects, 562 PharmaJet Stratis Needle-Free Injection System group, 568 needle and syringe group). The co-primary endpoints were HI GMT ratios for each vaccine strain and the absolute difference in seroconversion rates for each vaccine strain 28 days after vaccination. As shown in Table 8, non-inferiority of administration of AFLURIA (trivalent formulation) by the PharmaJet Stratis Needle-Free Injection System compared to administration of AFLURIA (trivalent formulation) by needle and syringe was demonstrated in the immunogenicity population for all strains. Post-hoc analyses of immunogenicity by age showed that younger subjects (18 through 49 years) elicited higher immunological responses than older subjects (50 through 64 years). Post-hoc analyses of immunogenicity according to sex and body mass index did not reveal significant influences of these variables on immune responses. The study population was not sufficiently diverse to assess immunogenicity by race or ethnicity.
| Baseline GMT | Post-vaccination GMT | GMT Ratio b | Seroconversion % c | Difference | Met both pre-defined non-inferiority criteria? d | ||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | Needle and Syringe
N=568 | PharmaJet Stratis Needle-Free Injection System N=562 | Needle and Syringe
N=568 | PharmaJet Stratis Needle-Free Injection System N=562 | Needle and Syringe over PharmaJet Stratis Needle-Free Injection System (95% CI) | Needle and Syringe
N=568 | PharmaJet Stratis Needle-Free Injection System N=562 | Needle and Syringe minus PharmaJet Stratis Needle-Free Injection System
(95% CI) |
|
|
Abbreviations: CI, confidence interval; GMT, geometric mean titer. |
|||||||||
|
a NCT01688921 |
|||||||||
|
b GMT ratio is defined as post-vaccination GMT for Needle and Syringe/PharmaJet Stratis Needle-Free Injection System. |
|||||||||
|
c Seroconversion rate is defined as a 4-fold increase in post-vaccination HI antibody titer from pre-vaccination titer ≥ 1:10 or an increase in titer from < 1:10 to ≥ 1:40. |
|||||||||
|
d Non-inferiority (NI) criterion for the GMT ratio: upper bound of 2-sided 95% CI on the GMT ratio of Needle and Syringe/PharmaJet Stratis Needle-Free Injection System should not exceed 1.5. NI criterion for the seroconversion rate (SCR) difference: upper bound of 2-sided 95% CI on the difference between SCR Needle and Syringe – SCR PharmaJet Stratis Needle-Free Injection System should not exceed 10%. |
|||||||||
| A(H1N1) | 79.5 | 83.7 | 280.6 | 282.9 | 0.99 (0.88, 1.12) | 38.4 | 37.5 | 0.8 (-4.8, 6.5) | Yes |
| A(H3N2) | 75.4 | 68.1 | 265.9 | 247.3 | 1.08 (0.96, 1.21) | 45.1 | 43.8 | 1.3 (-4.5, 7.1) | Yes |
| B | 12.6 | 13.5 | 39.7 | 42.5 | 0.94 (0.83, 1.06) | 35.2 | 34.9 | 0.3 (-5.2, 5.9) | Yes |
14.4 Immunogenicity of AFLURIA QUADRIVALENT in Children 5 through 17 Years Administered by Needle and Syringe
Study 3 was a randomized, observer-blinded, comparator-controlled trial conducted in the U.S. in children 5 through 17 years of age. A total of 2278 subjects were randomized 3:1 to receive one or two doses of AFLURIA QUADRIVALENT (N=1709) or a U.S.-licensed comparator quadrivalent influenza vaccine (N=569). Subjects 5 through 8 years of age were eligible to receive a second dose at least 28 days after the first dose depending on their influenza vaccination history, consistent with the 2015-2016 recommendations of the Advisory Committee on Immunization Practices (ACIP) for Prevention and Control of Seasonal Influenza with Vaccines. Approximately 25% of subjects in each treatment group in the 5 through 8 years of age sub-group received two vaccine doses.
Baseline serology for HI assessment was collected prior to vaccination. Post-vaccination immunogenicity was evaluated by HI assay on sera obtained 28 days after the last vaccination dose.
The primary objective was to demonstrate that vaccination with AFLURIA QUADRIVALENT elicits an immune response that is not inferior to that of a comparator vaccine containing the same recommended virus strains. The Per Protocol Population (AFLURIA QUADRIVALENT n=1605, Comparator n=528) was used for the primary endpoint analyses. The co-primary endpoints were HI Geometric Mean Titer (GMT) ratios (adjusted for baseline HI titers and other covariates) and seroconversion rates for each vaccine strain, 28 days after the last vaccination. Pre-specified non-inferiority criteria required that the upper bound of the 2-sided 95% CI of the GMT ratio (Comparator/AFLURIA QUADRIVALENT) did not exceed 1.5 and the upper bound of the 2-sided 95% CI of the seroconversion rate difference (Comparator minus AFLURIA QUADRIVALENT) did not exceed 10.0% for each strain. Serum HI antibody responses to AFLURIA QUADRIVALENT were non-inferior for both GMT ratio and seroconversion rates relative to the comparator vaccine for all influenza strains (Table 9). Analyses of immunogenicity endpoints by gender did not demonstrate meaningful differences between males and females. The study population was not sufficiently diverse to assess differences among races or ethnicities.
| Post-vaccination GMT | GMT Ratio c | Seroconversion % d | SCR Difference e | Met both pre-defined non-inferiority criteria? f | |||
|---|---|---|---|---|---|---|---|
| Strain | AFLURIA
Quadrivalent N=1605 | Comparator
N=528 | Comparator over AFLURIA
Quadrivalent (95% CI) | AFLURIA
Quadrivalent N=1605 (95% CI) | Comparator N=528 (95% CI) | Comparator minus AFLURIA
Quadrivalent (95% CI) |
|
|
Abbreviations: CI, confidence interval; Comparator, Comparator quadrivalent influenza vaccine (Fluarix® Quadrivalent [GlaxoSmithKline Biologicals]); GMT (adjusted), geometric mean titer; SCR, seroconversion rate. |
|||||||
|
a NCT02545543 |
|||||||
|
b The Per-Protocol Population comprised all subjects in the Evaluable Population who did not have any protocol deviations that were medically assessed as potentially impacting on immunogenicity results. |
|||||||
|
c GMT Ratio = Comparator /AFLURIA QUADRIVALENT. Adjusted analysis model: Log-transformed Post-Vaccination HI Titer=Vaccine + Age Strata [5-8, 9-17] + Gender + Vaccination History [y/n] + Log-transformed Pre-Vaccination HI Titer + Site + Number of Doses (1 vs 2) + Age Strata*Vaccine. The Age Strata*Vaccine interaction term was excluded from the model fit for the strains B/Yamagata and B/Victoria as the interaction result was non-significant (p>0.05). Least square means were back transformed. |
|||||||
|
d Seroconversion rate was defined as the percentage of subjects with either a prevaccination HI titer < 1:10 and a postvaccination HI titer ≥ 1:40 or a prevaccination HI titer ≥ 1:10 and a 4-fold increase in postvaccination HI titer. |
|||||||
|
e Seroconversion rate difference = Comparator SCR percentage minus AFLURIA QUADRIVALENT SCR percentage. |
|||||||
|
f Non-inferiority (NI) criterion for the GMT ratio: upper bound of two-sided 95% CI on the GMT ratio of Comparator /AFLURIA QUADRIVALENT should not exceed 1.5. NI criterion for the SCR difference: upper bound of two-sided 95% CI on the difference between SCR Comparator – AFLURIA QUADRIVALENT should not exceed 10%. |
|||||||
|
g Subject 8400394-0046 was excluded from the Per-Protocol Population for the adjusted GMT analysis for the GMT ratio since the subject did not have information on all covariates (unknown prevaccination history). |
|||||||
| A(H1N1) | 952.6 (n=1604 g) | 958.8 | 1.01 (0.93, 1.09) | 66.4 (64.0, 68.7) | 63.3 (59.0, 67.4) | -3.1 (-8.0, 1.8) | Yes |
| A(H3N2) | 886.4 (n=1604 g) | 930.6 | 1.05 (0.96, 1.15) | 82.9 (81.0, 84.7) | 83.3 (79.9, 86.4) | 0.4 (-4.5, 5.3) | Yes |
| B/Phuket/3073/ 2013 (B Yamagata) | 60.9 (n=1604 g) | 54.3 | 0.89 (0.81, 0.98) | 58.5 (56.0, 60.9) | 55.1 (50.8, 59.4) | -3.4 (-8.3, 1.5) | Yes |
| B/Brisbane/60/ 2008 (B Victoria) | 145.0 (n=1604 g) | 133.4 | 0.92 (0.83, 1.02) | 72.1 (69.8, 74.3) | 70.1 (66.0, 74.0) | -2.0 (-6.9, 2.9) | Yes |
14.5 Immunogenicity of AFLURIA QUADRIVALENT in Children 6 Months through 59 Months Administered by Needle and Syringe
Study 4 was a randomized, observer-blind, comparator-controlled trial conducted in the U.S. in children 6 months through 59 months of age. A total of 2247 subjects were randomized 3:1 to receive AFLURIA QUADRIVALENT (N=1684) or a U.S.-licensed comparator quadrivalent influenza vaccine (N=563). Children 6 months through 35 months received one or two 0.25 mL doses and children 36 months through 59 months received one or two 0.5 mL doses. Subjects were eligible to receive a second dose at least 28 days after the first dose depending on their influenza vaccination history, consistent with the 2016-2017 recommendations of the Advisory Committee on Immunization Practices (ACIP) for Prevention and Control of Seasonal Influenza with Vaccines. Approximately 40% of subjects in each treatment group received two vaccine doses.
Baseline serology for HI assessment was collected prior to vaccination. Postvaccination immunogenicity was evaluated by HI assay on sera obtained 28 days after the last vaccination dose.
The primary objective was to demonstrate that vaccination with AFLURIA QUADRIVALENT elicits an immune response that is not inferior to that of a comparator vaccine containing the same recommended virus strains. The Per Protocol Population (AFLURIA QUADRIVALENT n=1456, Comparator QIV n=484) was used for the primary endpoint analyses. The co-primary endpoints were HI Geometric Mean Titer (GMT) ratios (adjusted for baseline HI titers and other covariates) and seroconversion rates for each vaccine strain, 28 days after the last vaccination. Pre-specified non-inferiority criteria required that the upper bound of the 2-sided 95% CI of the GMT ratio (Comparator QIV/AFLURIA QUADRIVALENT) did not exceed 1.5 and the upper bound of the 2-sided 95% CI of the seroconversion rate difference (Comparator QIV minus AFLURIA QUADRIVALENT) did not exceed 10.0% for each strain. Serum HI antibody responses to AFLURIA QUADRIVALENT were non-inferior for both GMT ratio and seroconversion rates relative to the comparator vaccine for all influenza strains (Table 10). Analyses of immunogenicity endpoints by gender did not demonstrate meaningful differences between males and females. The study population was not sufficiently diverse to assess differences among races or ethnicities.
| Post-vaccination GMT | GMT Ratio c | Seroconversion % d | SCR Difference e | Met both pre-defined non-inferiority criteria? f | |||
|---|---|---|---|---|---|---|---|
| Strain | AFLURIA
Quadrivalent N=1456 | Comparator
N=484 | Comparator over AFLURIA
Quadrivalent (95% CI) | AFLURIA
Quadrivalent N=1456 (95% CI) | Comparator
N=484 (95% CI) | Comparator minus AFLURIA
Quadrivalent (95% CI) |
|
|
Abbreviations: CI, confidence interval; Comparator, Comparator quadrivalent influenza vaccine (Fluzone Quadrivalent [Sanofi Aventis]); GMT (adjusted), geometric mean titer; SCR, seroconversion rate. |
|||||||
|
a NCT02914275 |
|||||||
|
b The Per-Protocol Population comprised all subjects (6 through 35 months of age receiving one or two 0.25 mL doses and 36 through 59 months of age receiving one or two 0.5 mL doses) in the Evaluable Population who did not have any protocol deviations that were medically assessed as potentially impacting on immunogenicity results. |
|||||||
|
c GMT Ratio = Comparator / AFLURIA QUADRIVALENT. Adjusted analysis model: Log-transformed Post-Vaccination HI Titer=Vaccine + Age Cohort [6 through 35 months or 36 through 59 months] + Gender + Vaccination History [y/n] + Log-transformed Pre-Vaccination HI Titer + Site + Number of Doses (1 vs 2) + Age Cohort*Vaccine. The Age Cohort*Vaccine interaction term was excluded from the model fit for the strains A(H1N1), A(H3N2) and B/Yamagata as the interaction result was non-significant (p>0.05). Least square means were back transformed. |
|||||||
|
d Seroconversion rate was defined as the percentage of subjects with either a prevaccination HI titer < 1:10 and a postvaccination HI titer ≥ 1:40 or a prevaccination HI titer ≥ 1:10 and a 4-fold increase in postvaccination HI titer. |
|||||||
|
e Seroconversion rate difference = Comparator SCR percentage minus AFLURIA QUADRIVALENT SCR percentage. |
|||||||
|
f Noninferiority (NI) criterion for the GMT ratio: upper bound of two-sided 95% CI on the GMT ratio of Comparator / AFLURIA QUADRIVALENT should not exceed 1.5. NI criterion for the SCR difference: upper bound of two-sided 95% CI on the difference between SCR Comparator – AFLURIA QUADRIVALENT should not exceed 10%. |
|||||||
|
g Subject 8400402-0073 was excluded from the Per-Protocol Population for the adjusted GMT analysis for the GMT ratio because the subject did not have information on all covariates (unknown prevaccination history). |
|||||||
|
h Subject 8400427-0070 had missing B/Victoria Antigen pre-vaccination titer. |
|||||||
| A(H1N1) | 353.5 (n=1455 g) | 281.0 (n=484) | 0.79 (0.72, 0.88) | 79.1 (76.9, 81.1) (n=1456) | 68.8 (64.5, 72.9) (n=484) | -10.3 (-15.4, -5.1) | Yes |
| A(H3N2) | 393.0 (n=1454 gi) | 500.5 (n=484) | 1.27 (1.15, 1.42) | 82.3 (80.2, 84.2) (n=1455 i) | 84.9 (81.4, 88.0) (n=484) | 2.6 (-2.5, 7.8) | Yes |
| B/Phuket/3073/ 2013 (B Yamagata) | 23.7 (n=1455 g) | 26.5 (n=484) | 1.12 (1.01, 1.24) | 38.9 (36.4, 41.4) (n=1456) | 41.9 (37.5, 46.5) (n=484) | 3.1 (-2.1, 8.2) | Yes |
| B/Brisbane/60/ 2008 (B Victoria) | 54.6 (n=1455 g) | 52.9 (n=483h) | 0.97 (0.86, 1.09) | 60.2 (57.6, 62.7) (n=1456) | 61.1 (56.6, 65.4) (n=483 h) | 0.9 (-4.2, 6.1) | Yes |
iSubject 8400402-0074 had missing A/H3N2 post-vaccination titer.
15. References
- Centers for Disease Control and Prevention. Prevention and Control of Influenza: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2010;59 (RR-8):1-62.
- Hannoun C, Megas F, Piercy J. Immunogenicity and Protective Efficacy of Influenza Vaccination. Virus Res 2004;103:133-138.
- Hobson D, Curry RL, Beare AS, et al. The Role of Serum Hemagglutination-Inhibiting Antibody in Protection against Challenge Infection with Influenza A2 and B Viruses. J Hyg Camb 1972;70:767-777.
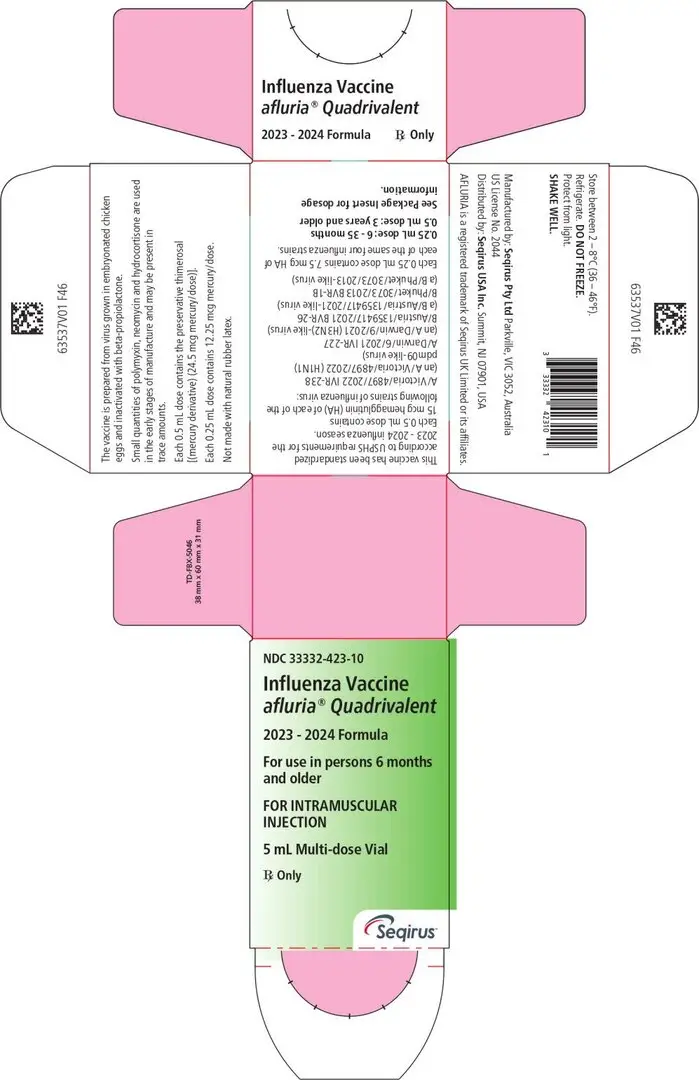
16. How is Afluria Quadrivalent supplied
16.1 How Supplied
Each product presentation includes a package insert and the following components:
| Presentation | Carton
NDC Number | Components |
| Pre-Filled Syringe | 33332-323-03 |
|
| Multi-Dose Vial | 33332-423-10 |
|
16.2 Storage and Handling
- Store refrigerated at 2-8°C (36-46°F).
- Do not freeze. Discard if product has been frozen.
- Protect from light.
- Do not use AFLURIA QUADRIVALENT beyond the expiration date printed on the label.
- Between uses, return the multi-dose vial to the recommended storage conditions.
- Once the stopper of the multi-dose vial has been pierced the vial must be discarded within 28 days.
- The number of needle punctures should not exceed 20 per multi-dose vial.
17. Patient Counseling Information
- Inform the vaccine recipient or guardian of the potential benefits and risks of immunization with AFLURIA QUADRIVALENT.
- Inform the vaccine recipient or guardian that AFLURIA QUADRIVALENT is an inactivated vaccine that cannot cause influenza but stimulates the immune system to produce antibodies that protect against influenza, and that the full effect of the vaccine is generally achieved approximately 3 weeks after vaccination.
- Instruct the vaccine recipient or guardian to report any severe or unusual adverse reactions to their healthcare provider.
- Encourage women who receive AFLURIA QUADRIVALENT while pregnant to enroll in the pregnancy registry. Pregnant women can enroll in the pregnancy registry by calling 1-855-358-8966 or sending an email to Seqirus at [email protected].
- Provide the vaccine recipient Vaccine Information Statements prior to immunization. These materials are available free of charge at the Centers for Disease Control and Prevention (CDC) website (www.cdc.gov/vaccines).
- Instruct the vaccine recipient that annual revaccination is recommended.
Manufactured by:
Seqirus Pty Ltd. Parkville, Victoria, 3052, Australia
U.S. License No. 2044
Distributed by:
Seqirus USA Inc. 25 Deforest Avenue, Summit, NJ 07901, USA
1-855-358-8966
AFLURIA and AFLURIA QUADRIVALENT are registered trademarks of Seqirus UK Limited or its affiliates.
PharmaJet® and Stratis® are trademarks of PharmaJet Inc.
Luer-Lok™ is a trademark of Becton, Dickinson and Company Corporation.
Principal Display Panel – 0.5 mL Carton Label
NDC 33332-323-03
Influenza Vaccine
afluria® Quadrivalent
2023-2024 Formula
For use in persons 3 years and older
FOR INTRAMUSCULAR INJECTION
10 Pre-Filled Syringes
each containing a single 0.5 mL dose
RX only
Seqirus™

Principal Display Panel – 0.5 mL Syringe Label
NDC 33332-323-04
Influenza Vaccine
afluria® Quadrivalent
2023-2024 Formula
0.5 mL Rx only
FOR IM USE ONLY
For 3 years and older


| AFLURIA QUADRIVALENT
influenza a virus a/victoria/4897/2022 ivr-238 (h1n1) antigen (propiolactone inactivated), influenza a virus a/darwin/6/2021 ivr-227 (h3n2) antigen (propiolactone inactivated), influenza b virus b/austria/1359417/2021 bvr-26 antigen (propiolactone inactivated), influenza b virus b/phuket/3073/2013 bvr-1b antigen (propiolactone inactivated) injection, suspension |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| AFLURIA QUADRIVALENT
influenza a virus a/victoria/4897/2022 ivr-238 (h1n1) antigen (propiolactone inactivated), influenza a virus a/darwin/6/2021 ivr-227 (h3n2) antigen (propiolactone inactivated), influenza b virus b/austria/1359417/2021 bvr-26 antigen (propiolactone inactivated), influenza b virus b/phuket/3073/2013 bvr-1b antigen (propiolactone inactivated) injection, suspension |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Seqirus PTY LTD. (747286735) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Seqirus Pty Ltd, Australia | 747286735 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon Manufacturing Services LLC, USA | 079415560 | MANUFACTURE | |
