Drug Detail:Arranon (Nelarabine [ nel-ar-a-been ])
Drug Class: Antimetabolites
Highlights of Prescribing Information
ARRANON® (nelarabine) injection, for intravenous use
Initial U.S. Approval: 2005
WARNING: NEUROLOGIC ADVERSE REACTIONS
See full prescribing information for complete boxed warning.
Severe neurologic adverse reactions have been reported with the use of ARRANON. These adverse reactions have included altered mental states including severe somnolence, central nervous system effects including convulsions, and peripheral neuropathy ranging from numbness and paresthesias to motor weakness and paralysis. There have also been reports of adverse reactions associated with demyelination, and ascending peripheral neuropathies similar in appearance to Guillain-Barré syndrome. (5.1)
Full recovery from these adverse reactions has not always occurred with cessation of therapy with ARRANON. Monitor frequently for signs and symptoms of neurologic toxicity. Discontinue ARRANON for neurologic adverse reactions of NCI Common Toxicity Criteria for Adverse Events (CTCAE) Grade 2 or greater. (5.1)
Recent Major Changes
|
Warnings and Precautions, Neurologic Adverse Reactions (5.1) |
11/2018 |
Indications and Usage for Arranon
ARRANON is a nucleoside metabolic inhibitor indicated for the treatment of patients with T-cell acute lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma (T-LBL) in adult and pediatric patients age 1 year and older whose disease has not responded to or has relapsed following treatment with at least two chemotherapy regimens. (1)
Arranon Dosage and Administration
- Adult Dose: 1500 mg/m2 administered intravenously over 2 hours on Days 1, 3, and 5 repeated every 21 days. (2.1)
- Pediatric Dose: 650 mg/m2 administered intravenously over 1 hour daily for 5 consecutive days repeated every 21 days. (2.1)
- Discontinue treatment for neurologic reactions greater than or equal to Grade 2. (2.2)
- Dosage may be delayed for hematologic reactions. (2.2)
- Take measures to prevent hyperuricemia. (2.4)
Dosage Forms and Strengths
Injection: 250 mg/50 mL (5 mg/mL) single-dose vial. (3)
Contraindications
None. (4)
Warnings and Precautions
-
Neurologic Adverse Reactions: Severe neurologic reactions have been reported. Monitor for signs and symptoms of neurologic toxicity. (5.1)
-
Hematologic Reactions: Complete blood counts including platelets should be monitored regularly. (5.2)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception; and advise males to use condoms. (5.3, 8.1, 8.3)
-
Effects on Ability to Drive and Use Machines: Somnolence may occur. Advise patients to refrain from these activities until somnolence has resolved. (5.6)
Adverse Reactions/Side Effects
The most common (≥ 20%) adverse reactions were:
-
Adult: anemia, thrombocytopenia, neutropenia, nausea, diarrhea, vomiting, constipation, fatigue, pyrexia, cough, and dyspnea. (6.1)
- Pediatric: anemia, neutropenia, thrombocytopenia, and leukopenia. (6.1)
The most common (> 10%) neurological adverse reactions were:
-
Adult: somnolence, dizziness, peripheral neurologic disorders, hypoesthesia, headache, and paresthesia. (6.1)
- Pediatric: headache and peripheral neurologic disorders. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Administration in combination with adenosine deaminase (ADA) inhibitors, such as pentostatin, is not recommended. (7, 12.3)
Use In Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
- Renal Impairment: Closely monitor patients with moderate or severe renal impairment for toxicities. (8.6)
- Hepatic Impairment: Closely monitor patients with severe hepatic impairment for toxicities. (8.7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 7/2019
Full Prescribing Information
1. Indications and Usage for Arranon
ARRANON is indicated for the treatment of T-cell acute lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma (T-LBL) in adult and pediatric patients age 1 year and older whose disease has not responded to or has relapsed following treatment with at least 2 chemotherapy regimens.
2. Arranon Dosage and Administration
2.1 Recommended Dosage
This product is for intravenous use only.
Adult Dosage: The recommended adult dose of ARRANON is 1500 mg/m2 administered intravenously over 2 hours on Days 1, 3, and 5 repeated every 21 days. Administer ARRANON undiluted.
Pediatric Dosage: The recommended pediatric dose of ARRANON is 650 mg/m2 administered intravenously over 1 hour daily for 5 consecutive days repeated every 21 days. Administer ARRANON undiluted.
The recommended duration of treatment for adult and pediatric patients has not been clearly established. In clinical trials, treatment was generally continued until there was evidence of disease progression, the patient experienced unacceptable toxicity, the patient became a candidate for hematopoietic stem cell transplantation (HSCT), or the patient no longer continued to benefit from treatment.
2.2 Dosage Modification
Discontinue ARRANON if the patient develops a neurologic adverse reaction of NCI CTCAE Grade 2 or greater. Dosage may be delayed for other toxicity, including hematologic toxicity [see Boxed Warning, Warnings and Precautions (5.1, 5.2)].
5. Warnings and Precautions
5.1 Neurologic Adverse Reactions
Nervous system adverse reactions of any grade were reported for 223 (76%) adult patients across the Phase I and Phase II trials, and Grade 3 or higher (severe, life-threatening, or fatal) adverse reactions were reported for 55 (19%) patients following initiation of ARRANON therapy [see Adverse Reactions (6.1)]. Based on patients with complete data, the median time to onset of first event is 5 days from start of first infusion (range: 1-166), and the median duration is 6 days (range: 1-393 days).
Nervous system adverse reactions of any grade were reported for 69 (42%) pediatric patients across the Phase I and Phase II trials, and Grade 3 or higher (severe, life-threatening, or fatal) adverse reactions were reported for 25 (15%) patients following initiation of ARRANON therapy [see Adverse Reactions (6.1)]. Based on patients with complete data, the median time to onset of first event is 8 days from start of first infusion (range: 1-269), and the median duration is 2 days (range: 1-82 days).
Common signs and symptoms of ARRANON-related neurotoxicity include somnolence, headache, paresthesia and dysesthesia, dizziness, neuropathy (sensory and motor), cerebellar disturbances and tremor. Severe neurologic toxicity can manifest as coma, status epilepticus, craniospinal demyelination, or ascending neuropathy similar in presentation to Guillain-Barré syndrome.
Full recovery from these adverse reactions has not always occurred with cessation of therapy with ARRANON. Patients treated previously or concurrently with intrathecal chemotherapy or previously with craniospinal irradiation may be at increased risk for neurologic adverse events.
Monitor patients frequently for signs and symptoms of neurologic toxicity during and for at least 24 hours after completion of treatment with ARRANON. Discontinue ARRANON for neurologic adverse reactions of NCI CTCAE Grade 2 or greater and provide supportive care [see Dosage and Administration (2.2), Adverse Reactions (6.1)].
5.2 Hematologic Adverse Reactions
Leukopenia, thrombocytopenia, anemia, and neutropenia, including febrile neutropenia, have been associated with ARRANON therapy. Complete blood counts including platelets should be monitored regularly [see Dosage and Administration (2.2), Adverse Reactions (6.1)].
5.3 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animal studies, ARRANON can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. In animal reproduction studies, intravenous administration of nelarabine to pregnant rabbits during the period of organogenesis resulted in teratogenicity at maternal doses below the recommended human adult dose of 1500 mg/m2/day (see Data).
Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with ARRANON. Advise males with female partners of reproductive potential to use condoms during treatment with ARRANON and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
5.6 Effects on Ability to Drive and Use Machines
Patients treated with ARRANON may experience somnolence during and for several days after treatment [see Adverse Reactions (6.1)]. Advise patients to refrain from driving or engaging in hazardous occupations or activities until somnolence has resolved.
6. Adverse Reactions/Side Effects
The following clinically-significant adverse reactions are discussed in greater detail in other sections of the label:
- Neurologic [see Boxed Warning, Warnings and Precautions (5.1)]
- Hematologic [see Warnings and Precautions (5.2)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.4)]
- Effects on Ability to Drive and Use Machines [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed or Refractory T-ALL and T-LBL
ARRANON was studied in 459 patients in Phase I and Phase II clinical trials.
Adult Patient: The safety profile of ARRANON is based on data from 103 adult patients treated with the recommended dose and schedule in 2 studies: an adult T-ALL/T-cell T-LBL trial and an adult chronic lymphocytic leukemia trial.
The most common adverse reactions in adults were fatigue; gastrointestinal disorders (nausea, diarrhea, vomiting, and constipation); hematologic disorders (anemia, neutropenia, and thrombocytopenia); respiratory disorders (cough and dyspnea); nervous system disorders (somnolence and dizziness); and pyrexia.
The most common adverse reactions in adults by Body System, including severe or life-threatening adverse reactions (NCI CTCAE Grade 3 or Grade 4) and fatal adverse reactions (Grade 5) are shown in Table 1.
| Abbreviation: AST, aspartate transaminase. a Five (5) patients had a fatal adverse reaction. Fatal adverse reactions included hypotension (n = 1), respiratory arrest (n = 1), pleural effusion/pneumothorax (n = 1), pneumonia (n = 1), and cerebral hemorrhage/coma/leukoencephalopathy (n = 1). |
|||
| Percentage of Patients (N = 103) | |||
| Toxicity Grade | |||
| Body System | Grade 3 | Grades 4 and 5a | All Grades |
| Adverse Reaction | % | % | % |
| Blood and Lymphatic System Disorders | |||
| Anemia | 20 | 14 | 99 |
| Thrombocytopenia | 37 | 22 | 86 |
| Neutropenia | 14 | 49 | 81 |
| Febrile neutropenia | 9 | 1 | 12 |
| Cardiac Disorders | |||
| Sinus tachycardia | 1 | 0 | 8 |
| Gastrointestinal Disorders | |||
| Nausea | 0 | 0 | 41 |
| Diarrhea | 1 | 0 | 22 |
| Vomiting | 1 | 0 | 22 |
| Constipation | 1 | 0 | 21 |
| Abdominal pain | 1 | 0 | 9 |
| Stomatitis | 1 | 0 | 8 |
| Abdominal distension | 0 | 0 | 6 |
| General Disorders and Administration Site Conditions | |||
| Fatigue | 10 | 2 | 50 |
| Pyrexia | 5 | 0 | 23 |
| Asthenia | 0 | 1 | 17 |
| Edema, peripheral | 0 | 0 | 15 |
| Edema | 0 | 0 | 11 |
| Pain | 3 | 0 | 11 |
| Rigors | 0 | 0 | 8 |
| Gait, abnormal | 0 | 0 | 6 |
| Chest pain | 0 | 0 | 5 |
| Noncardiac chest pain | 0 | 1 | 5 |
| Infections | |||
| Infection | 2 | 1 | 9 |
| Pneumonia | 4 | 1 | 8 |
| Sinusitis | 1 | 0 | 7 |
| Hepatobiliary Disorders | |||
| AST increased | 1 | 1 | 6 |
| Metabolism and Nutrition Disorders | |||
| Anorexia | 0 | 0 | 9 |
| Dehydration | 3 | 1 | 7 |
| Hyperglycemia | 1 | 0 | 6 |
| Musculoskeletal and Connective Tissue Disorders | |||
| Myalgia | 1 | 0 | 13 |
| Arthralgia | 1 | 0 | 9 |
| Back pain | 0 | 0 | 8 |
| Muscular weakness | 5 | 0 | 8 |
| Pain in extremity | 1 | 0 | 7 |
| Nervous System Disorders (see Table 2) | |||
| Psychiatric Disorders | |||
| Confusional state | 2 | 0 | 8 |
| Insomnia | 0 | 0 | 7 |
| Depression | 1 | 0 | 6 |
| Respiratory, Thoracic, and Mediastinal Disorders | |||
| Cough | 0 | 0 | 25 |
| Dyspnea | 4 | 2 | 20 |
| Pleural effusion | 5 | 1 | 10 |
| Epistaxis | 0 | 0 | 8 |
| Dyspnea, exertional | 0 | 0 | 7 |
| Wheezing | 0 | 0 | 5 |
| Vascular Disorders | |||
| Petechiae | 2 | 0 | 12 |
| Hypotension | 1 | 1 | 8 |
Other Adverse Reactions: Blurred vision was also reported in 4% of adult patients.
There was a single report of biopsy-confirmed progressive multifocal leukoencephalopathy in the adult patient population.
Neurologic Adverse Reactions: Nervous system adverse reactions, were reported for 76% of adult patients across the Phase I and Phase II trials. The most common neurologic adverse reactions (≥ 2%) in adult patients including all grades (NCI CTCAE) are shown in Table 2.
| Percentage of Patients (N =103) | |||||
| Nervous System Disorders | Grade 1 | Grade 2 | Grade 3 | Grade 4 | All Grades |
| Adverse Reaction | % | % | % | % | % |
| Somnolence | 20 | 3 | 0 | 0 | 23 |
| Dizziness | 14 | 8 | 0 | 0 | 21 |
| Peripheral neurologic disorders, any adverse reaction | 8 | 12 | 2 | 0 | 21 |
| Neuropathy | 0 | 4 | 0 | 0 | 4 |
| Peripheral neuropathy | 2 | 2 | 1 | 0 | 5 |
| Peripheral motor neuropathy | 3 | 3 | 1 | 0 | 7 |
| Peripheral sensory neuropathy | 7 | 6 | 0 | 0 | 13 |
| Hypoesthesia | 5 | 10 | 2 | 0 | 17 |
| Headache | 11 | 3 | 1 | 0 | 15 |
| Paresthesia | 11 | 4 | 0 | 0 | 15 |
| Ataxia | 1 | 6 | 2 | 0 | 9 |
| Depressed level of consciousness | 4 | 1 | 0 | 1 | 6 |
| Tremor | 2 | 3 | 0 | 0 | 5 |
| Amnesia | 2 | 1 | 0 | 0 | 3 |
| Dysgeusia | 2 | 1 | 0 | 0 | 3 |
| Balance disorder | 1 | 1 | 0 | 0 | 2 |
| Sensory loss | 0 | 2 | 0 | 0 | 2 |
One patient had a fatal neurologic adverse reaction, cerebral hemorrhage/coma/leukoencephalopathy.
Most nervous system adverse reactions in the adult patients were evaluated as Grade 1 or 2. The additional Grade 3 adverse reactions in adult patients, were aphasia, convulsion, hemiparesis, and loss of consciousness, each reported in 1 patient (1%). The additional Grade 4 adverse reactions were cerebral hemorrhage, coma, intracranial hemorrhage, leukoencephalopathy, and metabolic encephalopathy, each reported in one patient (1%).
The other neurologic adverse reactions reported as Grade 1, 2, or unknown in adult patients were abnormal coordination, burning sensation, disturbance in attention, dysarthria, hyporeflexia, neuropathic pain, nystagmus, peroneal nerve palsy, sciatica, sensory disturbance, sinus headache, and speech disorder, each reported in one patient (1%).
Pediatric Patient: The safety profile for children is based on data from 84 pediatric patients treated with the recommended dose and schedule in a T-ALL/T-LBL treatment trial.
The most common adverse reactions in pediatric patients were hematologic disorders (anemia, leukopenia, neutropenia, and thrombocytopenia). Of the non-hematologic adverse reactions in pediatric patients, the most frequent adverse reactions reported were headache, increased transaminase levels, decreased blood potassium, decreased blood albumin, increased blood bilirubin, and vomiting.
The most common adverse reactions in pediatric patients by System Organ Class including severe or life threatening adverse reactions (NCI CTCAE Grade 3 or Grade 4) and fatal adverse reactions (Grade 5) are shown in Table 3.
| a Three (3) patients had a fatal adverse reaction. Fatal adverse reactions included neutropenia and pyrexia (n = 1), status epilepticus/seizure (n = 1), and fungal pneumonia (n = 1). | |||
| Percentage of Patients (N = 84) | |||
| Toxicity Grade | |||
| Body System | Grade 3 | Grade 4 and 5a | All Grades |
| Adverse Reaction | % | % | % |
| Blood and Lymphatic System Disorders | |||
| Anemia | 45 | 10 | 95 |
| Neutropenia | 17 | 62 | 94 |
| Thrombocytopenia | 27 | 32 | 88 |
| Leukopenia | 14 | 7 | 38 |
| Hepatobiliary Disorders | |||
| Transaminases increased | 4 | 0 | 12 |
| Blood albumin decreased | 5 | 1 | 10 |
| Blood bilirubin increased | 7 | 2 | 10 |
| Metabolic/Laboratory | |||
| Blood potassium decreased | 4 | 2 | 11 |
| Blood calcium decreased | 1 | 1 | 8 |
| Blood creatinine increased | 0 | 0 | 6 |
| Blood glucose decreased | 4 | 0 | 6 |
| Blood magnesium decreased | 2 | 0 | 6 |
| Nervous System Disorders (see Table 4) | |||
| Gastrointestinal Disorders | |||
| Vomiting | 0 | 0 | 10 |
| General Disorders & Administration Site Conditions | |||
| Asthenia | 1 | 0 | 6 |
| Infections & Infestations | |||
| Infection | 2 | 1 | 5 |
Neurologic Adverse Reactions: Nervous system adverse reactions were reported for 42% of pediatric patients across the Phase I and Phase II trials. The most common neurologic adverse reactions (≥ 2%) in pediatric patients including all grades (NCI CTCAE) are shown in Table 4.
| a One (1) patient had a fatal neurologic adverse reaction, status epilepticus. | |||||
| Percentage of Patients (N = 84) | |||||
| Nervous System Disorders | Grade 1 | Grade 2 | Grade 3 | Grade 4 and 5a | All Grades |
| Adverse Reaction | % | % | % | % | % |
| Headache | 8 | 2 | 4 | 2 | 17 |
| Peripheral neurologic disorders, any adverse reaction | 1 | 4 | 7 | 0 | 12 |
| Peripheral neuropathy | 0 | 4 | 2 | 0 | 6 |
| Peripheral motor neuropathy | 1 | 0 | 2 | 0 | 4 |
| Peripheral sensory neuropathy | 0 | 0 | 6 | 0 | 6 |
| Somnolence | 1 | 4 | 1 | 1 | 7 |
| Hypoesthesia | 1 | 1 | 4 | 0 | 6 |
| Seizures | 0 | 0 | 0 | 6 | 6 |
| Convulsions | 0 | 0 | 0 | 3 | 4 |
| Grand mal convulsions | 0 | 0 | 0 | 1 | 1 |
| Status epilepticus | 0 | 0 | 0 | 1 | 1 |
| Motor dysfunction | 1 | 1 | 1 | 0 | 4 |
| Nervous system disorder | 1 | 2 | 0 | 0 | 4 |
| Paresthesia | 0 | 2 | 1 | 0 | 4 |
| Tremor | 1 | 2 | 0 | 0 | 4 |
| Ataxia | 1 | 0 | 1 | 0 | 2 |
The other Grade 3 neurologic adverse reaction in pediatric patients was hypertonia reported in 1 patient (1%). The additional Grade 4 neurologic adverse reactions, were third nerve paralysis, and sixth nerve paralysis, each reported in 1 patient (1%).
The other neurologic adverse reactions reported as Grade 1, 2, or unknown in pediatric patients were dysarthria, encephalopathy, hydrocephalus, hyporeflexia, lethargy, mental impairment, paralysis, and sensory loss, each reported in 1 patient (1%).
ARRANON in Combination with Multi-Agent Chemotherapy in T-ALL and T-LBL
ARRANON was studied in combination with multi-agent chemotherapy in a randomized clinical trial [NCT00408005]. The safety population in this trial included 804 patients with newly-diagnosed T-ALL (85%) or T-LBL (15%) treated with (n = 411) or without (n =393) ARRANON in combination with the augmented Berlin-Frankfurt-Münster chemotherapy regimen (aBFM) after initial induction therapy. Patients assigned to ARRANON received 650 mg/m2 intravenously over 1 hour daily for 5 consecutive days, during consolidation Days 1 to 5 and 43 to 47, delayed intensification Days 29 to 33, and during the initial 3 courses of maintenance Days 29 to 33. The median age on enrollment was 9.5 years (range: 1-29), the majority of patients were male (73%) and white (69%). Sixty-five percent of patients assigned to the ARRANON arms received at least 85% of the planned dose through the third course of maintenance therapy compared to 79% of patients on the control arms who received 3 courses of maintenance therapy.
There was one fatal neurological adverse reaction in the ARRANON arm. The incidence of the following Grades 3 and 4 adverse reactions were higher in the ARRANON treated arms compared to the control arms: abnormal transaminases, motor and sensory neuropathy, nausea and vomiting, and dehydration. The incidence of seizures of any grade was 3% (14 of 411). Rhabdomyolysis was diagnosed in 2% (7 of 411) of ARRANON treated patients and occurred after the first course of ARRANON during the consolidation phase of therapy.
7. Drug Interactions
Administration of ARRANON in combination with adenosine deaminase (ADA) inhibitors, such as pentostatin, is not recommended [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings in animal studies, ARRANON can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Limited available data with ARRANON use in pregnant women are insufficient to determine a drug-associated risk for major birth defects, miscarriage or adverse maternal, or fetal outcomes. There are risks to the pregnant woman associated with untreated leukemia or lymphoma (see Clinical Considerations). In animal reproduction studies, intravenous administration of nelarabine to pregnant rabbits during the period of organogenesis resulted in teratogenicity at maternal doses below the recommended human adult dose of 1500 mg/m2/day (see Data). Advise pregnant women of the potential risk to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo-fetal Risk
There are risks to the mother from untreated leukemia or lymphoma, including anemia, thrombocytopenia, and death.
Data
Animal Data
In an embryo-fetal development study in which pregnant rabbits were administered daily doses of nelarabine during organogenesis, increased incidences of fetal malformations, anomalies, and variations were observed at doses greater than or equal to 360 mg/m2/day (8-hour IV infusion; approximately 25% of the recommended human adult dose compared on a mg/m2 basis), which was the lowest dose tested. Cleft palate was seen in rabbits given 3600 mg/m2/day (approximately 2-fold the adult dose), absent pollices (digits) in rabbits given greater than or equal to 1200 mg/m2/day (approximately 75% of the recommended adult dose), while absent gall bladder, absent accessory lung lobes, fused or extra sternebrae, and delayed ossification was seen at all doses. Maternal body weight gain and fetal body weights were reduced in rabbits given 3600 mg/m2/day (approximately 2-fold the adult dose), but could not account for the increased incidence of malformations seen at this or lower administered doses.
8.2 Lactation
Risk Summary
There are no data on the presence of nelarabine or ara-G in human or animal milk, the effect on the breastfed child, or the effect on milk production. Because of the potential for serious adverse reactions in the breastfed child from ARRANON, such as severe neurological reactions, advise women not to breastfeed during treatment with ARRANON.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
ARRANON can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Verify the pregnancy status of females of reproductive potential prior to starting treatment with ARRANON.
Contraception
Females
ARRANON can cause fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.3), Use in Specific Populations (8.1)]. Because of the potential for genotoxicity, advise females of reproductive potential to use effective contraception during treatment with ARRANON.
Males
Because of the potential for genotoxicity, advise males (including those who have had vasectomies) with female partners of reproductive potential to use condoms during treatment with ARRANON and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
8.6 Renal Impairment
Ara-G clearance decreased as renal function decreased [see Clinical Pharmacology (12.3)]. Because the risk of adverse reactions to this drug may be greater in patients with moderate (CLCr 30 to 50 mL/min) or severe (CLCr less than 30 mL/min) renal impairment, these patients should be closely monitored for toxicities when treated with ARRANON [see Dosage and Administration (2.3)].
8.7 Hepatic Impairment
The influence of hepatic impairment on the pharmacokinetics of nelarabine has not been evaluated. Because the risk of adverse reactions to this drug may be greater in patients with severe hepatic impairment (total bilirubin greater than 3 times upper limit of normal), these patients should be closely monitored for toxicities when treated with ARRANON.
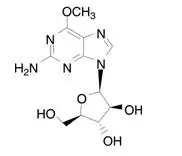
12. Arranon - Clinical Pharmacology
12.3 Pharmacokinetics
Absorption: Following intravenous administration of nelarabine to adult patients with refractory leukemia or lymphoma, plasma ara-G Cmax values generally occurred at the end of the nelarabine infusion and were generally higher than nelarabine Cmax values, suggesting rapid and extensive conversion of nelarabine to ara-G. Mean plasma nelarabine and ara-G Cmax values were 5.0 ± 3.0 mcg/mL and 31.4 ± 5.6 mcg/mL, respectively, after a 1500 mg/m2 nelarabine dose infused over 2 hours in adult patients. The area under the concentration-time curve (AUC) of ara-G is 37 times higher than that for nelarabine on Day 1 after nelarabine IV infusion of 1500 mg/m2 dose (162 ± 49 mcg.h/mL versus 4.4 ± 2.2 mcg.h/mL, respectively). Comparable Cmax and AUC values were obtained for nelarabine between Days 1 and 5 at the nelarabine adult dosage of 1500 mg/m2, indicating that nelarabine does not accumulate after multiple-dosing. There are not enough ara-G data to make a comparison between Day 1 and Day 5. After a nelarabine adult dose of 1500 mg/m2, intracellular Cmax for ara-GTP appeared within 3 to 25 hours on Day 1. Exposure (AUC) to intracellular ara-GTP was 532 times higher than that for nelarabine and 14 times higher than that for ara-G (2,339 ± 2,628 mcg.h/mL versus 4.4 ± 2.2 mcg.h/mL and 162 ± 49 mcg.h/mL, respectively). Because the intracellular levels of ara-GTP were so prolonged, its elimination half-life could not be accurately estimated.
Distribution: Nelarabine and ara-G are extensively distributed throughout the body. For nelarabine, VSS values were 197 ± 216 L/m2 in adult patients. For ara-G, VSS/F values were 50 ± 24 L/m2 in adult patients.
Nelarabine and ara-G are not substantially bound to human plasma proteins (< 25%) in vitro, and binding is independent of nelarabine or ara-G concentrations up to 600 μM.
Metabolism: The principal route of metabolism for nelarabine is O-demethylation by ADA to form ara-G, which undergoes hydrolysis to form guanine. In addition, some nelarabine is hydrolyzed to form methylguanine, which is O-demethylated to form guanine. Guanine is N-deaminated to form xanthine, which is further oxidized to yield uric acid.
Excretion: Nelarabine and ara-G are partially eliminated by the kidneys. Mean urinary excretion of nelarabine and ara-G was 6.6 ± 4.7% and 27 ± 15% of the administered dose, respectively, in 28 adult patients over the 24 hours after nelarabine infusion on Day 1. Renal clearance averaged 24 ± 23 L/h for nelarabine and 6.2 ± 5.0 L/h for ara-G in 21 adult patients. Combined Phase I pharmacokinetic data at nelarabine doses of 199 to 2900 mg/m2 (n = 66 adult patients) indicate that the mean clearance (CL) of nelarabine is 197 ± 189 L/h/m2 on Day 1. The apparent clearance of ara-G (CL/F) is 10.5 ± 4.5 L/h/m2 on Day 1. Nelarabine and ara-G are rapidly eliminated from plasma with a mean half-life of 18 minutes and 3.2 hours, respectively, in adult patients.
Pediatrics: No pharmacokinetic data are available in pediatric patients at the once-daily 650 mg/m2 nelarabine dosage. Combined Phase I pharmacokinetic data at nelarabine doses of 104 to 2900 mg/m2 indicate that the mean clearance (CL) of nelarabine is about 30% higher in pediatric patients than in adult patients (259 ± 409 L/h/m2 versus 197 ± 189 L/h/m2, respectively) (n = 66 adults, n = 22 pediatric patients) on Day 1. The apparent clearance of ara-G (CL/F) is comparable between the 2 groups (10.5 ± 4.5 L/h/m2 in adult patients and 11.3 ± 4.2 L/h/m2 in pediatric patients) on Day 1. Nelarabine and ara-G are extensively distributed throughout the body. For nelarabine, VSS values were 213 ± 358 L/m2 in pediatric patients. For ara-G, VSS/F values were 33 ± 9.3 L/m2 in pediatric patients. Nelarabine and ara-G are rapidly eliminated from plasma in pediatric patients, with a half-life of 13 minutes and 2 hours, respectively.
Effect of Age: Age has no effect on the pharmacokinetics of nelarabine or ara-G in adults. Decreased renal function, which is more common in the elderly, may reduce ara-G clearance [see Use in Specific Populations (8.5)].
Effect of Gender: Gender has no effect on nelarabine or ara-G pharmacokinetics.
Effect of Race: In general, nelarabine mean clearance and volume of distribution values tend to be higher in whites (n = 63) than in blacks (by about 10%) (n = 15). The opposite is true for ara-G; mean apparent clearance and volume of distribution values tend to be lower in whites than in blacks (by about 15% to 20%). No differences in safety or effectiveness were observed between these groups.
Effect of Renal Impairment: The pharmacokinetics of nelarabine and ara-G have not been specifically studied in renally impaired or hemodialyzed patients. Nelarabine is excreted by the kidney to a small extent (5% to 10% of the administered dose). Ara-G is excreted by the kidney to a greater extent (20% to 30% of the administered nelarabine dose). In the combined Phase I trials, patients were categorized into 3 groups: normal with CLCr greater than 80 mL/min (n = 67), mild with CLCr = 50 to 80 mL/min (n = 15), and moderate with CLCr less than 50 mL/min (n = 3). The mean apparent clearance (CL/F) of ara-G was about 15% and 40% lower in patients with mild and moderate renal impairment, respectively, than in patients with normal renal function [see Use in Specific Populations (8.6), Dosage and Administration (2.3)]. No differences in safety or effectiveness were observed.
Effect of Hepatic Impairment: The influence of hepatic impairment on the pharmacokinetics of nelarabine has not been evaluated [see Use in Specific Populations (8.7)].
Drug Interactions: Cytochrome P450: Nelarabine and ara-G did not significantly inhibit the activities of the human hepatic cytochrome P450 isoenzymes 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4 in vitro at concentrations of nelarabine and ara-G up to 100 μM.
Fludarabine: Administration of fludarabine 30 mg/m2 as a 30-minute infusion 4 hours before a 1200-mg/m2 infusion of nelarabine did not affect the pharmacokinetics of nelarabine, ara-G, or ara-GTP in 12 patients with refractory leukemia.
Pentostatin: There is in vitro evidence that pentostatin is a strong inhibitor of ADA. Inhibition of ADA may result in a reduction in the conversion of the prodrug nelarabine to its active moiety and consequently in a reduction in efficacy of nelarabine and/or change in adverse reaction profile of either drug [see Drug Interactions (7)].

| ARRANON
nelarabine injection |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
