Drug Detail:Dalvance (Dalbavancin [ dal-ba-van-sin ])
Drug Class: Glycopeptide antibiotics
Highlights of Prescribing Information
DALVANCE (dalbavancin) for injection, for intravenous use
Initial U.S. Approval: 2014
Recent Major Changes
| Indications and Usage (1) | 7/2021 |
| Dosage and Administration (2) | 7/2021 |
| Contraindications (4) | 7/2021 |
| Warnings and Precautions (5) | 7/2021 |
Indications and Usage for Dalvance
DALVANCE is a lipoglycopeptide antibacterial indicated for the treatment of adult and pediatric patients with acute bacterial skin and skin structure infections (ABSSSI) caused by designated susceptible strains of Gram-positive microorganisms. (1.1)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of DALVANCE and other antibacterial drugs, DALVANCE should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. (1.2)
Dalvance Dosage and Administration
| Dosage in Adult Patients (2.1, 2.3): | ||
| Estimated Creatinine Clearance (CLcr) | Single Dose
Regimen | Two-Dose Regimen |
| 30 mL/min and above or on regular hemodialysis | 1500 mg | 1000 mg followed one week later by 500 mg |
| Less than 30 mL/min and not on regular hemodialysis | 1125 mg | 750 mg followed one week later by 375 mg |
- Administer by intravenous infusion over 30 minutes (2.1, 2.4)
- See Full Prescribing Information for instructions on reconstitution of lyophilized powder and preparation of injection (2.4).
| Dosage in Pediatric Patients with CLcr 30 mL/min/1.73m2
and above (2.2) |
|
| Age Range | Dosage (Single Dose Regimen) |
| Birth to less than 6 years | 22.5 mg/kg (maximum of 1500 mg) |
| 6 to less than 18 years | 18 mg/kg (maximum of 1500 mg) |
- Dosage adjustment in pediatric patients with CLcr less than 30 mL/min has not been studied.
Dosage Forms and Strengths
For injection: 500 mg of lyophilized powder in a vial for reconstitution (3)
Contraindications
Known hypersensitivity to dalbavancin (4)
Warnings and Precautions
- Serious hypersensitivity (anaphylactic) and skin reactions have been reported in patients treated with DALVANCE. If an allergic reaction occurs, discontinue treatment with DALVANCE and institute appropriate therapy for the allergic reaction. Carefully monitor patients with known hypersensitivity to glycopeptides. (5.1)
- Rapid intravenous infusion of DALVANCE can cause flushing of the upper body, urticaria, pruritus, rash, and/or back pain. Stopping or slowing the infusion may result in cessation of these reactions. (5.2)
- Alanine Aminotransferase (ALT) elevations with DALVANCE treatment were reported in clinical trials. (5.3, 6.1)
- Clostridioides difficile-associated diarrhea (CDAD) has been reported with nearly all systemic antibacterial agents, including DALVANCE. Evaluate if diarrhea occurs. (5.4)
Adverse Reactions/Side Effects
The most common adverse reactions occurring in >4% of adult patients treated with DALVANCE were nausea, headache, and diarrhea. The most common adverse reaction that occured in >1% of pediatric patients was pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Allergan at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2021
Full Prescribing Information
1. Indications and Usage for Dalvance
1.1 Acute Bacterial Skin and Skin Structure Infections
DALVANCE® is indicated for the treatment of adult and pediatric patients with acute bacterial skin and skin structure infections (ABSSSI) caused by designated susceptible strains of the following Gram-positive microorganisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant isolates), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including S. anginosus, S. intermedius, S. constellatus) and Enterococcus faecalis (vancomycin susceptible isolates).
1.2 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of DALVANCE and other antibacterial agents, DALVANCE should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2. Dalvance Dosage and Administration
2.1 Recommended Dosage Regimen in Adult Patients with CLcr 30 mL/min and Above
The recommended dosage regimen of DALVANCE in adult patients with CLcr 30 mL/min and above is 1500 mg, administered either as a single dose regimen, or as a two-dose regimen of DALVANCE 1000 mg followed one week later by 500 mg. Administer DALVANCE over 30 minutes by intravenous infusion. For adult patients with CLcr less than 30 mL/min, dosage adjustment is required [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
2.2 Recommended Dosage Regimen in Pediatric Patients with CLcr 30 mL/min/1.73m2 and Above
The recommended dosage regimen of DALVANCE in pediatric patients with CLcr 30 mL/min/1.73m2 and above is a single dose regimen based on the age and weight of the pediatric patient (Table 1). Administer DALVANCE over 30 minutes by intravenous infusion.
There is insufficient information to recommend dosage adjustment for pediatric patients younger than 18 years with CLcr less than 30 mL/min/1.73m2 [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.3)].
| Age Range | Dosage (Single Dose Regimen) |
| Birth to less than 6 years | 22.5 mg/kg (maximum 1500 mg) |
| 6 to less than 18 years | 18 mg/kg (maximum 1500 mg) |
*Estimate CLcr or glomerular filtration rate (GFR) using an age-appropriate equation accepted for pediatric
patients (birth to less than 18 years old) to define renal function impairment.
2.3 Dosage Adjustments in Adult Patients with CLcr less than 30 mL/min
In adult patients with renal impairment whose known CLcr is less than 30 mL/min and who are not receiving regularly scheduled hemodialysis, the recommended dosage regimen of DALVANCE is 1125 mg, administered either as a single dose regimen, or as a two-dose regimen of DALVANCE 750 mg followed one week later by 375 mg.
No dosage adjustment is recommended for adult patients receiving regularly scheduled hemodialysis, and DALVANCE can be administered without regard to the timing of hemodialysis [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.4 Preparation and Administration
DALVANCE (dalbavancin) for injection must be reconstituted with either Sterile Water for Injection, USP, or 5% Dextrose Injection, USP, and subsequently diluted only with 5% Dextrose Injection, USP, to a final concentration of 1 mg/mL to 5 mg/mL.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Reconstitution: DALVANCE must be reconstituted under aseptic conditions, using 25 mL of either Sterile Water for Injection, USP, or 5% Dextrose Injection, USP, for each 500 mg vial. To avoid foaming, alternate between gentle swirling and inversion of the vial until its contents are completely dissolved. Do not shake. The reconstituted vial contains 20 mg/mL dalbavancin as a clear, colorless to yellow solution.
Reconstituted vials may be stored either refrigerated at 2°C to 8 °C (36°F to 46 °F), or at controlled room temperature 20°C to 25 °C (68°F to 77 °F). Do not freeze.
Dilution:
Adult Patients: Aseptically transfer the required dose of reconstituted DALVANCE solution from the vial(s) to an intravenous bag or bottle containing 5% Dextrose Injection, USP. The diluted solution must have a final dalbavancin concentration of 1 mg/mL to 5 mg/mL. Discard any unused portion of the reconstituted solution.
Pediatric Patients: For pediatric patients, the dose of DALVANCE will vary according to the age and weight of the child up to a maximum of 1500 mg [see Dosage and Administration (2.2)]. Aseptically transfer the required dose of reconstituted DALVANCE solution, based on the child’s weight, from the vial(s) to an intravenous bag or bottle containing 5% Dextrose Injection, USP. The diluted solution must have a final dalbavancin concentration of 1 mg/mL to 5 mg/mL. Discard any unused portion of the reconstituted solution.
Once diluted into an intravenous bag or bottle as described above, DALVANCE may be stored either refrigerated at 2 °C to 8 °C (36 °F to 46 °F) or at a controlled room temperature of 20°C to 25 °C (68 °F to 77 °F). Do not freeze.
The total time from reconstitution to dilution to administration should not exceed 48 hours.
Like all parenteral drug products, diluted DALVANCE should be inspected visually for particulate matter prior to infusion. If particulate matter is identified, do not use.
Administration: After reconstitution and dilution, administer DALVANCE via intravenous infusion, using a total infusion time of 30 minutes.
Do not co-infuse DALVANCE with other medications or electrolytes. Saline-based infusion solutions may cause precipitation and should not be used. The compatibility of reconstituted DALVANCE with intravenous medications, additives, or substances other than 5% Dextrose Injection, USP has not been established.
If a common intravenous line is being used to administer other drugs in addition to DALVANCE, the line should be flushed before and after each DALVANCE infusion with 5% Dextrose Injection, USP.
3. Dosage Forms and Strengths
DALVANCE is supplied in clear glass vials containing sterile powder (white/off-white to pale yellow) equivalent to 500 mg of dalbavancin.
4. Contraindications
DALVANCE is contraindicated in patients with known hypersensitivity to dalbavancin.
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Serious hypersensitivity (anaphylactic) and skin reactions have been reported in patients treated with DALVANCE. If an allergic reaction to DALVANCE occurs, discontinue treatment with DALVANCE and institute appropriate therapy for the allergic reaction. Before using DALVANCE, inquire carefully about previous hypersensitivity reactions to other glycopeptides. Due to the possibility of cross-sensitivity, carefully monitor for signs of hypersensitivity during treatment with DALVANCE in patients with a history of glycopeptide allergy [see Patient Counseling Information (17)].
5.2 Infusion-Related Reactions
DALVANCE is administered via intravenous infusion, using a total infusion time of 30 minutes to minimize the risk of infusion-related reactions. Rapid intravenous infusions of DALVANCE can cause flushing of the upper body, urticaria, pruritus, rash, and/or back pain. Stopping or slowing the infusion may result in cessation of these reactions.
5.3 Hepatic Effects
In Phase 2 and 3 clinical trials, more DALVANCE than comparator-treated subjects with normal baseline transaminase levels had post-baseline alanine aminotransferase (ALT) elevation greater than 3 times the upper limit of normal (ULN). Overall, abnormalities in liver tests (ALT, AST, bilirubin) were reported with similar frequency in the DALVANCE and comparator arms [see Adverse Reactions (6.1)].
5.4 Clostridioides difficile-Associated Diarrhea
Clostridioides difficile-associated diarrhea (CDAD) has been reported in users of nearly all systemic antibacterial drugs, including DALVANCE, with severity ranging from mild diarrhea to fatal colitis. Treatment with antibacterial agents can alter the normal flora of the colon, and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin-producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antibacterial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial use not directed against C. difficile should be discontinued, if possible. Appropriate measures such as fluid and electrolyte management, protein supplementation, antibacterial treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are also discussed elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Infusion Related Reactions [see Warnings and Precautions (5.2)]
- Hepatic Effects [see Warnings and Precautions (5.3)]
- Clostridioides difficile-associated Diarrhea [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of DALVANCE cannot be directly compared to rates in the clinical trials of another drug and may not reflect rates observed in practice.
Clinical Trials Experience in Adult Patients
Adverse reactions were evaluated for 2473 patients treated with DALVANCE: 1778 patients were treated with DALVANCE in seven Phase 2/3 trials comparing DALVANCE to comparator antibacterial drugs and 695 patients were treated with DALVANCE in one Phase 3 trial comparing DALVANCE single and two-dose regimens. The median age of patients treated with DALVANCE was 48 years, ranging between 16 and 93 years. Patients treated with DALVANCE were predominantly male (59.5%) and White (81.2%).
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
Serious adverse reactions occurred in 121/2473 (4.9%) of patients treated with any regimen of DALVANCE. In the Phase 2/3 trials comparing DALVANCE to comparator, serious adverse reactions occurred in 109/1778 (6.1%) of patients in the DALVANCE group and 80/1224 (6.5%) of patients in the comparator group. In a Phase 3 trial comparing DALVANCE single and two-dose regimens, serious adverse reactions occurred in 7/349 (2.0%) of patients in the DALVANCE single dose group and 5/346 (1.4%) of patients in the DALVANCE two-dose group. DALVANCE was discontinued due to an adverse reaction in 64/2473 (2.6%) patients treated with any regimen of DALVANCE. In the Phase 2/3 trials comparing DALVANCE to comparator, DALVANCE was discontinued due to an adverse reaction in 53/1778 (3.0%) of patients in the DALVANCE group and 35/1224 (2.9%) of patients in the comparator group. In a Phase 3 trial comparing DALVANCE single and two-dose regimens, DALVANCE was discontinued due to an adverse reaction in 6/349 (1.7%) of patients in the DALVANCE single dose group and 5/346 (1.4%) of patients in the DALVANCE two-dose group.
Most Common Adverse Reactions
The most common adverse reactions in patients treated with DALVANCE in Phase 2/3 trials were nausea (5.5%), headache (4.7%), and diarrhea (4.4%). The median duration of adverse reactions was 3.0 days in patients treated with DALVANCE. In the Phase 2/3 trials comparing DALVANCE to comparator, the median duration of adverse reactions was 3.0 days for patients in the DALVANCE group and 4.0 days in patients in the comparator group. In a Phase 3 trial comparing DALVANCE single and two-dose regimens, the median duration of adverse reactions was 3.0 days for patients in the DALVANCE single and two-dose group.
Table 2 lists selected adverse reactions occurring in 2% or more of patients treated with DALVANCE in Phase 2/3 clinical trials.
| Table 2. Selected Adverse Reactions Occurring in ≥ 2% of Patients Receiving DALVANCE in Phase 2/3 Trials
(Number (%) of Patients) |
||
| Adverse Reactions | DALVANCE | Comparator* |
| (N = 1778) | (N = 1224) | |
| Nausea | 98 (5.5) | 78 (6.4) |
| Diarrhea | 79 (4.4) | 72 (5.9) |
| Headache | 83 (4.7) | 59 (4.8) |
| Vomiting | 50 (2.8) | 37 (3) |
| Rash | 48 (2.7) | 30 (2.4) |
| Pruritus | 38 (2.1) | 41 (3.3) |
| * Comparators included linezolid, cefazolin, cephalexin, and vancomycin. | ||
In the Phase 3 trial comparing the single and two-dose regimen of DALVANCE, the adverse reaction that occurred in 2% or more of patients treated with DALVANCE was nausea (3.4% in the DALVANCE single dose group and 2% in the DALVANCE two-dose group).
The following selected adverse reactions were reported in DALVANCE treated patients at a rate of less than 2% in these clinical trials:
Blood and lymphatic system disorders: anemia, hemorrhagic anemia, leucopenia, neutropenia, thrombocytopenia, petechiae, eosinophilia, thrombocytosis
Gastrointestinal disorders: gastrointestinal hemorrhage, melena, hematochezia, abdominal pain
General disorders and administration site conditions: infusion-related reactions
Hepatobiliary disorders: hepatotoxicity
Immune system disorders: anaphylactic reaction
Infections and infestations: Clostridioides difficile colitis, oral candidiasis, vulvovaginal mycotic infection
Investigations: hepatic transaminases increased, blood alkaline phosphatase increased, international normalized ratio increased, blood lactate dehydrogenase increased, gamma-glutamyl transferase increased
Metabolism and nutrition disorders: hypoglycemia
Nervous system disorders: dizziness
Respiratory, thoracic and mediastinal disorders: bronchospasm
Skin and subcutaneous tissue disorders: rash, pruritus, urticaria
Vascular disorders: flushing, phlebitis, wound hemorrhage, spontaneous hematoma
Alanine Aminotransferase (ALT) Elevations
Among patients with normal baseline ALT levels treated with DALVANCE 17 (0.8%) had post baseline ALT elevations greater than 3 times the upper limit of normal (ULN) including five subjects with post-baseline ALT values greater than 10 times ULN. Among patients with normal baseline ALT levels treated with non-DALVANCE comparators 2 (0.2%) had post-baseline ALT elevations greater than 3 times the upper limit of normal. Fifteen of the 17 patients treated with DALVANCE and one comparator patient had underlying conditions which could affect liver enzymes, including chronic viral hepatitis, history of alcohol abuse and metabolic syndrome. In addition, one DALVANCE-treated subject in a Phase 1 trial had post-baseline ALT elevations greater than 20 times ULN. ALT elevations were reversible in all subjects with follow-up assessments. No comparator-treated subject with normal baseline transaminases had post-baseline ALT elevation greater than 10 times ULN.
Clinical Trials Experience in Pediatric Patients
Adverse reactions were evaluated in one Phase 3 pediatric clinical trial which included 161 pediatric patients from birth to less than 18 years of age with ABSSSI treated with DALVANCE (83 patients treated with a single dose of DALVANCE and 78 patients treated with a two-dose regimen of DALVANCE) and 30 patients treated with comparator agents for a treatment period up to 14 days. The median age of pediatric patients treated with DALVANCE was 9 years, ranging from birth to <18 years. The majority of patients were male (62.3%) and White (89.0%).
The safety findings of DALVANCE in pediatric patients were similar to those observed in adults.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
Serious adverse reactions (SARs) occurred in 3/161 (1.9%) of patients treated with DALVANCE, all in the single-dose arm. There were no adverse reactions leading to DALVANCE discontinuation.
Most Common Adverse Reactions
Most common adverse reaction occurring in more than 1% of pediatric patients 2/161 (1.2%) was pyrexia.
Other Adverse Reactions
The following selected adverse reactions were reported in DALVANCE-treated patients at a rate of less than 1% in this pediatric clinical trial:
Gastrointestinal disorders: diarrhea
Nervous system disorders: dizziness
Skin and subcutaneous tissue disorders: pruritus
8. Use In Specific Populations
8.4 Pediatric Use
The safety and effectiveness of DALVANCE for the treatment of ABSSSI has been established in pediatric patients aged birth to less than 18 years. Use of DALVANCE for this indication is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients aged birth to less than 18 years [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)].
There is insufficient information to recommend dosage adjustment for pediatric patients with ABSSSI and CLcr less than 30 mL/min/1.73m2 [see Dosage and Administration (2.2)].
8.6 Renal Impairment
In patients with renal impairment whose known CLcr is less than 30 mL/min and who are not receiving regularly scheduled hemodialysis, the recommended regimen for DALVANCE is 1125 mg, administered as a single dose, or 750 mg followed one week later by 375 mg. No dosage adjustment is recommended for patients receiving regularly scheduled hemodialysis, and DALVANCE can be administered without regard to the timing of hemodialysis. There is insufficient information to recommend dosage adjustment for pediatric patients younger than 18 years with CLcr less than 30 mL/min/1.73m2 [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
10. Overdosage
Specific information is not available on the treatment of overdose with DALVANCE, as dose-limiting toxicity has not been observed in clinical studies. In Phase 1 studies, healthy volunteers have been administered cumulative doses of up to 4500 mg over a period of up to 8 weeks (not an approved dosing regimen), with no signs of toxicity or laboratory results of clinical concern.
Treatment of overdose with DALVANCE should consist of observation and general supportive measures. Although no information is available specifically regarding the use of hemodialysis to treat overdose, in a Phase 1 study in patients with renal impairment less than 6% of the recommended dalbavancin dose was removed [see Clinical Pharmacology (12.3)].
12. Dalvance - Clinical Pharmacology
12.3 Pharmacokinetics
General Pharmacokinetic Properties
Dalbavancin pharmacokinetic parameters have been characterized in healthy subjects, patients, and specific populations. Pharmacokinetic parameters following administration of single intravenous 1000 mg and 1500 mg doses were as shown in Table 4. The pharmacokinetics of dalbavancin can be described using a three-compartment model.
| Table 4. Dalbavancin Pharmacokinetic Parameters in Healthy Subjects | ||
| Parameter | Single 1000 mg Dose | Single 1500 mg Dose |
| Cmax (mg/L) | 287 (13.9)1 | 423 (13.2)4 |
| AUC0-24 (mg•h/L) | 3185 (12.8)1 | 4837 (13.7)4 |
| AUC0-Day7 (mg•h/L) | 11160 (41.1)2 | ND |
| AUC0-inf (mg•h/L) | 23443 (40.9)2 | ND |
| Terminal t½ (h) | 346 (16.5)2,3 | ND |
| CL (L/h) | 0.0513 (46.8)2 | ND |
| All values are presented as mean (% coefficient of variation) 1 Data from 50 healthy subjects. 2 Data from 12 healthy subjects. 3 Based upon population pharmacokinetic analyses of data from patients, the effective half-life is approximately 8.5 days (204 hours). 4 Data from 49 healthy subjects. Abbreviation: ND – not determined |
||
In healthy subjects, dalbavancin AUC0-24h and Cmax both increased proportionally to dose following single IV dalbavancin doses ranging from 140 mg to 1500 mg, indicating linear pharmacokinetics.
The mean plasma concentration-time profile for dalbavancin following the recommended two-dose regimen of 1000 mg followed one week later by 500 mg is shown in Figure 2.
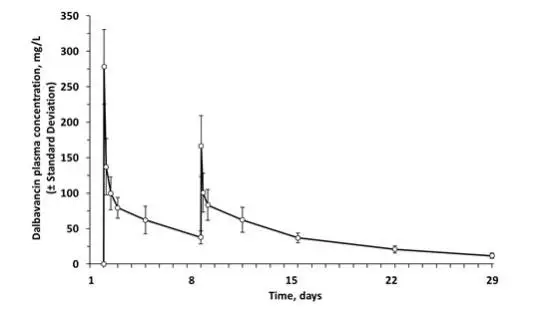
Figure 2. Mean (± standard deviation) dalbavancin plasma concentrations versus time in healthy subjects (n=10) following IV administration over 30 minutes of 1000 mg dalbavancin (Day 1) and 500 mg dalbavancin (Day 8).
No apparent accumulation of dalbavancin was observed following multiple IV infusions administered once weekly for up to eight weeks, with 1000 mg on Day 1 followed by up to seven weekly 500 mg doses, in healthy adults with normal renal function.
Distribution
Dalbavancin is reversibly bound to human plasma proteins, primarily to albumin. The plasma protein binding of dalbavancin is approximately 93% and is not altered as a function of drug concentration, renal impairment, or hepatic impairment. The mean concentrations of dalbavancin achieved in skin blister fluid remain above 30 mg/L up to 7 days (approximately 146 hours) post dose, following 1000 mg IV dalbavancin. The mean ratio of the AUC0-144 hrs in skin blister fluid/AUC0-144 hrs in plasma is 0.60 (range 0.44 to 0.64).
Metabolism
In vitro studies using human microsomal enzymes and hepatocytes indicate that dalbavancin is not a substrate, inhibitor, or inducer of CYP450 isoenzymes. A minor metabolite of dalbavancin (hydroxy-dalbavancin) has been observed in the urine of healthy subjects. Quantifiable concentrations of the hydroxy-dalbavancin metabolite have not been observed in human plasma (lower limit of quantitation = 0.4 µg/mL) [see Drug Interactions (7.2)].
Excretion
Following administration of a single 1000 mg dose in healthy subjects, 20% of the dose was excreted in feces through 70 days post dose. An average of 33% of the administered dalbavancin dose was excreted in urine as unchanged dalbavancin and approximately 12% of the administered dose was excreted in urine as the metabolite hydroxy-dalbavancin through 42 days post dose.
Specific Populations
Renal Impairment
The pharmacokinetics of dalbavancin were evaluated in 28 subjects with varying degrees of renal impairment and in 15 matched control subjects with normal renal function.
Following a single dose of 500 mg or 1000 mg dalbavancin, the mean plasma clearance (CLT) was reduced 11%, 35%, and 47% in subjects with CLcr 50 to 79 mL/min, CLcr 30 to 49 mL/min, and CLcr less than 30 mL/min, respectively, compared to subjects with normal renal function. The clinical significance of the decrease in mean plasma CLT, and the associated increase in AUC0-∞ noted in these pharmacokinetic studies of dalbavancin in subjects with CLcr less than 30 mL/min has not been established [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Dalbavancin pharmacokinetic parameters in subjects with end-stage renal disease receiving regularly scheduled hemodialysis (three times/week) are similar to those observed in subjects with mild to moderate renal impairment, and less than 6% of an administered dose is removed after three hours of hemodialysis.
Therefore, no dosage adjustment is recommended for patients receiving regularly scheduled hemodialysis, and dalbavancin may be administered without regard to the timing of hemodialysis in such patients [see Dosage and Administration (2.1), Overdosage (10)].
Hepatic Impairment
The pharmacokinetics of dalbavancin were evaluated in 17 subjects with mild, moderate, or severe hepatic impairment (Child-Pugh class A, B or C) and compared to those in nine matched healthy subjects with normal hepatic function. The mean AUC0-336 hrs was unchanged in subjects with mild hepatic impairment compared to subjects with normal hepatic function; however, the mean AUC0-336 hrs decreased 28% and 31% in subjects with moderate and severe hepatic impairment respectively, compared to subjects with normal hepatic function. The clinical significance of the decreased AUC0-336 hrs in subjects with moderate and severe hepatic function is unknown.
No dosage adjustment is recommended for patients with mild hepatic impairment. Caution should be exercised when prescribing dalbavancin to patients with moderate or severe hepatic impairment as no data are available to determine the appropriate dosing.
Gender
Clinically significant gender-related differences in dalbavancin pharmacokinetics have not been observed either in healthy subjects or in patients with infections. No dosage adjustment is recommended based on gender.
Geriatric Patients
Clinically significant age-related differences in dalbavancin pharmacokinetics have not been observed in patients with infections. No dosage adjustment is recommended based solely on age.
Pediatric Patients
The pharmacokinetics of dalbavancin has been evaluated in 211 individual pediatric patients [4 days to 17.9 years of age, including a preterm neonate (gestational age 36 weeks; n=1) and term neonates (gestational age 37 to 40 weeks; n=4)] with CLcr 30 mL/min/1.73 m2 and above. There is insufficient information to assess the exposure of DALVANCE in the pediatric patients with CLcr less than 30 mL/min/1.73 m2. No clinically important differences in drug exposure between pediatric age groups (including preterm neonates) and adults are expected following administration of the age-dependent recommended single dose of DALVANCE. The median plasma AUC from 0 to 120 hours (AUC0-120h) of dalbavancin in pediatric patient age groups from term neonates at birth to less than 18 years is expected to be comparable to that in adult patients (AUC0-120h, 10400 mg*h/L). The expected median plasma AUC0-120h of dalbavancin in preterm neonates at birth (gestational age 26 weeks to <37 weeks) was approximately 62% of that in adult patients. The expected median maximum plasma concentrations (Cmax) of dalbavancin for pediatric patient age groups ranged between approximately 53% to 73% of that in adult patients (Cmax, 412 mg/L). However, in all pediatric age groups, the percentage of patients attaining PK/PD targets related to in vivo drug activity were above 90% or higher for MICs up to 0.25 mg/L.
Drug Interactions
Nonclinical studies demonstrated that dalbavancin is not a substrate, inhibitor, or inducer of CYP450 isoenzymes. In a population pharmacokinetic analysis, dalbavancin pharmacokinetics were not affected by co-administration with known CYP450 substrates, inducers or inhibitors, nor by individual medications including acetaminophen, aztreonam, fentanyl, metronidazole, furosemide, proton pump inhibitors (omeprazole, esomeprazole, pantoprazole, lansoprazole), midazolam, and simvastatin.
12.4 Microbiology
Mechanism of Action
Dalbavancin, a semisynthetic lipoglycopeptide, interferes with cell wall synthesis by binding to the D-alanyl-D-alanine terminus of the stem pentapeptide in nascent cell wall peptidoglycan, thus preventing cross-linking. Dalbavancin is bactericidal in vitro against Staphylococcus aureus and Streptococcus pyogenes at concentrations similar to those sustained throughout treatment in humans treated according to the recommended dosage regimen.
Resistance
The development of bacterial isolates resistant to dalbavancin has not been observed, either in vitro, in studies using serial passage, or in animal infection experiments.
Interaction with Other Antimicrobials
When tested in vitro, dalbavancin demonstrated synergistic interactions with oxacillin and did not demonstrate antagonistic or synergistic interactions with any of the following antibacterial agents of various classes: gentamicin, vancomycin, levofloxacin, clindamycin, quinupristin/dalfopristin, linezolid, aztreonam, rifampin or daptomycin. The clinical significance of these in vitro findings is unknown.
Antimicrobial Activity
Dalbavancin has been shown to be active against the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1)].
Aerobic bacteria
Gram-positive bacteria
Staphylococcus aureus (including methicillin-resistant isolates)
Streptococcus pyogenes
Streptococcus agalactiae
Streptococcus dysgalactiae
Streptococcus anginosus group (including S. anginosus, S. intermedius, S. constellatus)
Enterococcus faecalis (vancomycin-susceptible isolates only)
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for dalbavancin against isolates of similar genus or organism group. However, the efficacy of dalbavancin in treating clinical infections caused by these bacteria has not been established in adequate well-controlled clinical trials.
Aerobic bacteria
Gram-positive bacteria
Enterococcus faecium (vancomycin-susceptible isolates only)
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
14. Clinical Studies
Clinical Studies of DALVANCE in Adult Patients with Acute Bacterial Skin and Skin Structure Infections
DALVANCE Two-dose Regimen (1000 mg Day 1; 500 mg Day 8)
Adult patients with ABSSSI were enrolled in two Phase 3, randomized, double-blind, double-dummy clinical trials of similar design (Trial 1 and Trial 2). The Intent-to-Treat (ITT) population included 1,312 randomized patients. Patients were treated for two weeks with either a two-dose regimen of intravenous DALVANCE (1000 mg followed one week later by 500 mg) or intravenous vancomycin (1000 mg or 15 mg/kg every 12 hours, with the option to switch to oral linezolid after 3 days). DALVANCE-treated patients with creatinine clearance of less than 30 mL/min received 750 mg followed one week later by 375 mg. Approximately 5% of patients also received a protocol-specified empiric course of treatment with intravenous aztreonam for coverage of Gram-negative pathogens.
The specific infections in these trials included cellulitis (approximately 50% of patients across treatment groups), major abscess (approximately 30%), and wound infection (approximately 20%). The median lesion area at baseline was 341 cm2. In addition to local signs and symptoms of infection, patients were also required to have at least one systemic sign of disease at baseline, defined as temperature 38°C or higher (approximately 85% of patients), white blood cell count greater than 12,000 cells/mm3 (approximately 40%), or 10% or more band forms on white blood cell differential (approximately 23%). Across both trials, 59% of patients were from Eastern Europe and 36% of patients were from North America. Approximately 89% of patients were Caucasian and 58% were males. The mean age was 50 years and the mean body mass index was 29.1 kg/m2.
The primary endpoint of these two ABSSSI trials was the clinical response rate where responders were defined as patients who had no increase from baseline in lesion area 48 to 72 hours after initiation of therapy, and had a temperature consistently at or below 37.6° C upon repeated measurement. Table 5 summarizes overall clinical response rates in these two ABSSSI trials using the pre-specified primary efficacy endpoint in the ITT population.
| DALVANCE
n/N (%) | Vancomycin/Linezolid
n/N (%) | Difference (95% CI)3 | |
| Trial 1 | 240/288 (83.3) | 233/285 (81.8) | 1.5 (-4.6, 7.9) |
| Trial 2 | 285/371 (76.8) | 288/368 (78.3) | -1.5 (-7.4, 4.6) |
1 There were 7 patients who did not receive treatment and were counted as non-responders: 6 DALVANCE patients (3 in each trial) and one vancomycin/linezolid patient in Trial 2.
2 Patients who died or used non-study antibacterial therapy or had missing measurements were classified as non-responders.
3 The 95% Confidence Interval (CI) is computed using the Miettinen and Nurminen approach, stratified by baseline fever status.
A key secondary endpoint in these two ABSSSI trials evaluated the percentage of ITT patients achieving a 20% or greater reduction in lesion area from baseline at 48-72 hours after initiation of therapy. Table 6 summarizes the findings for this endpoint in these two ABSSSI trials.
| DALVANCE
n/N (%) | Vancomycin/Linezolid
n/N (%) | Difference (95% CI)3 | |
| Trial 1 | 259/288 (89.9) | 259/285 (90.9) | -1.0 (-5.7, 4.0) |
| Trial 2 | 325/371 (87.6) | 316/368 (85.9) | 1.7 (-3.2, 6.7) |
1 There were 7 patients (as described in Table 5) who did not receive treatment and were counted as non-responders.
2 Patients who died or used non-study antibacterial therapy or had missing measurements were classified as non-responders.
3 The 95% CI is computed using the Miettinen and Nurminen approach, stratified by baseline fever status.
Another secondary endpoint in these two ABSSSI trials was the clinical success rate assessed at a follow-up visit occurring between Days 26 to 30. Clinical Success at this visit was defined as having a decrease in lesion size (both length and width measurements), a temperature of 37.6° C or lower, and meeting pre-specified criteria for local signs: purulent discharge and drainage absent or mild and improved from baseline, heat/warmth & fluctuance absent, swelling/induration & tenderness to palpation absent or mild.
Table 7 summarizes clinical success rates at a follow-up visit for the ITT and clinically evaluable population in these two ABSSSI trials. Note that there are insufficient historical data to establish the magnitude of drug effect for antibacterial drugs compared with placebo at the follow-up visits. Therefore, comparisons of DALVANCE to vancomycin/linezolid based on clinical success rates at these visits cannot be utilized to establish non-inferiority.
| DALVANCE
n/N (%) | Vancomycin/Linezolid
n/N (%) |
Difference (95% CI)3 |
|
| Trial 1 | |||
| ITT | 241/288 (83.7%) | 251/285 (88.1%) | -4.4% (-10.1, 1.4) |
| CE | 212/226 (93.8%) | 220/229 (96.1%) | -2.3% (-6.6, 2.0) |
| Trial 2 | |||
| ITT | 327/371 (88.1%) | 311/368 (84.5%) | 3.6% (-1.3, 8.7) |
| CE | 283/294 (96.3%) | 257/272 (94.5%) | 1.8% (-1.8, 5.6) |
1 There were 7 patients (as described in Table 5) who did not receive treatment and were counted as failures in the analysis.
2 Patients who died, used non-study antibacterial therapy, or had an unplanned surgical intervention 72 hours after the start of therapy were classified as Clinical Failures.
3 The 95% CI is computed using the Miettinen and Nurminen approach, stratified by baseline fever status.
Table 8 shows outcomes in patients with an identified baseline pathogen, using pooled data from Trials 1 and 2 in the microbiological ITT (microITT) population. The outcomes shown in the table are clinical response rates at 48 to 72 hours and clinical success rates at follow-up (Day 26 to 30), as defined above.
| Early Clinical Response at 48-72 hours | ||||||
| Early Responder2 | ≥ 20% reduction in lesion size | Clinical Success at Day 26 to 30 | ||||
| Pathogen | DALVANCE
n/N (%) | Comparator
n/N (%) | DALVANCE
n/N (%) | Comparator
n/N (%) | DALVANCE
n/N (%) | Comparator
n/N (%) |
| Staphylococcus aureus
Methicillin-susceptible Methicillin-resistant | 206/257 (80.2) 134/167 (80.2) 72/90 (80.0) | 219/256 (85.5) 163/189 (86.2) 56/67 (83.6) | 239/257 (93.0) 156/167 (93.4) 83/90 (92.2) | 232/256 (90.6) 173/189 (91.5) 59/67 (88.1) | 217/257 (84.4) 142/167 (85.0) 75/90 (83.3) | 229/256 (89.5) 171/189 (90.5) 57/67 (85.1) |
| Streptococcus agalactiae | 6/12 (50.0) | 11/14 (78.6) | 10/12 (83.3) | 10/14 (71.4) | 10/12 (83.3) | 11/14 (78.6) |
| Streptococcus pyogenes | 28/37 (75.7) | 24/36 (66.7) | 32/37 (86.5) | 27/36 (75.0) | 33/37 (89.2) | 32/36 (88.9) |
| Streptococcus
anginosus group | 18/22 (81.8) | 23/ 25 (92.0) | 21/22 (95.5) | 25/25 (100.0) | 21/22 (95.5) | 23/25 (92.0) |
| Enterococcus faecalis | 8/12 (66.7) | 10/13 (76.9) | 12/12 (100.0) | 12/13 (92.3) | 12/12 (100.0) | 11/13 (84.6) |
All DALVANCE dosing regimens in Trials 1 and 2 consisted of two doses.
1 There were 2 patients in the DALVANCE arm with methicillin-susceptible S. aureus at baseline who did not receive treatment and were counted as non-responders/failures.
2 Early Responders are patients who had no increase from baseline in lesion area 48 to 72 hours after initiation of therapy, and had a temperature consistently at or below 37.6° C upon repeated measurement.
DALVANCE 1500 mg Single Dose Regimen
Adult patients with ABSSSI were enrolled in a Phase 3, double-blind, clinical trial. The ITT population included 698 patients who were randomized to DALVANCE treatment with either a single 1500 mg dose or a two-dose regimen of 1000 mg followed one week later by 500 mg (Trial 3). Patients with creatinine clearance less than 30 mL/min had their dose adjusted (Section 2.2). Approximately 5% of patients also received a protocol-specified empiric course of treatment with intravenous aztreonam for coverage of Gram-negative pathogens. The specific infections and other patient characteristics in this trial were similar to those described above for previous ABSSSI trials.
The primary endpoint in this ABSSSI trial was the clinical response rate where responders were defined as patients who had at least a 20% decrease from baseline in lesion area 48 to 72 hours after randomization without receiving any rescue antibacterial therapy. The secondary endpoint was the clinical success rate at a follow-up visit occurring between Days 26 and 30, with clinical success defined as having at least a 90% decrease from baseline in lesion size, a temperature of 37.6° C or lower, and meeting pre-specified criteria for local signs: purulent discharge and drainage absent or mild and improved from baseline (for patients with wound infections), heat/warmth and fluctuance absent, swelling/induration and tenderness to palpation absent or mild. Table 9 summarizes results for these two endpoints in the ITT population. Note that there are insufficient historical data to establish the magnitude of drug effect for antibacterial drugs compared with placebo at the follow-up visit. Therefore, comparisons between treatment groups based on clinical success rates at this visit cannot be utilized to establish non-inferiority.
| DALVANCE, n/N (%) | |||
| Single Dose
(1500 mg) | Two doses
(1000 mg Day 1/500 mg Day 8) | Difference (95% CI)3 | |
| Clinical Responders at 48-72 Hours (ITT) | 284/349 (81.4) | 294/349 (84.2) | -2.9 (-8.5, 2.8) |
| Clinical Success at Day 26-30 (ITT) | 295/349 (84.5) | 297/349 (85.1) | -0.6 (-6.0, 4.8) |
| Clinical Success at Day 26-30 (CE) | 250/271 (92.3) | 247/267 (92.5) | -0.3 (-4.9, 4.4) |
1 There were 3 patients in the two-dose group who did not receive treatment and were counted as non-responders.
2 Patients who died or used non-study antibacterial therapy or had missing measurements were classified as non-responders.
3 The 95% Confidence Interval (CI) is computed using the Miettinen and Nurminen approach.
Abbreviations: ITT-intent to treat; CE-clinically evaluable
Table 10 shows outcomes in patients with an identified baseline pathogen from Trial 3 in the microbiological ITT (microITT) population. The outcomes shown in the table are clinical response rates at 48 to 72 hours and clinical success rates at follow-up (Day 26 to 30), as defined above.
| Early Clinical Response at 48-72 hours | ||||
| ≥ 20% reduction in lesion size | Clinical Success at Day 26 to 30 | |||
| Pathogen | Single dose
(1500 mg) n/N (%) | Two doses
(1000 mg Day 1/ 500 mg Day 8) n/N (%) | Single dose
(1500 mg) n/N (%) | Two doses
(1000 mg Day 1/ 500 mg Day 8) n/N (%) |
| Staphylococcus aureus
Methicillin-susceptible Methicillin-resistant | 123/139 (88.5) 92/103 (89.3) 31/36 (86.1) | 133/156 (85.3) 89/96 (89.6) 48/61 (78.7) | 124/139 (89.2) 93/103 (90.3) 31/36 (86.1) | 140/156 (89.7) 86/96 (89.6) 55/61 (90.2) |
| Streptococcus agalactiae | 6/6(100.0) | 4/6 (66.7) | 5/6 (83.3) | 5/6 (83.3) |
| Streptococcus anginosus group | 31/33 (93.9) | 19/19 (100.0) | 29/33 (87.9) | 17/19 (89.5) |
| Streptococcus pyogenes | 14/14 (100.0) | 18/22 (81.8) | 13/14 (92.9) | 19/22 (86.4) |
| Enterococcus faecalis | 4/4 (100.0) | 8/10 (80.0) | 4/4 (100.0) | 9/10 (90.0) |
In Trials 1, 2, and 3, all patients had blood cultures obtained at baseline. A total of 40 ABSSSI patients who received DALVANCE had bacteremia at baseline caused by one or more of the following bacteria: 26 S. aureus (21 MSSA and 5 MRSA), 6 S. agalactiae, 7 S. pyogenes, 2 S. anginosus group, and 1 E. faecalis. In patients who received DALVANCE, a total of 34/40 (85%) were clinical responders at 48-72 hours and 32/40 (80%) were clinical successes at Day 26 to 30.
Clinical Study of DALVANCE in Pediatric Patients with Acute Bacterial Skin and Skin Structure Infections
The pediatric trial was a multicenter, open-label, randomized, actively controlled trial (NCT02814916, Trial 4) conducted in pediatric patients 3 months of age to less than 18 years with ABSSSI, not known or expected to be caused exclusively by Gram-negative organisms. Patients were randomized in a 3:3:1 ratio to receive either DALVANCE single-dose regimen, DALVANCE two-dose regimen, or comparator. The comparator regimens included IV vancomycin for methicillin-resistant Gram-positive infections, or IV oxacillin or flucloxacillin for methicillin-susceptible Gram-positive infections. Patients in the comparator arm received IV treatment for a minimum of 72 hours before an optional switch to oral therapy to complete a total of 10-14 days of antibacterial drug therapy. Additional 5 patients from birth to < 3 months of age were enrolled and assigned to the DALVANCE single-dose regimen.
A study population of 191 pediatric patients received study medication (DALVANCE single dose regimen n=83, DALVANCE two-dose regimen n=78, comparator n=30); 62% of the patients were male and 89% were white, and 83% were from Eastern Europe. The pediatric age groups who received DALVANCE were as follows: 12 to < 18 years (n=58), 6 to < 12 years (n=49), 2 to < 6 years (n=35), 3 months to < 2 years (n=14), and birth < 3 months (n=5). Patients had diagnoses of major cutaneous abscess (53%), cellulitis (29%), or surgical site/traumatic wound infection (18%). The predominant pathogen at baseline was Staphylococcus aureus (84%).
The primary objective was to evaluate the safety and tolerability of DALVANCE. The trial was not powered for a comparative inferential efficacy analysis. Efficacy was assessed in the modified intent-to-treat population (n=183) which included all randomized patients who received any dose of study drug and had a diagnosis of ABSSSI caused by Gram-positive organism(s). Patients with ABSSSI only caused by Gram-negative organisms were excluded. The five patients in the age group birth to < 3 months of age were not included in efficacy analyses since they were enrolled with expanded inclusion criteria and only received the single dose DALVANCE regimen. An early clinical response at 48–72 hours was defined as ≥ 20% reduction in lesion size compared to baseline and no receipt of rescue antibacterial therapy. The proportion of patients with early clinical response, was 97.3% (73/75) in the DALVANCE single-dose arm, 93.6% (73/78) in the DALVANCE two-dose arm, and 86.7% (26/30) in the comparator arm. The difference in responder rates between the dalbavancin single-dose and comparator arms was 10.7%, with an exact 97.5% confidence interval (CI) of (-1.7%, 31.6%). The difference in responder rates between the dalbavancin two-dose and comparator arms was 6.9%, with an exact 97.5% CI of (-6.4%, 27.7%).
Clinical cure was defined as resolution of the clinical signs and symptoms of infection, when compared to baseline, and no additional antibacterial treatment for the disease under study. In patients, 3 months of age or older in the mITT population, the clinical cure rate at the test of cure (TOC) visit (28 ± 2 days) was 94.7% (71/75) in the DALVANCE single-dose arm, 92.3% (72/78) in the DALVANCE two-dose arm and 100% (30/30) in the comparator arm. The difference in cure rates between the dalbavancin single-dose and comparator arms was -5.3%, with an exact 97.5% CI of (-15.1%, 10.5%). The difference in cure rates between the dalbavancin two-dose and comparator arms was -7.7%, with an exact 97.5% CI of (-17.9%, 8.3%).
16. How is Dalvance supplied
DALVANCE (dalbavancin) for injection is supplied in the following packaging configuration:
500 mg/vial: package of 1 (NDC 57970-100-01)
DALVANCE (dalbavancin) for injection should be stored at 25ºC (77ºF); excursions permitted to 15 to 30ºC (59 to 86ºF) [see USP Controlled Room Temperature]. Storage of the reconstituted and diluted solutions of DALVANCE are described elsewhere in the prescribing information [see Dosage and Administration (2.4)].
17. Patient Counseling Information
Allergic Reactions
Advise patients that allergic reactions, including serious allergic reactions, could occur with DALVANCE, and that serious allergic reactions require immediate treatment. Patients should inform their healthcare provider about any previous hypersensitivity reactions to DALVANCE, or other glycopeptides [see Warnings and Precautions (5.1)].
Diarrhea
Advise patients that diarrhea is a common problem caused by antibacterial drugs, including DALVANCE, and usually resolves when the drug is discontinued. Sometimes, frequent watery or bloody diarrhea may occur and may be a sign of a more serious intestinal infection. If severe watery or bloody diarrhea develops, patients should contact their healthcare provider [see Warnings and Precautions (5.4)].
Development of Drug-Resistant Bacteria
Patients should be counseled that antibacterial drugs including DALVANCE should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When DALVANCE is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment, and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by DALVANCE or other antibacterial drugs in the future [see Warnings and Precautions (5.5)].
Distributed by:
Allergan USA, Inc.
Madison, NJ 07940
Patented. See www.allergan.com/patents.
DALVANCE® is a registered trademark of Allergan Pharmaceuticals International Limited.
© 2021 Allergan. All rights reserved.
v2.0USPI0100
| DALVANCE
dalbavancin injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Allergan, Inc. (144796497) |
