Drug Detail:Invokana (Canagliflozin [ kan-a-gli-floe-zin ])
Drug Class: SGLT-2 inhibitors
Highlights of Prescribing Information
INVOKANA (canagliflozin) tablets, for oral use
Initial U.S. Approval: 2013
Indications and Usage for Invokana
INVOKANA is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated:
- •
- As an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus (1)
- •
- To reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes mellitus and established cardiovascular disease (1)
- •
- To reduce the risk of end-stage kidney disease, doubling of serum creatinine, cardiovascular death, and hospitalization for heart failure in adults with type 2 diabetes mellitus and diabetic nephropathy with albuminuria (1).
Limitations of Use:
- •
- Not recommended in patients with type 1 diabetes mellitus. It may increase the risk of diabetic ketoacidosis in these patients (1)
- •
- Not recommended for use to improve glycemic control in adults with type 2 diabetes mellitus with an eGFR less than 30 mL/min/1.73 m2 (1)
Invokana Dosage and Administration
- •
- The recommended starting dose is 100 mg once daily, taken before the first meal of the day (2.2)
- •
- Dose can be increased to 300 mg once daily in patients tolerating 100 mg once daily who have an eGFR of 60 mL/min/1.73 m2 or greater and require additional glycemic control (2.2)
- •
- Assess renal function before initiating and as clinically indicated (2.1)
- •
- Dose adjustment for patients with renal impairment may be required (2.2)
- •
- See full prescribing information for INVOKANA dosage modifications due to drug interactions. (2.3)
Dosage Forms and Strengths
Tablets: 100 mg, 300 mg (3)
Contraindications
- •
- Serious hypersensitivity reaction to INVOKANA (4, 5.8)
- •
- On dialysis (4)
Warnings and Precautions
- •
- Lower Limb Amputation: Consider factors that may increase the risk of amputation before initiating INVOKANA. Monitor patients for infection or ulcers of lower limb and discontinue if these occur (5.1)
- •
- Ketoacidosis: Assess patients who present with signs and symptoms of metabolic acidosis for ketoacidosis, regardless of blood glucose level. If suspected, discontinue INVOKANA, evaluate and treat promptly. Before initiating INVOKANA, consider risk factors for ketoacidosis. Patients on INVOKANA may require monitoring and temporary discontinuation of therapy in clinical situations known to predispose to ketoacidosis (5.2)
- •
- Volume Depletion: May result in acute kidney injury. Before initiating INVOKANA, assess and correct volume status in patients with renal impairment, elderly patients, or patients on loop diuretics. Monitor for signs and symptoms during therapy (5.3, 6.1)
- •
- Urosepsis and pyelonephritis: Evaluate patients for signs and symptoms of urinary tract infections and treat promptly, if indicated (5.4)
- •
- Hypoglycemia: Consider a lower dose of insulin or the insulin secretagogue to reduce the risk of hypoglycemia when used in combination with INVOKANA (5.5)
- •
- Necrotizing fasciitis of the perineum (Fournier's gangrene): Serious, life-threatening cases have occurred in both females and males. Assess patients presenting with pain or tenderness, erythema, or swelling in the genital or perineal area, along with fever or malaise. If suspected, institute prompt treatment (5.6)
- •
- Genital mycotic infections: Monitor and treat if indicated (5.7)
- •
- Hypersensitivity reactions: Discontinue INVOKANA and monitor until signs and symptoms resolve (5.8)
- •
- Bone fracture: Consider factors that contribute to fracture risk before initiating INVOKANA (5.9)
Adverse Reactions/Side Effects
Most common adverse reactions (5% or greater incidence): female genital mycotic infections, urinary tract infection, and increased urination (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Pharmaceuticals, Inc. at 1-800-526-7736 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
See full prescribing information for information on drug interactions and interference of INVOKANA with laboratory tests. (7)
Use In Specific Populations
- •
- Pregnancy: Advise females of the potential risk to a fetus especially during the second and third trimesters (8.1)
- •
- Lactation: Not recommended when breastfeeding (8.2)
- •
- Geriatrics: Higher incidence of adverse reactions related to reduced intravascular volume (5.3, 8.5)
- •
- Renal impairment: Higher incidence of adverse reactions related to hypotension and renal function (2.3, 5.3, 8.6)
- •
- Hepatic impairment: Not recommended in patients with severe hepatic impairment (8.7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2023
Full Prescribing Information
1. Indications and Usage for Invokana
INVOKANA (canagliflozin) is indicated:
- •
- as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
- •
- to reduce the risk of major adverse cardiovascular events (cardiovascular death, nonfatal myocardial infarction and nonfatal stroke) in adults with type 2 diabetes mellitus and established cardiovascular disease (CVD).
- •
- to reduce the risk of end-stage kidney disease (ESKD), doubling of serum creatinine, cardiovascular (CV) death, and hospitalization for heart failure in adults with type 2 diabetes mellitus and diabetic nephropathy with albuminuria greater than 300 mg/day.
Limitations of Use
INVOKANA is not recommended in patients with type 1 diabetes mellitus. It may increase the risk of diabetic ketoacidosis in these patients [see Warnings and Precautions (5.2)].
INVOKANA is not recommended for use to improve glycemic control in adults with type 2 diabetes mellitus with an eGFR less than 30 mL/min/1.73 m2. INVOKANA is likely to be ineffective in this setting based upon its mechanism of action.
2. Invokana Dosage and Administration
2.1 Prior to Initiation of INVOKANA
Assess renal function before initiating INVOKANA and as clinically indicated [see Warnings and Precautions (5.3)].
- In patients with volume depletion, correct this condition before initiating INVOKANA [see Warnings and Precautions (5.3), Use in Specific Populations (8.5, 8.6)].
2.2 Recommended Dosage
See Table 1 for dosage recommendations based on estimated glomerular filtration rate (eGFR).
| estimated glomerular filtration rate
eGFR (mL/min/1.73 m2) | Recommended Dosage |
|---|---|
|
100 mg orally once daily, taken before the first meal of the day. Dose can be increased to 300 mg once daily for additional glycemic control. |
|
eGFR 30 to less than 60 |
100 mg once daily. |
|
eGFR less than 30 |
Initiation is not recommended, however patients with albuminuria greater than 300 mg/day may continue 100 mg once daily to reduce the risk of ESKD, doubling of serum creatinine, CV death, and hospitalization for heart failure [see Indications and Usage (1), Use in Specific Populations (8.6)]. |
|
On dialysis |
Contraindicated [see Contraindications (4)]. |
2.3 Concomitant Use with UDP-Glucuronosyl Transferase (UGT) Enzyme Inducers
Patients with eGFR 60 mL/min/1.73 m2 or greater
If an inducer of UGTs (e.g., rifampin, phenytoin, phenobarbital, ritonavir) is co-administered with INVOKANA, increase the dose to 200 mg (taken as two 100 mg tablets) once daily in patients currently tolerating INVOKANA 100 mg. The dose may be increased to 300 mg once daily in patients currently tolerating INVOKANA 200 mg and who require additional glycemic control [see Drug Interactions (7)].
Patients with eGFR less than 60 mL/min/1.73 m2
If an inducer of UGTs (e.g., rifampin, phenytoin, phenobarbital, ritonavir) is co-administered with INVOKANA, increase the dose to 200 mg (taken as two 100 mg tablets) once daily in patients currently tolerating INVOKANA 100 mg. Consider adding another antihyperglycemic agent in patients who require additional glycemic control.
3. Dosage Forms and Strengths
- •
- INVOKANA 100 mg tablets are yellow, capsule-shaped, tablets with "CFZ" on one side and "100" on the other side.
- •
- INVOKANA 300 mg tablets are white, capsule-shaped, tablets with "CFZ" on one side and "300" on the other side.
4. Contraindications
- •
- Serious hypersensitivity reaction to INVOKANA, such as anaphylaxis or angioedema [see Warnings and Precautions (5.8) and Adverse Reactions (6.1, 6.2)].
- •
- Patients on dialysis [see Use in Specific Populations (8.6)].
5. Warnings and Precautions
5.1 Lower Limb Amputation
An increased risk of lower limb amputations associated with INVOKANA use versus placebo was observed in CANVAS (5.9 vs 2.8 events per 1000 patient-years) and CANVAS-R (7.5 vs 4.2 events per 1000 patient-years), two randomized, placebo-controlled trials evaluating patients with type 2 diabetes who had either established cardiovascular disease or were at risk for cardiovascular disease. The risk of lower limb amputations was observed at both the 100 mg and 300 mg once daily dosage regimens. The amputation data for CANVAS and CANVAS-R are shown in Tables 3 and 4, respectively [see Adverse Reactions (6.1)].
Amputations of the toe and midfoot (99 out of 140 patients with amputations receiving INVOKANA in the two trials) were the most frequent; however, amputations involving the leg, below and above the knee, were also observed (41 out of 140 patients with amputations receiving INVOKANA in the two trials). Some patients had multiple amputations, some involving both lower limbs.
Lower limb infections, gangrene, and diabetic foot ulcers were the most common precipitating medical events leading to the need for an amputation. The risk of amputation was highest in patients with a baseline history of prior amputation, peripheral vascular disease, and neuropathy.
Before initiating INVOKANA, consider factors in the patient history that may predispose to the need for amputations, such as a history of prior amputation, peripheral vascular disease, neuropathy and diabetic foot ulcers. Counsel patients about the importance of routine preventative foot care. Monitor patients receiving INVOKANA for signs and symptoms of infection (including osteomyelitis), new pain or tenderness, sores or ulcers involving the lower limbs, and discontinue INVOKANA if these complications occur.
5.2 Ketoacidosis
Reports of ketoacidosis, a serious life-threatening condition requiring urgent hospitalization have been identified in clinical trials and postmarketing surveillance in patients with type 1 and type 2 diabetes mellitus receiving sodium glucose co-transporter-2 (SGLT2) inhibitors, including INVOKANA. In placebo-controlled trials of patients with type 1 diabetes, the risk of ketoacidosis was increased in patients who received SGLT2 inhibitors compared to patients who received placebo. The risk of ketoacidosis may be greater with higher doses. Fatal cases of ketoacidosis have been reported in patients taking INVOKANA. INVOKANA is not indicated for the treatment of patients with type 1 diabetes mellitus [see Indications and Usage (1)].
Patients treated with INVOKANA who present with signs and symptoms consistent with severe metabolic acidosis should be assessed for ketoacidosis regardless of presenting blood glucose levels, as ketoacidosis associated with INVOKANA may be present even if blood glucose levels are less than 250 mg/dL. If ketoacidosis is suspected, INVOKANA should be discontinued, patient should be evaluated, and prompt treatment should be instituted. Treatment of ketoacidosis may require insulin, fluid and carbohydrate replacement.
In many of the postmarketing reports, and particularly in patients with type 1 diabetes, the presence of ketoacidosis was not immediately recognized and institution of treatment was delayed because presenting blood glucose levels were below those typically expected for diabetic ketoacidosis (often less than 250 mg/dL). Signs and symptoms at presentation were consistent with dehydration and severe metabolic acidosis and included nausea, vomiting, abdominal pain, generalized malaise, and shortness of breath. In some but not all cases, factors predisposing to ketoacidosis such as insulin dose reduction, acute febrile illness, reduced caloric intake, surgery, pancreatic disorders suggesting insulin deficiency (e.g., type 1 diabetes, history of pancreatitis or pancreatic surgery), and alcohol abuse were identified.
Before initiating INVOKANA, consider factors in the patient history that may predispose to ketoacidosis including pancreatic insulin deficiency from any cause, caloric restriction, and alcohol abuse.
For patients who undergo scheduled surgery, consider temporarily discontinuing INVOKANA for at least 3 days prior to surgery [see Clinical Pharmacology (12.2, 12.3)].
Consider monitoring for ketoacidosis and temporarily discontinuing INVOKANA in other clinical situations known to predispose to ketoacidosis (e.g., prolonged fasting due to acute illness or post-surgery). Ensure risk factors for ketoacidosis are resolved prior to restarting INVOKANA.
Educate patients on the signs and symptoms of ketoacidosis and instruct patients to discontinue INVOKANA and seek medical attention immediately if signs and symptoms occur.
5.3 Volume Depletion
INVOKANA can cause intravascular volume contraction which may sometimes manifest as symptomatic hypotension or acute transient changes in creatinine [see Adverse Reactions (6.1)]. There have been post-marketing reports of acute kidney injury which are likely related to volume depletion, some requiring hospitalizations and dialysis, in patients with type 2 diabetes mellitus receiving SGLT2 inhibitors, including INVOKANA. Patients with impaired renal function (eGFR less than 60 mL/min/1.73 m2), elderly patients, or patients on loop diuretics may be at increased risk for volume depletion or hypotension. Before initiating INVOKANA in patients with one or more of these characteristics, assess and correct volume status. Monitor for signs and symptoms of volume depletion after initiating therapy.
5.4 Urosepsis and Pyelonephritis
There have been postmarketing reports of serious urinary tract infections including urosepsis and pyelonephritis requiring hospitalization in patients receiving SGLT2 inhibitors, including INVOKANA. Treatment with SGLT2 inhibitors increases the risk for urinary tract infections. Evaluate patients for signs and symptoms of urinary tract infections and treat promptly, if indicated [see Adverse Reactions (6)].
5.5 Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
Insulin and insulin secretagogues are known to cause hypoglycemia. INVOKANA may increase the risk of hypoglycemia when combined with insulin or an insulin secretagogue [see Adverse Reactions (6.1)]. Therefore, a lower dose of insulin or insulin secretagogue may be required to minimize the risk of hypoglycemia when used in combination with INVOKANA.
5.6 Necrotizing Fasciitis of the Perineum (Fournier's Gangrene)
Reports of necrotizing fasciitis of the perineum (Fournier's gangrene), a rare but serious and life-threatening necrotizing infection requiring urgent surgical intervention, have been identified in postmarketing surveillance in patients with diabetes mellitus receiving SGLT2 inhibitors, including INVOKANA. Cases have been reported in both females and males. Serious outcomes have included hospitalization, multiple surgeries, and death.
Patients treated with INVOKANA presenting with pain or tenderness, erythema, or swelling in the genital or perineal area, along with fever or malaise, should be assessed for necrotizing fasciitis. If suspected, start treatment immediately with broad-spectrum antibiotics and, if necessary, surgical debridement. Discontinue INVOKANA, closely monitor blood glucose levels, and provide appropriate alternative therapy for glycemic control.
5.7 Genital Mycotic Infections
INVOKANA increases the risk of genital mycotic infections. Patients with a history of genital mycotic infections and uncircumcised males were more likely to develop genital mycotic infections [see Adverse Reactions (6.1)]. Monitor and treat appropriately.
5.8 Hypersensitivity Reactions
Hypersensitivity reactions, including angioedema and anaphylaxis, have been reported with INVOKANA. These reactions generally occurred within hours to days after initiating INVOKANA. If hypersensitivity reactions occur, discontinue use of INVOKANA; treat and monitor until signs and symptoms resolve [see Contraindications (4) and Adverse Reactions (6.1, 6.2)].
5.9 Bone Fracture
An increased risk of bone fracture, occurring as early as 12 weeks after treatment initiation, was observed in patients using INVOKANA in the CANVAS trial [see Clinical Studies (14.2)]. Consider factors that contribute to fracture risk prior to initiating INVOKANA [see Adverse Reactions (6.1)].
6. Adverse Reactions/Side Effects
The following important adverse reactions are described below and elsewhere in the labeling:
- •
- Lower Limb Amputation [see Warnings and Precautions (5.1)]
- •
- Ketoacidosis [see Warnings and Precautions (5.2)]
- •
- Volume Depletion [see Warnings and Precautions (5.3)]
- •
- Urosepsis and Pyelonephritis [see Warnings and Precautions (5.4)]
- •
- Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues [see Warnings and Precautions (5.5)]
- •
- Necrotizing Fasciitis of the Perineum (Fournier's gangrene) [see Warnings and Precautions (5.6)]
- •
- Genital Mycotic Infections [see Warnings and Precautions (5.7)]
- •
- Hypersensitivity Reactions [see Warnings and Precautions (5.8)]
- •
- Bone Fracture [see Warnings and Precautions (5.9)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to the rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Pool of Placebo-Controlled Trials for Glycemic Control
The data in Table 2 is derived from four 26-week placebo-controlled trials where INVOKANA was used as monotherapy in one trial and as add-on therapy in three trials. These data reflect exposure of 1,667 patients to INVOKANA and a mean duration of exposure to INVOKANA of 24 weeks. Patients received INVOKANA 100 mg (N=833), INVOKANA 300 mg (N=834) or placebo (N=646) once daily. The mean age of the population was 56 years and 2% were older than 75 years of age. Fifty percent (50%) of the population was male and 72% were Caucasian, 12% were Asian, and 5% were Black or African American. At baseline the population had diabetes for an average of 7.3 years, had a mean HbA1C of 8.0% and 20% had established microvascular complications of diabetes. Baseline renal function was normal or mildly impaired (mean eGFR 88 mL/min/1.73 m2).
Table 2 shows common adverse reactions associated with the use of INVOKANA. These adverse reactions were not present at baseline, occurred more commonly on INVOKANA than on placebo, and occurred in at least 2% of patients treated with either INVOKANA 100 mg or INVOKANA 300 mg.
| Note: Percentages were weighted by studies. Study weights were proportional to the harmonic mean of the three treatment sample sizes. | |||
|
|||
|
Adverse Reaction |
Placebo
|
INVOKANA
|
INVOKANA
|
|
Urinary tract infections† |
3.8% |
5.9% |
4.4% |
|
Increased urination‡ |
0.7% |
5.1% |
4.6% |
|
Thirst§ |
0.1% |
2.8% |
2.4% |
|
Constipation |
0.9% |
1.8% |
2.4% |
|
Nausea |
1.6% |
2.1% |
2.3% |
|
N=312 |
N=425 |
N=430 |
|
|
Female genital mycotic infections¶ |
2.8% |
10.6% |
11.6% |
|
Vulvovaginal pruritus |
0.0% |
1.6% |
3.2% |
|
N=334 |
N=408 |
N=404 |
|
|
Male genital mycotic infections# |
0.7% |
4.2% |
3.8% |
Abdominal pain was also more commonly reported in patients taking INVOKANA 100 mg (1.8%), 300 mg (1.7%) than in patients taking placebo (0.8%).
Placebo-Controlled Trial in Diabetic Nephropathy
The occurrence of adverse reactions for INVOKANA was evaluated in patients participating in CREDENCE, a study in patients with type 2 diabetes mellitus and diabetic nephropathy with albuminuria > 300 mg/day [see Clinical Studies (14.3)]. These data reflect exposure of 2,201 patients to INVOKANA and a mean duration of exposure to INVOKANA of 137 weeks.
- •
- The rate of lower limb amputations associated with the use of INVOKANA 100 mg relative to placebo was 12.3 vs 11.2 events per 1000 patient-years, respectively, with 2.6 years mean duration of follow-up.
- •
- Incidence rates of adjudicated events of diabetic ketoacidosis (DKA) were 0.21 (0.5%, 12/2,200) and 0.03 (0.1%, 2/2,197) per 100 patient-years of follow-up with INVOKANA 100 mg and placebo, respectively.
- •
- The incidence of hypotension was 2.8% and 1.5% on INVOKANA 100 mg and placebo, respectively.
Pool of Placebo- and Active-Controlled Trials for Glycemic Control and Cardiovascular Outcomes
The occurrence of adverse reactions for INVOKANA was evaluated in patients participating in placebo- and active-controlled trials and in an integrated analysis of two cardiovascular trials, CANVAS and CANVAS-R.
The types and frequency of common adverse reactions observed in the pool of eight clinical trials (which reflect an exposure of 6,177 patients to INVOKANA) were consistent with those listed in Table 2. Percentages were weighted by studies. Study weights were proportional to the harmonic mean of the three treatment sample sizes. In this pool, INVOKANA was also associated with the adverse reactions of fatigue (1.8%, 2.2%, and 2.0% with comparator, INVOKANA 100 mg, and INVOKANA 300 mg, respectively) and loss of strength or energy (i.e., asthenia) (0.6%, 0.7%, and 1.1% with comparator, INVOKANA 100 mg, and INVOKANA 300 mg, respectively).
In the pool of eight clinical trials, the incidence rate of pancreatitis (acute or chronic) was 0.1%, 0.2%, and 0.1% receiving comparator, INVOKANA 100 mg, and INVOKANA 300 mg, respectively.
In the pool of eight clinical trials, hypersensitivity-related adverse reactions (including erythema, rash, pruritus, urticaria, and angioedema) occurred in 3.0%, 3.8%, and 4.2% of patients receiving comparator, INVOKANA 100 mg, and INVOKANA 300 mg, respectively. Five patients experienced serious adverse reactions of hypersensitivity with INVOKANA, which included 4 patients with urticaria and 1 patient with a diffuse rash and urticaria occurring within hours of exposure to INVOKANA. Among these patients, 2 patients discontinued INVOKANA. One patient with urticaria had recurrence when INVOKANA was re-initiated.
Photosensitivity-related adverse reactions (including photosensitivity reaction, polymorphic light eruption, and sunburn) occurred in 0.1%, 0.2%, and 0.2% of patients receiving comparator, INVOKANA 100 mg, and INVOKANA 300 mg, respectively.
Other adverse reactions occurring more frequently on INVOKANA than on comparator were:
Lower Limb Amputation
An increased risk of lower limb amputations associated with INVOKANA use versus placebo was observed in CANVAS (5.9 vs 2.8 events per 1000 patient-years) and CANVAS-R (7.5 vs 4.2 events per 1000 patient-years), two randomized, placebo-controlled trials evaluating patients with type 2 diabetes who had either established cardiovascular disease or were at risk for cardiovascular disease. Patients in CANVAS and CANVAS-R were followed for an average of 5.7 and 2.1 years, respectively [see Clinical Studies (14.2)]. The amputation data for CANVAS and CANVAS-R are shown in Tables 3 and 4, respectively.
| Placebo
N=1441 | INVOKANA
100 mg N=1445 | INVOKANA
300 mg N=1441 | INVOKANA
(Pooled) N=2886 |
|
|---|---|---|---|---|
| Note: Incidence is based on the number of patients with at least one amputation, and not the total number of amputation events. A patient's follow-up is calculated from Day 1 to the first amputation event date. Some patients had more than one amputation. | ||||
|
Patients with an amputation, n (%) |
22 (1.5) |
50 (3.5) |
45 (3.1) |
95 (3.3) |
|
Total amputations |
33 |
83 |
79 |
162 |
|
Amputation incidence rate |
2.8 |
6.2 |
5.5 |
5.9 |
|
Hazard Ratio (95% CI) |
-- |
2.24 (1.36, 3.69) |
2.01 (1.20, 3.34) |
2.12 (1.34, 3.38) |
| Placebo
N=2903 | INVOKANA
100 mg (with up-titration to 300 mg) N=2904 |
||||||
|---|---|---|---|---|---|---|---|
| Note: Incidence is based on the number of patients with at least one amputation, and not the total number of amputation events. A patient's follow-up is calculated from Day 1 to the first amputation event date. Some patients had more than one amputation. | |||||||
|
Patients with an amputation, n (%) |
25 (0.9) |
45 (1.5) |
|||||
|
Total amputations |
36 |
59 |
|||||
|
Amputation incidence rate |
4.2 |
7.5 |
|||||
|
Hazard Ratio (95% CI) |
-- |
1.80 (1.10, 2.93) |
|||||
Renal Cell Carcinoma
In the CANVAS trial (mean duration of follow-up of 5.7 years) [see Clinical Studies (14.2)], the incidence of renal cell carcinoma was 0.15% (2/1331) and 0.29% (8/2716) for placebo and INVOKANA, respectively, excluding patients with less than 6 months of follow-up, less than 90 days of treatment, or a history of renal cell carcinoma. A causal relationship to INVOKANA could not be established due to the limited number of cases.
Volume Depletion-Related Adverse Reactions
INVOKANA results in an osmotic diuresis, which may lead to reductions in intravascular volume. In clinical trials for glycemic control, treatment with INVOKANA was associated with a dose-dependent increase in the incidence of volume depletion-related adverse reactions (e.g., hypotension, postural dizziness, orthostatic hypotension, syncope, and dehydration). An increased incidence was observed in patients on the 300 mg dose. The three factors associated with the largest increase in volume depletion-related adverse reactions in these trials were the use of loop diuretics, moderate renal impairment (eGFR 30 to less than 60 mL/min/1.73 m2), and age 75 years and older (Table 5) [see Use in Specific Populations (8.5 and 8.6)].
| Baseline Characteristic | Comparator Group*
% | INVOKANA 100 mg
% | INVOKANA 300 mg
% |
|---|---|---|---|
|
|||
|
Overall population |
1.5% |
2.3% |
3.4% |
|
75 years of age and older† |
2.6% |
4.9% |
8.7% |
|
eGFR less than 60 mL/min/1.73 m2† |
2.5% |
4.7% |
8.1% |
|
Use of loop diuretic† |
4.7% |
3.2% |
8.8% |
Falls
In a pool of nine clinical trials with mean duration of exposure to INVOKANA of 85 weeks, the proportion of patients who experienced falls was 1.3%, 1.5%, and 2.1% with comparator, INVOKANA 100 mg, and INVOKANA 300 mg, respectively. The higher risk of falls for patients treated with INVOKANA was observed within the first few weeks of treatment.
Genital Mycotic Infections
In the pool of four placebo-controlled clinical trials for glycemic control, female genital mycotic infections (e.g., vulvovaginal mycotic infection, vulvovaginal candidiasis, and vulvovaginitis) occurred in 2.8%, 10.6%, and 11.6% of females treated with placebo, INVOKANA 100 mg, and INVOKANA 300 mg, respectively. Patients with a history of genital mycotic infections were more likely to develop genital mycotic infections on INVOKANA. Female patients who developed genital mycotic infections on INVOKANA were more likely to experience recurrence and require treatment with oral or topical antifungal agents and anti-microbial agents. In females, discontinuation due to genital mycotic infections occurred in 0% and 0.7% of patients treated with placebo and INVOKANA, respectively.
In the pool of four placebo-controlled clinical trials, male genital mycotic infections (e.g., candidal balanitis, balanoposthitis) occurred in 0.7%, 4.2%, and 3.8% of males treated with placebo, INVOKANA 100 mg, and INVOKANA 300 mg, respectively. Male genital mycotic infections occurred more commonly in uncircumcised males and in males with a prior history of balanitis or balanoposthitis. Male patients who developed genital mycotic infections on INVOKANA were more likely to experience recurrent infections (22% on INVOKANA versus none on placebo), and require treatment with oral or topical antifungal agents and anti-microbial agents than patients on comparators. In males, discontinuations due to genital mycotic infections occurred in 0% and 0.5% of patients treated with placebo and INVOKANA, respectively.
In the pooled analysis of 8 randomized trials evaluating glycemic control, phimosis was reported in 0.3% of uncircumcised male patients treated with INVOKANA and 0.2% required circumcision to treat the phimosis.
Hypoglycemia
In all glycemic control trials, hypoglycemia was defined as any event regardless of symptoms, where biochemical hypoglycemia was documented (any glucose value below or equal to 70 mg/dL). Severe hypoglycemia was defined as an event consistent with hypoglycemia where the patient required the assistance of another person to recover, lost consciousness, or experienced a seizure (regardless of whether biochemical documentation of a low glucose value was obtained). In individual clinical trials of glycemic control [see Clinical Studies (14.1)], episodes of hypoglycemia occurred at a higher rate when INVOKANA was co-administered with insulin or sulfonylureas (Table 6).
|
|||
|
Monotherapy
|
Placebo
|
INVOKANA 100 mg
|
INVOKANA 300 mg
|
|
Overall [N (%)] |
5 (2.6) |
7 (3.6) |
6 (3.0) |
|
In Combination with Metformin
|
Placebo + Metformin
|
INVOKANA 100 mg + Metformin
|
INVOKANA 300 mg + Metformin
|
|
Overall [N (%)] |
3 (1.6) |
16 (4.3) |
17 (4.6) |
|
Severe [N (%)]† |
0 (0) |
1 (0.3) |
1 (0.3) |
|
In Combination with Metformin
|
Glimepiride + Metformin
|
INVOKANA 100 mg + Metformin
|
INVOKANA 300 mg + Metformin
|
|
Overall [N (%)] |
165 (34.2) |
27 (5.6) |
24 (4.9) |
|
Severe [N (%)]† |
15 (3.1) |
2 (0.4) |
3 (0.6) |
|
In Combination with Sulfonylurea
|
Placebo + Sulfonylurea
|
INVOKANA 100 mg + Sulfonylurea
|
INVOKANA 300 mg + Sulfonylurea
|
|
Overall [N (%)] |
4 (5.8) |
3 (4.1) |
9 (12.5) |
|
In Combination with Metformin + Sulfonylurea
|
Placebo + Metformin + Sulfonylurea
|
INVOKANA 100 mg + Metformin + Sulfonylurea
|
INVOKANA 300 mg + Metformin + Sulfonylurea
|
|
Overall [N (%)] |
24 (15.4) |
43 (27.4) |
47 (30.1) |
|
Severe [N (%)]† |
1 (0.6) |
1 (0.6) |
0 |
|
In Combination with Metformin + Sulfonylurea
|
Sitagliptin + Metformin + Sulfonylurea
|
INVOKANA 300 mg + Metformin + Sulfonylurea
|
|
|
Overall [N (%)] |
154 (40.7) |
163 (43.2) |
|
|
Severe [N (%)]† |
13 (3.4) |
15 (4.0) |
|
|
In Combination with Metformin + Pioglitazone
|
Placebo + Metformin + Pioglitazone
|
INVOKANA 100 mg + Metformin + Pioglitazone
|
INVOKANA 300 mg + Metformin + Pioglitazone
|
|
Overall [N (%)] |
3 (2.6) |
3 (2.7) |
6 (5.3) |
|
In Combination with Insulin
|
Placebo
|
INVOKANA 100 mg
|
INVOKANA 300 mg
|
|
Overall [N (%)] |
208 (36.8) |
279 (49.3) |
285 (48.6) |
|
Severe [N (%)]† |
14 (2.5) |
10 (1.8) |
16 (2.7) |
Bone Fracture
In the CANVAS trial [see Clinical Studies (14.2)], the incidence rates of all adjudicated bone fracture were 1.09, 1.59, and 1.79 events per 100 patient-years of follow-up to placebo, INVOKANA 100 mg, and INVOKANA 300 mg, respectively. The fracture imbalance was observed within the first 26 weeks of therapy and remained through the end of the trial. Fractures were more likely to be low trauma (e.g., fall from no more than standing height), and affect the distal portion of upper and lower extremities.
Laboratory and Imaging Tests
Increases in Serum Creatinine and Decreases in eGFR
Initiation of INVOKANA causes an increase in serum creatinine and decrease in estimated GFR. In patients with moderate renal impairment, the increase in serum creatinine generally does not exceed 0.2 mg/dL, occurs within the first 6 weeks of starting therapy, and then stabilizes. Increases that do not fit this pattern should prompt further evaluation to exclude the possibility of acute kidney injury [see Clinical Pharmacology (12.1)]. The acute effect on eGFR reverses after treatment discontinuation suggesting acute hemodynamic changes may play a role in the renal function changes observed with INVOKANA.
Increases in Serum Potassium
In a pooled population of patients (N=723) in glycemic control trials with moderate renal impairment (eGFR 45 to less than 60 mL/min/1.73 m2), increases in serum potassium to greater than 5.4 mEq/L and 15% above baseline occurred in 5.3%, 5.0%, and 8.8% of patients treated with placebo, INVOKANA 100 mg, and INVOKANA 300 mg, respectively. Severe elevations (greater than or equal to 6.5 mEq/L) occurred in 0.4% of patients treated with placebo, no patients treated with INVOKANA 100 mg, and 1.3% of patients treated with INVOKANA 300 mg.
In these patients, increases in potassium were more commonly seen in those with elevated potassium at baseline. Among patients with moderate renal impairment, approximately 84% were taking medications that interfere with potassium excretion, such as potassium-sparing diuretics, angiotensin-converting-enzyme inhibitors, and angiotensin-receptor blockers [see Use in Specific Populations (8.6)].
In CREDENCE, no difference in serum potassium, no increase in adverse events of hyperkalemia, and no increase in absolute (> 6.5 mEq/L) or relative (> upper limit of normal and > 15% increase from baseline) increases in serum potassium were observed with INVOKANA 100 mg relative to placebo.
Increases in Low-Density Lipoprotein Cholesterol (LDL-C) and non-High-Density Lipoprotein Cholesterol (non-HDL-C)
In the pool of four glycemic control placebo-controlled trials, dose-related increases in LDL-C with INVOKANA were observed. Mean changes (percent changes) from baseline in LDL-C relative to placebo were 4.4 mg/dL (4.5%) and 8.2 mg/dL (8.0%) with INVOKANA 100 mg and INVOKANA 300 mg, respectively. The mean baseline LDL-C levels were 104 to 110 mg/dL across treatment groups.
Dose-related increases in non-HDL-C with INVOKANA were observed. Mean changes (percent changes) from baseline in non-HDL-C relative to placebo were 2.1 mg/dL (1.5%) and 5.1 mg/dL (3.6%) with INVOKANA 100 mg and 300 mg, respectively. The mean baseline non-HDL-C levels were 140 to 147 mg/dL across treatment groups.
Increases in Hemoglobin
In the pool of four placebo-controlled trials of glycemic control, mean changes (percent changes) from baseline in hemoglobin were -0.18 g/dL (-1.1%) with placebo, 0.47 g/dL (3.5%) with INVOKANA 100 mg, and 0.51 g/dL (3.8%) with INVOKANA 300 mg. The mean baseline hemoglobin value was approximately 14.1 g/dL across treatment groups. At the end of treatment, 0.8%, 4.0%, and 2.7% of patients treated with placebo, INVOKANA 100 mg, and INVOKANA 300 mg, respectively, had hemoglobin above the upper limit of normal.
Decreases in Bone Mineral Density
Bone mineral density (BMD) was measured by dual-energy X-ray absorptiometry in a clinical trial of 714 older adults (mean age 64 years) [see Clinical Studies (14.1)]. At 2 years, patients randomized to INVOKANA 100 mg and INVOKANA 300 mg had placebo-corrected declines in BMD at the total hip of 0.9% and 1.2%, respectively, and at the lumbar spine of 0.3% and 0.7%, respectively. Additionally, placebo-adjusted BMD declines were 0.1% at the femoral neck for both INVOKANA doses and 0.4% at the distal forearm for patients randomized to INVOKANA 300 mg. The placebo-adjusted change at the distal forearm for patients randomized to INVOKANA 100 mg was 0%.
6.2 Postmarketing Experience
Additional adverse reactions have been identified during post-approval use of INVOKANA. Because these reactions are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Ketoacidosis
Acute Kidney Injury
Anaphylaxis, Angioedema
Urosepsis and Pyelonephritis
Necrotizing Fasciitis of the Perineum (Fournier's gangrene)
7. Drug Interactions
|
UGT Enzyme Inducers |
|
|
Clinical Impact: |
UGT enzyme inducers decrease canagliflozin exposure which may reduce the effectiveness of INVOKANA. |
|
Intervention: |
For patients with eGFR 60 mL/min/1.73 m2 or greater, if an inducer of UGTs is administered with INVOKANA, increase the dosage to 200 mg daily in patients currently tolerating INVOKANA 100 mg daily. The total daily dosage may be increased to 300 mg daily in patients currently tolerating INVOKANA 200 mg daily who require additional glycemic control. |
|
Examples: |
Rifampin, phenytoin, phenobarbital, ritonavir |
|
Insulin or Insulin Secretagogues |
|
|
Clinical Impact: |
The risk of hypoglycemia is increased when INVOKANA is used concomitantly with insulin secretagogues (e.g., sulfonylurea) or insulin. |
|
Intervention: |
Concomitant use may require a lower dosage of the insulin secretagogue or insulin to reduce the risk of hypoglycemia. |
|
Digoxin |
|
|
Clinical Impact: |
Canagliflozin increases digoxin exposure [see Clinical Pharmacology (12.3)]. |
|
Intervention: |
Monitor patients taking INVOKANA with concomitant digoxin for a need to adjust the dosage of digoxin. |
|
Lithium |
|
|
Clinical Impact: |
Concomitant use of an SGLT2 inhibitor with lithium may decrease serum lithium concentrations. |
|
Intervention: |
Monitor serum lithium concentration more frequently during INVOKANA initiation and dosage changes. |
|
Drug/Laboratory Test Interference |
|
|
Positive Urine Glucose Test |
|
|
Clinical Impact: |
SGLT2 inhibitors increase urinary glucose excretion which will lead to positive urine glucose tests. |
|
Intervention: |
Monitoring glycemic control with urine glucose tests is not recommended in patients taking SGLT2 inhibitors. Use alternative methods to monitor glycemic control. |
|
Interference with 1,5-anhydroglucitol (1,5-AG) Assay |
|
|
Clinical Impact: |
Measurements of 1,5-AG are unreliable in assessing glycemic control in patients taking SGLT2 inhibitors. |
|
Intervention: |
Monitoring glycemic control with 1,5-AG assay is not recommended in patients taking SGLT2 inhibitors. Use alternative methods to monitor glycemic control. |
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on animal data showing adverse renal effects, INVOKANA is not recommended during the second and third trimesters of pregnancy.
Limited data with INVOKANA in pregnant women are not sufficient to determine a drug-associated risk for major birth defects or miscarriage. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy [see Clinical Considerations].
In animal studies, adverse renal pelvic and tubule dilatations that were not reversible were observed in rats when canagliflozin was administered during a period of renal development corresponding to the late second and third trimesters of human pregnancy, at an exposure 0.5-times the 300 mg clinical dose, based on AUC.
The estimated background risk of major birth defects is 6–10% in women with pre-gestational diabetes with a HbA1C >7 and has been reported to be as high as 20–25% in women with a HbA1C >10. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia related morbidity.
Animal Data
Canagliflozin dosed directly to juvenile rats from postnatal day (PND) 21 until PND 90 at doses of 4, 20, 65, or 100 mg/kg increased kidney weights and dose dependently increased the incidence and severity of renal pelvic and tubular dilatation at all doses tested. Exposure at the lowest dose was greater than or equal to 0.5-times the 300 mg clinical dose, based on AUC. These outcomes occurred with drug exposure during periods of renal development in rats that correspond to the late second and third trimester of human renal development. The renal pelvic dilatations observed in juvenile animals did not fully reverse within a 1-month recovery period.
In embryo-fetal development studies in rats and rabbits, canagliflozin was administered for intervals coinciding with the first trimester period of organogenesis in humans. No developmental toxicities independent of maternal toxicity were observed when canagliflozin was administered at doses up to 100 mg/kg in pregnant rats and 160 mg/kg in pregnant rabbits during embryonic organogenesis or during a study in which maternal rats were dosed from gestation day (GD) 6 through PND 21, yielding exposures up to approximately 19-times the 300 mg clinical dose, based on AUC.
8.2 Lactation
Risk Summary
There is no information regarding the presence of INVOKANA in human milk, the effects on the breastfed infant, or the effects on milk production. Canagliflozin is present in the milk of lactating rats [see Data]. Since human kidney maturation occurs in utero and during the first 2 years of life when lactational exposure may occur, there may be risk to the developing human kidney.
Because of the potential for serious adverse reactions in a breastfed infant, advise women that use of INVOKANA is not recommended while breastfeeding.
Data
Animal Data
Radiolabeled canagliflozin administered to lactating rats on day 13 post-partum was present at a milk/plasma ratio of 1.40, indicating that canagliflozin and its metabolites are transferred into milk at a concentration comparable to that in plasma. Juvenile rats directly exposed to canagliflozin showed a risk to the developing kidney (renal pelvic and tubular dilatations) during maturation.
8.4 Pediatric Use
Safety and effectiveness of INVOKANA in pediatric patients under 18 years of age have not been established.
8.5 Geriatric Use
In 13 clinical trials of INVOKANA, 2,294 patients 65 years and older, and 351 patients 75 years and older were exposed to INVOKANA [see Clinical Studies (14.1)].
Patients 65 years and older had a higher incidence of adverse reactions related to reduced intravascular volume with INVOKANA (such as hypotension, postural dizziness, orthostatic hypotension, syncope, and dehydration), particularly with the 300 mg daily dose, compared to younger patients; a more prominent increase in the incidence was seen in patients who were 75 years and older [see Dosage and Administration (2.1) and Adverse Reactions (6.1)]. Smaller reductions in HbA1C with INVOKANA relative to placebo were seen in older (65 years and older; -0.61% with INVOKANA 100 mg and -0.74% with INVOKANA 300 mg relative to placebo) compared to younger patients (-0.72% with INVOKANA 100 mg and -0.87% with INVOKANA 300 mg relative to placebo).
8.6 Renal Impairment
The efficacy and safety of INVOKANA for glycemic control were evaluated in a trial that included patients with moderate renal impairment (eGFR 30 to less than 50 mL/min/1.73 m2) [see Clinical Studies (14.1)]. These patients had less overall glycemic efficacy, and patients treated with 300 mg per day had increases in serum potassium, which were transient and similar by the end of study. Patients with renal impairment using INVOKANA for glycemic control may also be more likely to experience hypotension and may be at higher risk for acute kidney injury [see Warnings and Precautions (5.3)].
Efficacy and safety studies with INVOKANA did not enroll patients with ESKD on dialysis or patients with an eGFR less than 30 mL/min/1.73 m2. INVOKANA is contraindicated in patients with ESKD on dialysis [see Contraindications (4) and Clinical Pharmacology (12.1)].
10. Overdosage
In the event of an overdose, contact the Poison Control Center. It is also reasonable to employ the usual supportive measures, e.g., remove unabsorbed material from the gastrointestinal tract, employ clinical monitoring, and institute supportive treatment as dictated by the patient's clinical status. Canagliflozin was negligibly removed during a 4-hour hemodialysis session. Canagliflozin is not expected to be dialyzable by peritoneal dialysis.
11. Invokana Description
INVOKANA® (canagliflozin) contains canagliflozin, an inhibitor of sodium-glucose co-transporter 2 (SGLT2), the transporter responsible for reabsorbing the majority of glucose filtered by the kidney. Canagliflozin, the active ingredient of INVOKANA, is chemically known as (1S)-1,5-anhydro-1-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol hemihydrate and its molecular formula and weight are C24H25FO5S∙1/2 H2O and 453.53, respectively. The structural formula for canagliflozin is:
Canagliflozin is practically insoluble in aqueous media from pH 1.1 to 12.9.
INVOKANA is supplied as film-coated tablets for oral administration, containing 102 and 306 mg of canagliflozin in each tablet strength, corresponding to 100 mg and 300 mg of canagliflozin (anhydrous), respectively.
Inactive ingredients of the core tablet are croscarmellose sodium, hydroxypropyl cellulose, lactose anhydrous, magnesium stearate, and microcrystalline cellulose. The magnesium stearate is vegetable-sourced. The tablets are finished with a commercially available film-coating consisting of the following excipients: polyvinyl alcohol (partially hydrolyzed), titanium dioxide, macrogol/PEG, talc, and iron oxide yellow, E172 (100 mg tablet only).
12. Invokana - Clinical Pharmacology
12.1 Mechanism of Action
Sodium-glucose co-transporter 2 (SGLT2), expressed in the proximal renal tubules, is responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Canagliflozin is an inhibitor of SGLT2. By inhibiting SGLT2, canagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose (RTG), and thereby increases urinary glucose excretion (UGE).
Canagliflozin increases the delivery of sodium to the distal tubule by blocking SGLT2-dependent glucose and sodium reabsorption. This is believed to increase tubuloglomerular feedback and reduce intraglomerular pressure.
12.2 Pharmacodynamics
Following single and multiple oral doses of canagliflozin in patients with type 2 diabetes, dose-dependent decreases in the renal threshold for glucose (RTG) and increases in urinary glucose excretion were observed. From a starting RTG value of approximately 240 mg/dL, canagliflozin at 100 mg and 300 mg once daily suppressed RTG throughout the 24-hour period. Data from single oral doses of canagliflozin in healthy volunteers indicate that, on average, the elevation in urinary glucose excretion approaches baseline by about 3 days for doses up to 300 mg once daily. Maximal suppression of mean RTG over the 24-hour period was seen with the 300 mg daily dose to approximately 70 to 90 mg/dL in patients with type 2 diabetes in Phase 1 trials. The reductions in RTG led to increases in mean UGE of approximately 100 g/day in subjects with type 2 diabetes treated with either 100 mg or 300 mg of canagliflozin. In patients with type 2 diabetes given 100 to 300 mg once daily over a 16-day dosing period, reductions in RTG and increases in urinary glucose excretion were observed over the dosing period. In this trial, plasma glucose declined in a dose-dependent fashion within the first day of dosing. In single-dose trials in healthy and type 2 diabetic subjects, treatment with canagliflozin 300 mg before a mixed-meal delayed intestinal glucose absorption and reduced postprandial glucose.
Cardiac Electrophysiology
In a randomized, double-blind, placebo-controlled, active-comparator, 4-way crossover trial, 60 healthy subjects were administered a single oral dose of canagliflozin 300 mg, canagliflozin 1,200 mg (4 times the maximum recommended dose), moxifloxacin, and placebo. No meaningful changes in QTc interval were observed with either the recommended dose of 300 mg or the 1,200 mg dose.
12.3 Pharmacokinetics
The pharmacokinetics of canagliflozin is similar in healthy subjects and patients with type 2 diabetes. Following single-dose oral administration of 100 mg and 300 mg of INVOKANA, peak plasma concentrations (median Tmax) of canagliflozin occurs within 1 to 2 hours post-dose. Plasma Cmax and AUC of canagliflozin increased in a dose-proportional manner from 50 mg to 300 mg. The apparent terminal half-life (t1/2) was 10.6 hours and 13.1 hours for the 100 mg and 300 mg doses, respectively. Steady-state was reached after 4 to 5 days of once-daily dosing with canagliflozin 100 mg to 300 mg. Canagliflozin does not exhibit time-dependent pharmacokinetics and accumulated in plasma up to 36% following multiple doses of 100 mg and 300 mg.
Absorption
The mean absolute oral bioavailability of canagliflozin is approximately 65%. Co-administration of a high-fat meal with canagliflozin had no effect on the pharmacokinetics of canagliflozin; therefore, INVOKANA may be taken with or without food. However, based on the potential to reduce postprandial plasma glucose excursions due to delayed intestinal glucose absorption, it is recommended that INVOKANA be taken before the first meal of the day [see Dosage and Administration (2.2)].
Distribution
The mean steady-state volume of distribution of canagliflozin following a single intravenous infusion in healthy subjects was 83.5 L, suggesting extensive tissue distribution. Canagliflozin is extensively bound to proteins in plasma (99%), mainly to albumin. Protein binding is independent of canagliflozin plasma concentrations. Plasma protein binding is not meaningfully altered in patients with renal or hepatic impairment.
Metabolism
O-glucuronidation is the major metabolic elimination pathway for canagliflozin, which is mainly glucuronidated by UGT1A9 and UGT2B4 to two inactive O-glucuronide metabolites.
CYP3A4-mediated (oxidative) metabolism of canagliflozin is minimal (approximately 7%) in humans.
Excretion
Following administration of a single oral [14C] canagliflozin dose to healthy subjects, 41.5%, 7.0%, and 3.2% of the administered radioactive dose was recovered in feces as canagliflozin, a hydroxylated metabolite, and an O-glucuronide metabolite, respectively. Enterohepatic circulation of canagliflozin was negligible.
Approximately 33% of the administered radioactive dose was excreted in urine, mainly as O-glucuronide metabolites (30.5%). Less than 1% of the dose was excreted as unchanged canagliflozin in urine. Renal clearance of canagliflozin 100 mg and 300 mg doses ranged from 1.30 to 1.55 mL/min.
Mean systemic clearance of canagliflozin was approximately 192 mL/min in healthy subjects following intravenous administration.
Specific Populations
Renal Impairment
A single-dose, open-label trial evaluated the pharmacokinetics of canagliflozin 200 mg in subjects with varying degrees of renal impairment (classified using the MDRD-eGFR formula) compared to healthy subjects.
Renal impairment did not affect the Cmax of canagliflozin. Compared to healthy subjects (N=3; eGFR greater than or equal to 90 mL/min/1.73 m2), plasma AUC of canagliflozin was increased by approximately 15%, 29%, and 53% in subjects with mild (N=10), moderate (N=9), and severe (N=10) renal impairment, respectively, (eGFR 60 to less than 90, 30 to less than 60, and 15 to less than 30 mL/min/1.73 m2, respectively), but was similar for ESKD (N=8) subjects and healthy subjects.
Increases in canagliflozin AUC of this magnitude are not considered clinically relevant. The glucose lowering pharmacodynamic response to canagliflozin declines with increasing severity of renal impairment [see Contraindications (4) and Warnings and Precautions (5.3)].
Canagliflozin was negligibly removed by hemodialysis.
Hepatic Impairment
Relative to subjects with normal hepatic function, the geometric mean ratios for Cmax and AUC∞ of canagliflozin were 107% and 110%, respectively, in subjects with Child-Pugh class A (mild hepatic impairment) and 96% and 111%, respectively, in subjects with Child-Pugh class B (moderate hepatic impairment) following administration of a single 300 mg dose of canagliflozin.
These differences are not considered to be clinically meaningful. There is no clinical experience in patients with Child-Pugh class C (severe) hepatic impairment [see Use in Specific Populations (8.7)].
Pharmacokinetic Effects of Age, Body Mass Index (BMI)/Weight, Gender and Race
Based on the population PK analysis with data collected from 1526 subjects, age, body mass index (BMI)/weight, gender, and race do not have a clinically meaningful effect on the pharmacokinetics of canagliflozin [see Use in Specific Populations (8.5)].
Drug Interaction Studies
In Vitro Assessment of Drug Interactions
Canagliflozin did not induce CYP450 enzyme expression (3A4, 2C9, 2C19, 2B6, and 1A2) in cultured human hepatocytes. Canagliflozin did not inhibit the CYP450 isoenzymes (1A2, 2A6, 2C19, 2D6, or 2E1) and weakly inhibited CYP2B6, CYP2C8, CYP2C9, and CYP3A4 based on in vitro studies with human hepatic microsomes. Canagliflozin is a weak inhibitor of P-gp.
Canagliflozin is also a substrate of drug transporters P-glycoprotein (P-gp) and MRP2.
In Vivo Assessment of Drug Interactions
| Co-Administered Drug | Dose of Co-Administered Drug* | Dose of Canagliflozin* | Geometric Mean Ratio
(Ratio With/Without Co-Administered Drug) No Effect = 1.0 |
|
|---|---|---|---|---|
| AUC†
(90% CI) | Cmax
(90% CI) |
|||
| QD = once daily; BID = twice daily | ||||
|
||||
|
See Drug Interactions (7) for the clinical relevance of the following: |
||||
|
Rifampin |
600 mg QD for 8 days |
300 mg |
0.49 |
0.72 |
|
No dose adjustments of INVOKANA required for the following: |
||||
|
Cyclosporine |
400 mg |
300 mg QD for 8 days |
1.23 |
1.01 |
|
Ethinyl estradiol and levonorgestrel |
0.03 mg ethinyl estradiol and 0.15 mg levonorgestrel |
200 mg QD for 6 days |
0.91 |
0.92 |
|
Hydrochlorothiazide |
25 mg QD for 35 days |
300 mg QD for 7 days |
1.12 |
1.15 |
|
Metformin |
2,000 mg |
300 mg QD for 8 days |
1.10 |
1.05 |
|
Probenecid |
500 mg BID for 3 days |
300 mg QD for 17 days |
1.21 |
1.13 |
| Co-Administered Drug | Dose of Co-Administered Drug* | Dose of Canagliflozin* | Geometric Mean Ratio
(Ratio With/Without Co-Administered Drug) No Effect = 1.0 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AUC†
(90% CI) | Cmax
(90% CI) |
|||||||||
| QD = once daily; BID = twice daily; INR = International Normalized Ratio | ||||||||||
|
||||||||||
|
See Drug Interactions (7) for the clinical relevance of the following: |
||||||||||
|
Digoxin |
0.5 mg QD first day followed by 0.25 mg QD for 6 days |
300 mg QD for 7 days |
Digoxin |
1.20 |
1.36 |
|||||
|
No dose adjustments of co-administered drug required for the following: |
||||||||||
|
Acetaminophen |
1,000 mg |
300 mg BID for 25 days |
Acetaminophen |
1.06‡
|
1.00 |
|||||
|
Ethinyl estradiol and levonorgestrel |
0.03 mg ethinyl estradiol and 0.15 mg levonorgestrel |
200 mg QD for 6 days |
ethinyl estradiol |
1.07 |
1.22 |
|||||
|
Levonorgestrel |
1.06 |
1.22 |
||||||||
|
Glyburide |
1.25 mg |
200 mg QD for 6 days |
Glyburide |
1.02 |
0.93 |
|||||
|
3-cis-hydroxy-glyburide |
1.01 |
0.99 |
||||||||
|
4-trans-hydroxy-glyburide |
1.03 |
0.96 |
||||||||
|
Hydrochlorothiazide |
25 mg QD for 35 days |
300 mg QD for 7 days |
Hydrochlorothiazide |
0.99 |
0.94 |
|||||
|
Metformin |
2,000 mg |
300 mg QD for 8 days |
Metformin |
1.20 |
1.06 |
|||||
|
Simvastatin |
40 mg |
300 mg QD for 7 days |
Simvastatin |
1.12 |
1.09 |
|||||
|
simvastatin acid |
1.18 |
1.26 |
||||||||
|
Warfarin |
30 mg |
300 mg QD for 12 days |
(R)-warfarin |
1.01 |
1.03 |
|||||
|
(S)-warfarin |
1.06 |
1.01 |
||||||||
|
INR |
1.00 |
1.05 |
||||||||
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity was evaluated in 2-year studies conducted in CD1 mice and Sprague-Dawley rats. Canagliflozin did not increase the incidence of tumors in mice dosed at 10, 30, or 100 mg/kg (less than or equal to 14 times exposure from a 300 mg clinical dose).
Testicular Leydig cell tumors, considered secondary to increased luteinizing hormone (LH), increased significantly in male rats at all doses tested (10, 30, and 100 mg/kg). In a 12-week clinical trial, LH did not increase in males treated with canagliflozin.
Renal tubular adenoma and carcinoma increased significantly in male and female rats dosed at 100 mg/kg, or approximately 12-times exposure from a 300 mg clinical dose. Also, adrenal pheochromocytoma increased significantly in males and numerically in females dosed at 100 mg/kg. Carbohydrate malabsorption associated with high doses of canagliflozin was considered a necessary proximal event in the emergence of renal and adrenal tumors in rats. Clinical trials have not demonstrated carbohydrate malabsorption in humans at canagliflozin doses of up to 2-times the recommended clinical dose of 300 mg.
Mutagenesis
Canagliflozin was not mutagenic with or without metabolic activation in the Ames assay. Canagliflozin was mutagenic in the in vitro mouse lymphoma assay with but not without metabolic activation. Canagliflozin was not mutagenic or clastogenic in an in vivo oral micronucleus assay in rats and an in vivo oral Comet assay in rats.
Impairment of Fertility
Canagliflozin had no effects on the ability of rats to mate and sire or maintain a litter up to the high dose of 100 mg/kg (approximately 14 times and 18 times the 300 mg clinical dose in males and females, respectively), although there were minor alterations in a number of reproductive parameters (decreased sperm velocity, increased number of abnormal sperm, slightly fewer corpora lutea, fewer implantation sites, and smaller litter sizes) at the highest dosage administered.
14. Clinical Studies
14.1 Glycemic Control Trials in Adults with Type 2 Diabetes Mellitus
INVOKANA (canagliflozin) has been studied as monotherapy, in combination with metformin, sulfonylurea, metformin and sulfonylurea, metformin and sitagliptin, metformin and a thiazolidinedione (i.e., pioglitazone), and in combination with insulin (with or without other anti-hyperglycemic agents). The efficacy of INVOKANA was compared to a dipeptidyl peptidase-4 (DPP-4) inhibitor (sitagliptin), both as add-on combination therapy with metformin and sulfonylurea, and a sulfonylurea (glimepiride), both as add-on combination therapy with metformin. INVOKANA was also evaluated in adults 55 to 80 years of age and patients with moderate renal impairment.
Monotherapy
A total of 584 patients with type 2 diabetes inadequately controlled on diet and exercise participated in a 26-week double-blind, placebo-controlled trial to evaluate the efficacy and safety of INVOKANA. The mean age was 55 years, 44% of patients were men, and the mean baseline eGFR was 87 mL/min/1.73 m2. Patients taking other antihyperglycemic agents (N=281) discontinued the agent and underwent an 8-week washout followed by a 2-week, single-blind, placebo run-in period. Patients not taking oral antihyperglycemic agents (N=303) entered the 2-week, single-blind, placebo run-in period directly. After the placebo run-in period, patients were randomized to INVOKANA 100 mg, INVOKANA 300 mg, or placebo, administered once daily for 26 weeks.
At the end of treatment, INVOKANA 100 mg and 300 mg once daily resulted in a statistically significant improvement in HbA1C (p<0.001 for both doses) compared to placebo. INVOKANA 100 mg and 300 mg once daily also resulted in a greater proportion of patients achieving an HbA1C less than 7%, in significant reduction in fasting plasma glucose (FPG), in improved postprandial glucose (PPG), and in percent body weight reduction compared to placebo (see Table 10). Statistically significant (p<0.001 for both doses) mean changes from baseline in systolic blood pressure relative to placebo were -3.7 mmHg and -5.4 mmHg with INVOKANA 100 mg and 300 mg, respectively.
| Efficacy Parameter | Placebo
(N=192) | INVOKANA
100 mg (N=195) | INVOKANA
300 mg (N=197) |
|---|---|---|---|
|
|||
|
HbA1C (%) |
|||
|
Baseline (mean) |
7.97 |
8.06 |
8.01 |
|
Change from baseline (adjusted mean) |
0.14 |
-0.77 |
-1.03 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-0.91‡
|
-1.16‡
|
|
|
Percent of Patients Achieving HbA1C < 7% |
21 |
45‡ |
62‡ |
|
Fasting Plasma Glucose (mg/dL) |
|||
|
Baseline (mean) |
166 |
172 |
173 |
|
Change from baseline (adjusted mean) |
8 |
-27 |
-35 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-36‡
|
-43‡
|
|
|
2-hour Postprandial Glucose (mg/dL) |
|||
|
Baseline (mean) |
229 |
250 |
254 |
|
Change from baseline (adjusted mean) |
5 |
-43 |
-59 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-48‡
|
-64‡
|
|
|
Body Weight | |||
|
Baseline (mean) in kg |
87.5 |
85.9 |
86.9 |
|
% change from baseline (adjusted mean) |
-0.6 |
-2.8 |
-3.9 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-2.2‡
|
-3.3‡
|
|
Add-on Combination Therapy with Metformin
A total of 1,284 patients with type 2 diabetes inadequately controlled on metformin monotherapy (greater than or equal to 2,000 mg/day, or at least 1,500 mg/day if higher dose not tolerated) participated in a 26-week, double-blind, placebo- and active-controlled trial to evaluate the efficacy and safety of INVOKANA in combination with metformin. The mean age was 55 years, 47% of patients were men, and the mean baseline eGFR was 89 mL/min/1.73 m2. Patients already on the required metformin dose (N=1009) were randomized after completing a 2-week, single-blind, placebo run-in period. Patients taking less than the required metformin dose or patients on metformin in combination with another antihyperglycemic agent (N=275) were switched to metformin monotherapy (at doses described above) for at least 8 weeks before entering the 2-week, single-blind, placebo run-in. After the placebo run-in period, patients were randomized to INVOKANA 100 mg, INVOKANA 300 mg, sitagliptin 100 mg, or placebo, administered once daily as add-on therapy to metformin.
At the end of treatment, INVOKANA 100 mg and 300 mg once daily resulted in a statistically significant improvement in HbA1C (p<0.001 for both doses) compared to placebo when added to metformin. INVOKANA 100 mg and 300 mg once daily also resulted in a greater proportion of patients achieving an HbA1C less than 7%, in significant reduction in fasting plasma glucose (FPG), in improved postprandial glucose (PPG), and in percent body weight reduction compared to placebo when added to metformin (see Table 11). Statistically significant (p<0.001 for both doses) mean changes from baseline in systolic blood pressure relative to placebo were -5.4 mmHg and -6.6 mmHg with INVOKANA 100 mg and 300 mg, respectively.
| Efficacy Parameter | Placebo + Metformin
(N=183) | INVOKANA 100 mg + Metformin
(N=368) | INVOKANA 300 mg + Metformin
(N=367) |
|---|---|---|---|
|
|||
|
HbA1C (%) |
|||
|
Baseline (mean) |
7.96 |
7.94 |
7.95 |
|
Change from baseline (adjusted mean) |
-0.17 |
-0.79 |
-0.94 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-0.62‡
|
-0.77‡
|
|
|
Percent of patients achieving HbA1C < 7% |
30 |
46‡ |
58‡ |
|
Fasting Plasma Glucose (mg/dL) |
|||
|
Baseline (mean) |
164 |
169 |
173 |
|
Change from baseline (adjusted mean) |
2 |
-27 |
-38 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-30‡
|
-40‡
|
|
|
2-hour Postprandial Glucose (mg/dL) |
|||
|
Baseline (mean) |
249 |
258 |
262 |
|
Change from baseline (adjusted mean) |
-10 |
-48 |
-57 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-38‡
|
-47‡
|
|
|
Body Weight |
|||
|
Baseline (mean) in kg |
86.7 |
88.7 |
85.4 |
|
% change from baseline (adjusted mean) |
-1.2 |
-3.7 |
-4.2 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-2.5‡
|
-2.9‡
|
|
Initial Combination Therapy with Metformin
A total of 1,186 patients with type 2 diabetes inadequately controlled with diet and exercise participated in a 26-week double-blind, active-controlled, parallel-group, 5-arm, multicenter trial to evaluate the efficacy and safety of initial therapy with INVOKANA in combination with metformin XR. The median age was 56 years, 48% of patients were men, and the mean baseline eGFR was 87.6 mL/min/1.73 m2. The median duration of diabetes was 1.6 years, and 72% of patients were treatment naïve. After completing a 2-week single-blind placebo run-in period, patients were randomly assigned for a double-blind treatment period of 26 weeks to 1 of 5 treatment groups (Table 12). The metformin XR dose was initiated at 500 mg/day for the first week of treatment and then increased to 1000 mg/day. Metformin XR or matching placebo was up-titrated every 2–3 weeks during the next 8 weeks of treatment to a maximum daily dose of 1500 to 2000 mg/day, as tolerated; about 90% of patients reached 2000 mg/day.
At the end of treatment, INVOKANA 100 mg and INVOKANA 300 mg in combination with metformin XR resulted in a statistically significant greater improvement in HbA1C compared to their respective INVOKANA doses (100 mg and 300 mg) alone or metformin XR alone.
| Efficacy Parameter | Metformin XR
(N=237) | INVOKANA 100 mg
(N=237) | INVOKANA 300 mg
(N=238) | INVOKANA 100 mg + Metformin XR
(N=237) | INVOKANA 300 mg + Metformin XR
(N=237) |
|---|---|---|---|---|---|
|
|||||
|
HbA1C (%) |
|||||
|
Baseline (mean) |
8.81 |
8.78 |
8.77 |
8.83 |
8.90 |
|
Change from baseline (adjusted mean)† |
-1.30 |
-1.37 |
-1.42 |
-1.77 |
-1.78 |
|
Difference from canagliflozin 100 mg (adjusted mean) (95% CI)‡ |
-0.40§
| ||||
|
Difference from canagliflozin 300 mg (adjusted mean) (95% CI)‡ |
-0.36§
|
||||
|
Difference from metformin XR (adjusted mean) (95% CI)‡ |
-0.06¶
|
-0.11¶
|
-0.46§
|
-0.48§
|
|
|
Percent of patients achieving HbA1C < 7% |
38 |
34 |
39 |
47# |
51# |
INVOKANA Compared to Glimepiride, Both as Add-on Combination With Metformin
A total of 1,450 patients with type 2 diabetes inadequately controlled on metformin monotherapy (greater than or equal to 2,000 mg/day, or at least 1,500 mg/day if higher dose not tolerated) participated in a 52-week, double-blind, active-controlled trial to evaluate the efficacy and safety of INVOKANA in combination with metformin.
The mean age was 56 years, 52% of patients were men, and the mean baseline eGFR was 90 mL/min/1.73 m2. Patients tolerating maximally required metformin dose (N=928) were randomized after completing a 2-week, single-blind, placebo run-in period. Other patients (N=522) were switched to metformin monotherapy (at doses described above) for at least 10 weeks, then completed a 2-week single-blind run-in period. After the 2-week run-in period, patients were randomized to INVOKANA 100 mg, INVOKANA 300 mg, or glimepiride (titration allowed throughout the 52-week trial to 6 or 8 mg), administered once daily as add-on therapy to metformin.
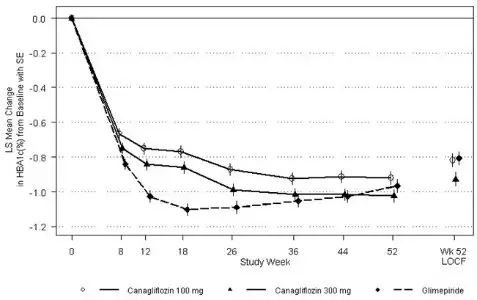
As shown in Table 13 and Figure 1, at the end of treatment, INVOKANA 100 mg provided similar reductions in HbA1C from baseline compared to glimepiride when added to metformin therapy. INVOKANA 300 mg provided a greater reduction from baseline in HbA1C compared to glimepiride, and the relative treatment difference was -0.12% (95% CI: –0.22; –0.02). As shown in Table 13, treatment with INVOKANA 100 mg and 300 mg daily provided greater improvements in percent body weight change, relative to glimepiride.
| Efficacy Parameter | INVOKANA 100 mg + Metformin
(N=483) | INVOKANA 300 mg + Metformin
(N=485) | Glimepiride (titrated) + Metformin
(N=482) |
|---|---|---|---|
|
|||
|
HbA1C (%) |
|||
|
Baseline (mean) |
7.78 |
7.79 |
7.83 |
|
Change from baseline (adjusted mean) |
-0.82 |
-0.93 |
-0.81 |
|
Difference from glimepiride (adjusted mean) (95% CI)† |
-0.01‡
|
-0.12‡
| |
|
Percent of patients achieving HbA1C < 7% |
54 |
60 |
56 |
|
Fasting Plasma Glucose (mg/dL) |
|||
|
Baseline (mean) |
165 |
164 |
166 |
|
Change from baseline (adjusted mean) |
-24 |
-28 |
-18 |
|
Difference from glimepiride (adjusted mean) (95% CI)† |
-6 |
-9 | |
|
Body Weight |
|||
|
Baseline (mean) in kg |
86.8 |
86.6 |
86.6 |
|
% change from baseline (adjusted mean) |
-4.2 |
-4.7 |
1.0 |
|
Difference from glimepiride (adjusted mean) (95% CI)† |
-5.2§
|
-5.7§
| |
| Figure 1: Mean HbA1C Change at Each Time Point (Completers) and at Week 52 Using Last Observation Carried Forward (mITT Population) |
|---|
 |
Add-on Combination Therapy with Sulfonylurea
A total of 127 patients with type 2 diabetes inadequately controlled on sulfonylurea monotherapy participated in an 18-week, double-blind, placebo-controlled sub-study to evaluate the efficacy and safety of INVOKANA in combination with sulfonylurea. The mean age was 65 years, 57% of patients were men, and the mean baseline eGFR was 69 mL/min/1.73 m2. Patients treated with sulfonylurea monotherapy on a stable protocol-specified dose (greater than or equal to 50% maximal dose) for at least 10 weeks completed a 2-week, single-blind, placebo run-in period. After the run-in period, patients with inadequate glycemic control were randomized to INVOKANA 100 mg, INVOKANA 300 mg, or placebo, administered once daily as add-on to sulfonylurea.
As shown in Table 14, at the end of treatment, INVOKANA 100 mg and 300 mg daily provided statistically significant (p<0.001 for both doses) improvements in HbA1C relative to placebo when added to sulfonylurea. INVOKANA 300 mg once daily compared to placebo resulted in a greater proportion of patients achieving an HbA1C less than 7%, (33% vs 5%), greater reductions in fasting plasma glucose (-36 mg/dL vs +12 mg/dL), and greater percent body weight reduction (-2.0% vs -0.2%).
| Efficacy Parameter | Placebo + Sulfonylurea
(N=45) | INVOKANA 100 mg + Sulfonylurea
(N=42) | INVOKANA 300 mg + Sulfonylurea
(N=40) |
|---|---|---|---|
|
|||
|
HbA1C (%) |
|||
|
Baseline (mean) |
8.49 |
8.29 |
8.28 |
|
Change from baseline (adjusted mean) |
0.04 |
-0.70 |
-0.79 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-0.74‡
|
-0.83‡
|
|
Add-on Combination Therapy with Metformin and Sulfonylurea
A total of 469 patients with type 2 diabetes inadequately controlled on the combination of metformin (greater than or equal to 2,000 mg/day or at least 1,500 mg/day if higher dose not tolerated) and sulfonylurea (maximal or near-maximal effective dose) participated in a 26-week, double-blind, placebo-controlled trial to evaluate the efficacy and safety of INVOKANA in combination with metformin and sulfonylurea. The mean age was 57 years, 51% of patients were men, and the mean baseline eGFR was 89 mL/min/1.73 m2. Patients already on the protocol-specified doses of metformin and sulfonylurea (N=372) entered a 2-week, single-blind, placebo run-in period. Other patients (N=97) were required to be on a stable protocol-specified dose of metformin and sulfonylurea for at least 8 weeks before entering the 2-week run-in period. Following the run-in period, patients were randomized to INVOKANA 100 mg, INVOKANA 300 mg, or placebo, administered once daily as add-on to metformin and sulfonylurea.
At the end of treatment, INVOKANA 100 mg and 300 mg once daily resulted in a statistically significant improvement in HbA1C (p<0.001 for both doses) compared to placebo when added to metformin and sulfonylurea. INVOKANA 100 mg and 300 mg once daily also resulted in a greater proportion of patients achieving an HbA1C less than 7%, in a significant reduction in fasting plasma glucose (FPG), and in percent body weight reduction compared to placebo when added to metformin and sulfonylurea (see Table 15).
| Efficacy Parameter | Placebo + Metformin and Sulfonylurea
(N=156) | INVOKANA 100 mg + Metformin and Sulfonylurea
(N=157) | INVOKANA 300 mg + Metformin and Sulfonylurea
(N=156) |
|---|---|---|---|
|
|||
|
HbA1C (%) |
|||
|
Baseline (mean) |
8.12 |
8.13 |
8.13 |
|
Change from baseline (adjusted mean) |
-0.13 |
-0.85 |
-1.06 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-0.71‡
|
-0.92‡
|
|
|
Percent of patients achieving A1C < 7% |
18 |
43‡ |
57‡ |
|
Fasting Plasma Glucose (mg/dL) |
|||
|
Baseline (mean) |
170 |
173 |
168 |
|
Change from baseline (adjusted mean) |
4 |
-18 |
-31 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-22‡
|
-35‡
|
|
|
Body Weight |
|||
|
Baseline (mean) in kg |
90.8 |
93.5 |
93.5 |
|
% change from baseline (adjusted mean) |
-0.7 |
-2.1 |
-2.6 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-1.4‡
|
-2.0‡
|
|
Add-on Combination Therapy with Metformin and Sitagliptin
A total of 217 patients with type 2 diabetes inadequately controlled on the combination of metformin (greater than or equal to 1,500 mg/day) and sitagliptin 100 mg/day (or equivalent fixed-dose combination) participated in a 26-week, double-blind, placebo-controlled trial to evaluate the efficacy and safety of INVOKANA in combination with metformin and sitagliptin. The mean age was 57 years, 58% of patients were men, 73% of patients were Caucasian, 15% were Asian, and 12% were Black or African-American. The mean baseline eGFR was 90 mL/min/1.73 m2 and the mean baseline BMI was 32 kg/m2. The mean duration of diabetes was 10 years. Eligible patients entered a 2-week, single-blind, placebo run-in period and were subsequently randomized to INVOKANA 100 mg or placebo, administered once daily as add-on to metformin and sitagliptin. Patients with a baseline eGFR of 70 mL/min/1.73 m2 or greater who were tolerating INVOKANA 100 mg and who required additional glycemic control (fasting finger stick 100 mg/dL or greater at least twice within 2 weeks) were up-titrated to INVOKANA 300 mg. While up-titration occurred as early as Week 4, most (90%) patients randomized to INVOKANA were up-titrated to INVOKANA 300 mg by 6 to 8 weeks.
At the end of 26 weeks, INVOKANA resulted in a statistically significant improvement in HbA1C (p<0.001) compared to placebo when added to metformin and sitagliptin.
| Efficacy Parameter | Placebo + Metformin and Sitagliptin
(N=108*) | INVOKANA + Metformin and Sitagliptin
(N=109*) |
|---|---|---|
|
||
|
HbA1C (%) |
||
|
Baseline (mean) |
8.40 |
8.50 |
|
Change from baseline (adjusted mean) |
-0.03 |
-0.83 |
|
Difference from placebo (adjusted mean) (95% CI)†‡ |
-0.81§
|
|
|
Percent of patients achieving HbA1C < 7%¶ |
9 |
28 |
|
Fasting Plasma Glucose (mg/dL)# |
||
|
Baseline (mean) |
180 |
185 |
|
Change from baseline (adjusted mean) |
-3 |
-28 |
|
Difference from placebo (adjusted mean) (95% CI) |
-25§
|
|
INVOKANA Compared to Sitagliptin, Both as Add-on Combination Therapy with Metformin and Sulfonylurea
A total of 755 patients with type 2 diabetes inadequately controlled on the combination of metformin (greater than or equal to 2,000 mg/day or at least 1,500 mg/day if higher dose not tolerated) and sulfonylurea (near-maximal or maximal effective dose) participated in a 52-week, double-blind, active-controlled trial to compare the efficacy and safety of INVOKANA 300 mg versus sitagliptin 100 mg in combination with metformin and sulfonylurea. The mean age was 57 years, 56% of patients were men, and the mean baseline eGFR was 88 mL/min/1.73 m2. Patients already on protocol-specified doses of metformin and sulfonylurea (N=716) entered a 2-week single-blind, placebo run-in period. Other patients (N=39) were required to be on a stable protocol-specified dose of metformin and sulfonylurea for at least 8 weeks before entering the 2-week run-in period. Following the run-in period, patients were randomized to INVOKANA 300 mg or sitagliptin 100 mg as add-on to metformin and sulfonylurea.
As shown in Table 17 and Figure 2, at the end of treatment, INVOKANA 300 mg provided greater HbA1C reduction compared to sitagliptin 100 mg when added to metformin and sulfonylurea (p<0.05). INVOKANA 300 mg resulted in a mean percent change in body weight from baseline of -2.5% compared to +0.3% with sitagliptin 100 mg. A mean change in systolic blood pressure from baseline of -5.06 mmHg was observed with INVOKANA 300 mg compared to +0.85 mmHg with sitagliptin 100 mg.
| Efficacy Parameter | INVOKANA 300 mg + Metformin and Sulfonylurea
(N=377) | Sitagliptin 100 mg + Metformin and Sulfonylurea
(N=378) |
|---|---|---|
|
||
|
HbA1C (%) |
||
|
Baseline (mean) |
8.12 |
8.13 |
|
Change from baseline (adjusted mean) |
-1.03 |
-0.66 |
|
Difference from sitagliptin (adjusted mean) (95% CI)† |
-0.37‡
| |
|
Percent of patients achieving HbA1C < 7% |
48 |
35 |
|
Fasting Plasma Glucose (mg/dL) |
||
|
Baseline (mean) |
170 |
164 |
|
Change from baseline (adjusted mean) |
-30 |
-6 |
|
Difference from sitagliptin (adjusted mean) (95% CI)† |
-24 | |
|
Body Weight |
||
|
Baseline (mean) in kg |
87.6 |
89.6 |
|
% change from baseline (adjusted mean) |
-2.5 |
0.3 |
|
Difference from sitagliptin (adjusted mean) (95% CI)† |
-2.8§
| |
Figure 2: Mean HbA1C Change at Each Time Point (Completers) and at Week 52 Using Last Observation Carried Forward (mITT Population)
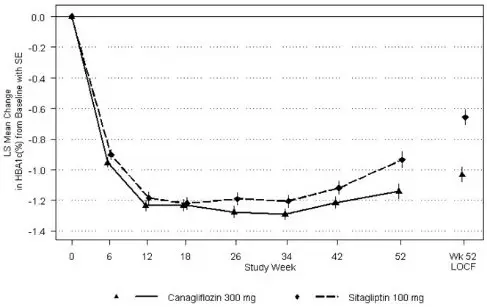
Add-on Combination Therapy with Metformin and Pioglitazone
A total of 342 patients with type 2 diabetes inadequately controlled on the combination of metformin (greater than or equal to 2,000 mg/day or at least 1,500 mg/day if higher dose not tolerated) and pioglitazone (30 or 45 mg/day) participated in a 26-week, double-blind, placebo-controlled trial to evaluate the efficacy and safety of INVOKANA in combination with metformin and pioglitazone. The mean age was 57 years, 63% of patients were men, and the mean baseline eGFR was 86 mL/min/1.73 m2. Patients already on protocol-specified doses of metformin and pioglitazone (N=163) entered a 2-week, single-blind, placebo run-in period. Other patients (N=181) were required to be on stable protocol-specified doses of metformin and pioglitazone for at least 8 weeks before entering the 2-week run-in period. Following the run-in period, patients were randomized to INVOKANA 100 mg, INVOKANA 300 mg, or placebo, administered once daily as add-on to metformin and pioglitazone.
At the end of treatment, INVOKANA 100 mg and 300 mg once daily resulted in a statistically significant improvement in HbA1C (p<0.001 for both doses) compared to placebo when added to metformin and pioglitazone. INVOKANA 100 mg and 300 mg once daily also resulted in a greater proportion of patients achieving an HbA1C less than 7%, in significant reduction in fasting plasma glucose (FPG) and in percent body weight reduction compared to placebo when added to metformin and pioglitazone (see Table 18). Statistically significant (p<0.05 for both doses) mean changes from baseline in systolic blood pressure relative to placebo were -4.1 mmHg and -3.5 mmHg with INVOKANA 100 mg and 300 mg, respectively.
| Efficacy Parameter | Placebo + Metformin and Pioglitazone
(N=115) | INVOKANA 100 mg + Metformin and Pioglitazone
(N=113) | INVOKANA 300 mg + Metformin and Pioglitazone
(N=114) |
|---|---|---|---|
|
|||
|
HbA1C (%) |
|||
|
Baseline (mean) |
8.00 |
7.99 |
7.84 |
|
Change from baseline (adjusted mean) |
-0.26 |
-0.89 |
-1.03 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-0.62‡
|
-0.76‡
|
|
|
Percent of patients achieving HbA1C < 7% |
33 |
47‡ |
64‡ |
|
Fasting Plasma Glucose (mg/dL) |
|||
|
Baseline (mean) |
164 |
169 |
164 |
|
Change from baseline (adjusted mean) |
3 |
-27 |
-33 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-29‡
|
-36‡
|
|
|
Body Weight |
|||
|
Baseline (mean) in kg |
94.0 |
94.2 |
94.4 |
|
% change from baseline (adjusted mean) |
-0.1 |
-2.8 |
-3.8 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-2.7‡
|
-3.7‡
|
|
Add-On Combination Therapy with Insulin (With or Without Other Antihyperglycemic Agents)
A total of 1,718 patients with type 2 diabetes inadequately controlled on insulin greater than or equal to 30 units/day or insulin in combination with other antihyperglycemic agents participated in an 18-week, double-blind, placebo-controlled substudy of a cardiovascular trial to evaluate the efficacy and safety of INVOKANA in combination with insulin. The mean age was 63 years, 66% of patients were men, and the mean baseline eGFR was 75 mL/min/1.73 m2. Patients on basal, bolus, or basal/bolus insulin for at least 10 weeks entered a 2-week, single-blind, placebo run-in period. Approximately 70% of patients were on a background basal/bolus insulin regimen. After the run-in period, patients were randomized to INVOKANA 100 mg, INVOKANA 300 mg, or placebo, administered once daily as add-on to insulin. The mean daily insulin dose at baseline was 83 units, which was similar across treatment groups.
At the end of treatment, INVOKANA 100 mg and 300 mg once daily resulted in a statistically significant improvement in HbA1C (p<0.001 for both doses) compared to placebo when added to insulin. INVOKANA 100 mg and 300 mg once daily also resulted in a greater proportion of patients achieving an HbA1C less than 7%, in significant reductions in fasting plasma glucose (FPG), and in percent body weight reductions compared to placebo (see Table 19). Statistically significant (p<0.001 for both doses) mean changes from baseline in systolic blood pressure relative to placebo were -2.6 mmHg and -4.4 mmHg with INVOKANA 100 mg and 300 mg, respectively.
| Efficacy Parameter | Placebo + Insulin
(N=565) | INVOKANA 100 mg + Insulin
(N=566) | INVOKANA 300 mg + Insulin
(N=587) |
|---|---|---|---|
|
|||
|
HbA1C (%) |
|||
|
Baseline (mean) |
8.20 |
8.33 |
8.27 |
|
Change from baseline (adjusted mean) |
0.01 |
-0.63 |
-0.72 |
|
Difference from placebo (adjusted mean) (95% CI)† |
-0.65‡
|
-0.73‡
|
|
|
Percent of patients achieving HbA1C < 7% |
8 |
20‡ |
25‡ |
|
Fasting Plasma Glucose (mg/dL) |
|||
|
Baseline |
169 |
170 |
168 |
|
Change from baseline (adjusted mean) |
4 |
-19 |
-25 |
|
Difference from placebo (adjusted mean) (97.5% CI)† |
-23‡
|
-29‡
|
|
|
Body Weight |
|||
|
Baseline (mean) in kg |
97.7 |
96.9 |
96.7 |
|
% change from baseline (adjusted mean) |
0.1 |
-1.8 |
-2.3 |
|
Difference from placebo (adjusted mean) (97.5% CI)† |
-1.9‡
|
-2.4‡
|
|
Study in Patients Ages 55 to 80
A total of 714 type 2 diabetes patients ages 55 to 80 years and inadequately controlled on current diabetes therapy (either diet and exercise alone or in combination with oral or parenteral agents) participated in a 26-week, double-blind, placebo-controlled trial to evaluate the efficacy and safety of INVOKANA in combination with current diabetes treatment. The mean age was 64 years, 55% of patients were men, and the mean baseline eGFR was 77 mL/min/1.73 m2. Patients were randomized in a 1:1:1 ratio to the addition of INVOKANA 100 mg, INVOKANA 300 mg, or placebo, administered once daily. At the end of treatment, INVOKANA provided statistically significant improvements from baseline relative to placebo in HbA1C (p<0.001 for both doses) of -0.57% (95% CI: -0.71%; -0.44%) for INVOKANA 100 mg and -0.70% (95% CI: -0.84%; -0.57%) for INVOKANA 300 mg. [see Use in Specific Populations (8.5)].
Glycemic Control in Patients with Moderate Renal Impairment
A total of 269 patients with type 2 diabetes and a baseline eGFR of 30 mL/min/1.73 m2 to less than 50 mL/min/1.73 m2 inadequately controlled on current diabetes therapy participated in a 26-week, double-blind, placebo-controlled clinical trial to evaluate the efficacy and safety of INVOKANA in combination with current diabetes treatment (diet or antihyperglycemic agent therapy, with 95% of patients on insulin and/or sulfonylurea). The mean age was 68 years, 61% of patients were men, and the mean baseline eGFR was 39 mL/min/1.73 m2. Patients were randomized in a 1:1:1 ratio to the addition of INVOKANA 100 mg, INVOKANA 300 mg, or placebo, administered once daily.
At the end of treatment, INVOKANA 100 mg and INVOKANA 300 mg daily provided greater reductions in HbA1C relative to placebo (-0.30% [95% CI: -0.53%; -0.07%] and -0.40%, [95% CI: -0.64%; -0.17%], respectively) [see Warnings and Precautions (5.3), Adverse Reactions (6.1), Use in Specific Populations (8.6), and Clinical Studies (14.3)].
14.2 Cardiovascular Outcomes in Patients with Type 2 Diabetes Mellitus and Atherosclerotic Cardiovascular Disease
The CANVAS and CANVAS-R trials were multicenter, multi-national, randomized, double-blind parallel group, with similar inclusion and exclusion criteria. Patients eligible for enrollment in both CANVAS and CANVAS-R trials were: 30 years of age or older and had established, stable, cardiovascular, cerebrovascular, peripheral artery disease (66% of the enrolled population) or were 50 years of age or older and had two or more other specified risk factors for cardiovascular disease (34% of the enrolled population).
The integrated analysis of the CANVAS and CANVAS-R trials compared the risk of Major Adverse Cardiovascular Event (MACE) between canagliflozin and placebo when these were added to and used concomitantly with standard of care treatments for diabetes and atherosclerotic cardiovascular disease. The primary endpoint, MACE, was the time to first occurrence of a three-part composite outcome which included cardiovascular death, non-fatal myocardial infarction and non-fatal stroke.
In CANVAS, patients were randomly assigned 1:1:1 to canagliflozin 100 mg, canagliflozin 300 mg, or matching placebo. In CANVAS-R, patients were randomly assigned 1:1 to canagliflozin 100 mg or matching placebo, and titration to 300 mg was permitted at the investigator's discretion (based on tolerability and glycemic needs) after Week 13. Concomitant antidiabetic and atherosclerotic therapies could be adjusted, at the discretion of investigators, to ensure participants were treated according to the standard care for these diseases.
A total of 10,134 patients were treated (4,327 in CANVAS and 5,807 in CANVAS-R; total of 4,344 randomly assigned to placebo and 5,790 to canagliflozin) for a mean exposure duration of 149 weeks (223 weeks [4.3 years] in CANVAS and 94 weeks [1.8 years] in CANVAS-R). Approximately 78% of the trial population was Caucasian, 13% was Asian, and 3% was Black. The mean age was 63 years and approximately 64% were male.
The mean HbA1C at baseline was 8.2% and mean duration of diabetes was 13.5 years with 70% of patients having had diabetes for 10 years or more. Approximately 31%, 21% and 17% reported a past history of neuropathy, retinopathy and nephropathy, respectively, and the mean eGFR 76 mL/min/1.73 m2. At baseline, patients were treated with one (19%) or more (80%) antidiabetic medications including metformin (77%), insulin (50%), and sulfonylurea (43%).
At baseline, the mean systolic blood pressure was 137 mmHg, the mean diastolic blood pressure was 78 mmHg, the mean LDL was 89 mg/dL, the mean HDL was 46 mg/dL, and the mean urinary albumin to creatinine ratio (UACR) was 115 mg/g. At baseline, approximately 80% of patients were treated with renin angiotensin system inhibitors, 53% with beta-blockers, 13% with loop diuretics, 36% with non-loop diuretics, 75% with statins, and 74% with antiplatelet agents (mostly aspirin). During the trial, investigators could modify anti-diabetic and cardiovascular therapies to achieve local standard of care treatment targets with respect to blood glucose, lipid, and blood pressure. More patients receiving canagliflozin compared to placebo initiated anti-thrombotics (5.2% vs 4.2%) and statins (5.8% vs 4.8%) during the trial.
For the primary analysis, a stratified Cox proportional hazards model was used to test for non-inferiority against a pre-specified risk margin of 1.3 for the hazard ratio of MACE.
In the integrated analysis of CANVAS and CANVAS-R trials, canagliflozin reduced the risk of first occurrence of MACE. The estimated hazard ratio (95% CI) for time to first MACE was 0.86 (0.75, 0.97). Refer to Table 20. Vital status was obtained for 99.6% of patients across the trials. The Kaplan-Meier curve depicting time to first occurrence of MACE is shown in Figure 3.
| Placebo
N=4347(%) | Canagliflozin
N=5795 (%) | Hazard ratio
(95% C.I.)† |
|
|---|---|---|---|
|
|||
|
Composite of cardiovascular death, non-fatal myocardial infarction, non-fatal stroke |
426 (10.4) |
585 (9.2) |
0.86 (0.75, 0.97) |
|
Non-fatal myocardial infarction§, ¶ |
159 (3.9) |
215 (3.4) |
0.85 (0.69, 1.05) |
|
Non-fatal Stroke§, ¶ |
116 (2.8) |
158 (2.5) |
0.90 (0.71, 1.15) |
|
Cardiovascular Death§, ¶ |
185 (4.6) |
268 (4.1) |
0.87 (0.72, 1.06) |
Figure 3: Time to First Occurrence of MACE
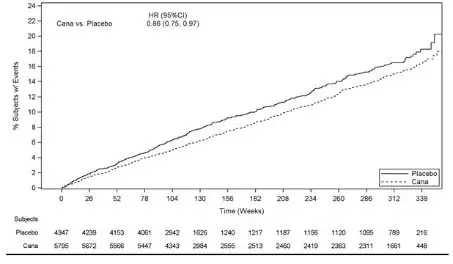
14.3 Renal and Cardiovascular Outcomes in Patients with Diabetic Nephropathy and Albuminuria
The Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation Trial (CREDENCE) was a multinational, randomized, double-blind, placebo-controlled trial comparing canagliflozin with placebo in patients with type 2 diabetes mellitus, an eGFR ≥ 30 to < 90 mL/min/1.73 m2 and albuminuria (urine albumin/creatinine > 300 to ≤ 5000 mg/g) who were receiving standard of care including a maximum-tolerated, labeled daily dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB).
The primary objective of CREDENCE was to assess the efficacy of canagliflozin relative to placebo in reducing the composite endpoint of end stage kidney disease (ESKD), doubling of serum creatinine, and renal or CV death.
Patients were randomized to receive canagliflozin 100 mg (N=2,202) or placebo (N=2,199) and treatment was continued until the initiation of dialysis or renal transplantation.
The median follow-up duration for the 4,401 randomized subjects was 137 weeks. Vital status was obtained for 99.9% of subjects.
The population was 67% White, 20% Asian, and 5% Black; 32% were of Hispanic or Latino ethnicity. The mean age was 63 years and 66% were male.
At randomization, the mean HbA1c was 8.3%, the median urine albumin/creatinine was 927 mg/g, the mean eGFR was 56.2 mL/min/1.73 m2, 50% had prior CV disease, and 15% reported a history of heart failure. The most frequent antihyperglycemic agents (AHA) medications used at baseline were insulin (66%), biguanides (58%), and sulfonylureas (29%). Nearly all subjects (99.9%) were on ACEi or ARB at randomization, approximately 60% were taking an anti-thrombotic agent (including aspirin), and 69% were on a statin.
The primary composite endpoint in the CREDENCE study was the time to first occurrence of ESKD (defined as an eGFR < 15 mL/min/1.73 m2, initiation of chronic dialysis or renal transplant), doubling of serum creatinine, and renal or CV death. Canagliflozin 100 mg significantly reduced the risk of the primary composite endpoint based on a time-to-event analysis [HR: 0.70; 95% CI: 0.59, 0.82; p<0.0001] (see Figure 4). The treatment effect reflected a reduction in progression to ESKD, doubling of serum creatinine and cardiovascular death as shown in Table 21 and Figure 4. There were few renal deaths during the trial. Canagliflozin 100 mg also significantly reduced the risk of hospitalization for heart failure [HR: 0.61; 95% CI: 0.47 to 0.80; p<0.001].
| Placebo | canagliflozin | ||||
|---|---|---|---|---|---|
| Endpoint | N=2,199 (%) | Event Rate* | N=2,202 (%) | Event Rate* | HR†
(95% CI) |
| Intent-To-Treat Analysis Set (time to first occurrence) | |||||
| The individual components do not represent a breakdown of the composite outcomes, but rather the total number of subjects experiencing an event during the course of the study. | |||||
|
|||||
|
Primary Composite Endpoint (ESKD, doubling of serum creatinine, renal death, or CV death) |
340 (15.5) |
6.1 |
245 (11.1) |
4.3 |
0.70 |
|
ESKD |
165 (7.5) |
2.9 |
116 (5.3) |
2.0 |
0.68 |
|
Doubling of serum creatinine |
188 (8.5) |
3.4 |
118 (5.4) |
2.1 |
0.60 |
|
Renal death |
5 (0.2) |
0.1 |
2 (0.1) |
0.0 | |
|
CV death |
140 (6.4) |
2.4 |
110 (5.0) |
1.9 |
0.78 |
|
CV death or hospitalization for heart failure |
253 (11.5) |
4.5 |
179 (8.1) |
3.1 |
0.69 |
|
CV death, non-fatal myocardial infarction or non-fatal stroke |
269 (12.2) |
4.9 |
217 (9.9) |
3.9 |
0.80 |
|
Non-fatal myocardial infarction |
87 (4.0) |
1.6 |
71 (3.2) |
1.3 |
0.81 |
|
Non-fatal stroke |
66 (3.0) |
1.2 |
53 (2.4) |
0.9 |
0.80 |
|
Hospitalization for heart failure |
141 (6.4) |
2.5 |
89 (4.0) |
1.6 |
0.61 |
|
ESKD, doubling of serum creatinine or renal death |
224 (10.2) |
4.0 |
153 (6.9) |
2.7 |
0.66 |
The Kaplan-Meier curve (Figure 4) shows time to first occurrence of the primary composite endpoint of ESKD, doubling of serum creatinine, renal death, or CV death. The curves begin to separate by Week 52 and continue to diverge thereafter.
Figure 4: CREDENCE: Time to First Occurrence of the Primary Composite Endpoint
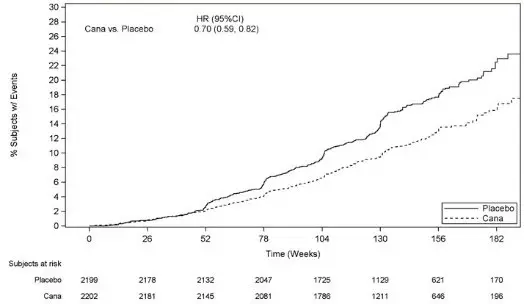
16. How is Invokana supplied
INVOKANA® (canagliflozin) tablets are available in the strengths and packages listed below:
100 mg tablets are yellow, capsule-shaped, film-coated tablets with "CFZ" on one side and "100" on the other side.
|
NDC 55154-1425-8 |
Bottle of Approximately 2070 Tablets |
300 mg tablets are white, capsule-shaped, film-coated tablets with "CFZ" on one side and "300" on the other side.
|
NDC 55154-1426-8 |
Bottle of Approximately 720 Tablets |
17. Patient Counseling Information
See FDA-approved patient labeling (Medication Guide).
Lower Limb Amputation
Inform patients that INVOKANA is associated with an increased risk of amputations. Counsel patients about the importance of routine preventative foot care. Instruct patients to monitor for new pain or tenderness, sores or ulcers, or infections involving the leg or foot and to seek medical advice immediately if such signs or symptoms develop [see Warnings and Precautions (5.1)].
Ketoacidosis
Inform patients that ketoacidosis is a serious life-threatening condition and that cases of ketoacidosis have been reported during use of INVOKANA, sometimes associated with illness or surgery among other risk factors. Instruct patients to check ketones (when possible) if symptoms consistent with ketoacidosis occur even if blood glucose is not elevated. If symptoms of ketoacidosis (including nausea, vomiting, abdominal pain, tiredness, and labored breathing) occur, instruct patients to discontinue INVOKANA and seek medical attention immediately [see Warnings and Precautions (5.2)].
Volume Depletion
Inform patients that symptomatic hypotension may occur with INVOKANA and advise them to contact their doctor if they experience such symptoms [see Warnings and Precautions (5.3)]. Inform patients that dehydration may increase the risk for hypotension, and to have adequate fluid intake.
Serious Urinary Tract Infections
Inform patients of the potential for urinary tract infections, which may be serious. Provide them with information on the symptoms of urinary tract infections. Advise them to seek medical advice if such symptoms occur [see Warnings and Precautions (5.4)].
Necrotizing Fasciitis of the Perineum (Fournier's Gangrene)
Inform patients that necrotizing infections of the perineum (Fournier's gangrene) have occurred with INVOKANA. Counsel patients to promptly seek medical attention if they develop pain or tenderness, redness, or swelling of the genitals or the area from the genitals back to the rectum, along with a fever above 100.4°F or malaise [see Warnings and Precautions (5.6)].
Genital Mycotic Infections in Females (e.g., Vulvovaginitis)
Inform female patients that vaginal yeast infection may occur and provide them with information on the signs and symptoms of vaginal yeast infection. Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.7)].
Genital Mycotic Infections in Males (e.g., Balanitis or Balanoposthitis)
Inform male patients that yeast infection of penis (e.g., balanitis or balanoposthitis) may occur, especially in uncircumcised males and patients with prior history. Provide them with information on the signs and symptoms of balanitis and balanoposthitis (rash or redness of the glans or foreskin of the penis). Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.7)].
Hypersensitivity Reactions
Inform patients that serious hypersensitivity reactions, such as urticaria, rash, anaphylaxis, and angioedema, have been reported with INVOKANA. Advise patients to report immediately any signs or symptoms suggesting allergic reaction, and to discontinue drug until they have consulted prescribing physicians [see Warnings and Precautions (5.8)].
Bone Fracture
Inform patients that bone fractures have been reported in patients taking INVOKANA. Provide them with information on factors that may contribute to fracture risk [see Warnings and Precautions (5.9)].
Pregnancy
Advise pregnant women, and females of reproductive potential of the potential risk to a fetus with treatment with INVOKANA [see Use in Specific Populations (8.1)]. Instruct females of reproductive potential to report pregnancies to their physicians as soon as possible.
Lactation
Advise women that breastfeeding is not recommended during treatment with INVOKANA [see Use in Specific Populations (8.2)].
Laboratory Tests
Inform patients that due to its mechanism of action, patients taking INVOKANA will test positive for glucose in their urine [see Drug Interactions (7)].
Missed Dose
If a dose is missed, advise patients to take it as soon as it is remembered unless it is almost time for the next dose, in which case patients should skip the missed dose and take the medicine at the next regularly scheduled time. Advise patients not to take two doses of INVOKANA at the same time.
Active ingredient made in Belgium
Manufactured for:
Janssen Pharmaceuticals, Inc.
Titusville, NJ 08560
Licensed from Mitsubishi Tanabe Pharma Corporation
© 2013 – 2019 Janssen Pharmaceutical Companies
Distributed by:
Cardinal Health
Dublin, OH 43017
L7296 Rev. A
L7297 Rev. A
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | |||||||
| Revised: 10/2022 | |||||||
|
Medication Guide
|
|||||||
|
What is the most important information I should know about INVOKANA? INVOKANA can cause serious side effects, including:
Call your doctor right away if you have new pain or tenderness, any sores, ulcers, or infections in your leg or foot. Your doctor may decide to stop your INVOKANA for a while if you have any of these signs or symptoms. Talk to your doctor about proper foot care.
|
|||||||
|
|
||||||
|
If you get any of these symptoms during treatment with INVOKANA, if possible, check for ketones in your urine, even if your blood sugar is less than 250 mg/dL. |
|||||||
Talk to your doctor about what you can do to prevent dehydration including how much fluid you should drink on a daily basis. Call your healthcare provider right away if you reduce the amount of food or liquid you drink, for example if you cannot eat or you start to lose liquids from your body, for example from vomiting, diarrhea, or being in the sun too long. |
|||||||
|
|||||||
|
|
||||||
|
Talk to your doctor about what to do if you get symptoms of a yeast infection of the vagina or penis. Your doctor may suggest you use an over-the-counter antifungal medicine. Talk to your doctor right away if you use an over-the-counter antifungal medication and your symptoms do not go away. |
|||||||
|
What is INVOKANA?
|
|||||||
|
Do not take INVOKANA if you:
|
|||||||
|
Before taking INVOKANA, tell your doctor about all of your medical conditions, including if you:
INVOKANA may affect the way other medicines work, and other medicines may affect how INVOKANA works. Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine. |
|||||||
|
How should I take INVOKANA?
|
|||||||
|
What are the possible side effects of INVOKANA? INVOKANA may cause serious side effects including: See "What is the most important information I should know about INVOKANA?"
|
|||||||
|
|
|
|||||
|
|||||||
|
|
|
|||||
The most common side effects of INVOKANA include:
These are not all the possible side effects of INVOKANA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Janssen Pharmaceuticals, Inc. at 1-800-526-7736. |
|||||||
|
How should I store INVOKANA?
|
|||||||
|
General information about the safe and effective use of INVOKANA. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use INVOKANA for a condition for which it was not prescribed. Do not give INVOKANA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about INVOKANA that is written for health professionals. |
|||||||
|
What are the ingredients in INVOKANA? Active ingredient: canagliflozin Inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, lactose anhydrous, magnesium stearate, and microcrystalline cellulose. In addition, the tablet coating contains iron oxide yellow E172 (100 mg tablet only), macrogol/PEG, polyvinyl alcohol, talc, and titanium dioxide. |
|||||||
|
Active ingredient made in Belgium. Manufactured for: Janssen Pharmaceuticals, Inc., Titusville, NJ 08560. Licensed from Mitsubishi Tanabe Pharma Corporation. © 2013 – 2019 Janssen Pharmaceutical Companies |
|||||||


| INVOKANA
canagliflozin tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| INVOKANA
canagliflozin tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - Cardinal Health 107, LLC (118546603) |
