Drug Detail:Kimmtrak (Tebentafusp-tebn)
Drug Class: Miscellaneous antineoplastics
Highlights of Prescribing Information
KIMMTRAK® (tebentafusp-tebn) injection, for intravenous use
Initial U.S. Approval: 2022
WARNING: CYTOKINE RELEASE SYNDROME
Cytokine Release Syndrome (CRS), which may be serious or life-threatening, occurred in patients receiving KIMMTRAK. Monitor for at least 16 hours following first three infusions and then as clinically indicated (2.2, 5.1).
Recent Major Changes
| Dosage and Administration (2.1) | 11/2022 |
Indications and Usage for Kimmtrak
KIMMTRAK is a bispecific gp100 peptide-HLA-directed CD3 T cell engager indicated for the treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma (1, 2.1).
Kimmtrak Dosage and Administration
- Recommended dosage: 20 mcg intravenously on Day 1, 30 mcg intravenously on Day 8, 68 mcg intravenously on Day 15, and 68 mcg intravenously once every week thereafter (2.2).
- Dilute and administer by intravenous infusion over 15-20 minutes (2.2, 2.4).
- See Full Prescribing Information for instructions on preparation and administration of the diluted solution for intravenous infusion (2.2, 2.4).
- Dosage interruption or permanent discontinuation may be required based on individual safety and tolerability (2.3).
Dosage Forms and Strengths
Injection: 100 mcg/0.5 mL solution in a single-dose vial (3).
Contraindications
None (4).
Warnings and Precautions
- Skin reactions: Rash, pruritus, and cutaneous edema occurred in patients treated with KIMMTRAK. If skin reactions occur, treat based on persistence and severity of symptoms (2.3, 5.2).
- Elevated liver enzymes: Elevations in liver enzymes occurred in patients treated with KIMMTRAK. Monitor ALT, AST, and total bilirubin (2.3, 5.3).
- Embryo-Fetal toxicity: May cause fetal harm. Advise patients of reproductive potential of the potential risk to the fetus and to use effective contraception (5.4, 8.1, 8.3).
Adverse Reactions/Side Effects
The most common adverse reactions (occurring in ≥ 30%) are cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache and vomiting (6.1). The most common laboratory abnormalities (occurring in ≥50%) are decreased lymphocyte count, increased creatinine, increased glucose, increased aspartate aminotransferase, increased alanine aminotransferase, decreased hemoglobin, and decreased phosphate (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Immunocore at 1-844-IMMUNO1 (1-844-466-8661) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2022
Full Prescribing Information
WARNING: CYTOKINE RELEASE SYNDROME
Cytokine Release Syndrome (CRS), which may be serious or life-threatening, occurred in patients receiving KIMMTRAK. Monitor for at least 16 hours following first three infusions and then as clinically indicated [(see Dosage and Administration (2.2), see Warnings and Precautions (5.1)].
1. Indications and Usage for Kimmtrak
KIMMTRAK is indicated for the treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.
2. Kimmtrak Dosage and Administration
2.1 Patient Selection
Select patients for treatment of unresectable or metastatic uveal melanoma with KIMMTRAK based on a positive HLA-A*02:01 genotyping test of a whole blood sample [see Clinical Studies (14)]. Information on FDA-approved tests is available at http://www.fda.gov/companiondiagnostics.
2.2 Recommended Dosage
The recommended dosage of KIMMTRAK administered intravenously is:
- 20 mcg on Day 1
- 30 mcg on Day 8
- 68 mcg on Day 15
- 68 mcg once every week thereafter
Treat patients until unacceptable toxicity or disease progression occur.
Administer the first three infusions of KIMMTRAK in an appropriate healthcare setting by intravenous infusion over 15-20 minutes. Monitor patients during the infusion and for at least 16 hours after the infusion is complete.
If the patient does not experience Grade 2 or worse hypotension (requiring medical intervention) during or after the third infusion, administer subsequent doses in an appropriate ambulatory care setting, and monitor patients for a minimum of 30 minutes following each of these infusions [see Warnings and Precautions (5.1)].
2.3 Dosage Modifications for Adverse Reactions
No dosage reduction for KIMMTRAK is recommended. Dosage modifications for KIMMTRAK for adverse reactions are summarized in Table 1.
Table 1: Dose Modifications for Adverse Reactions
| a Based on National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03 (NCI CTCAEv4.03). | ||
| Adverse Reaction | Severity | KIMMTRAK Dosage Modifications |
| Cytokine Release Syndrome (CRS)
[see Warnings and Precautions (5.1)] | Moderate defined as temperature ≥ 38°C with
|
|
Severe defined as temperature ≥ 38°C with
|
|
|
Life threatening defined as temperature ≥ 38°C with
|
|
|
| Skin Reactions
[see Warnings and Precautions (5.2)] | Grade 2 or 3a |
|
| Grade 4a |
|
|
| Elevated Liver Enzymes
[see Warnings and Precautions (5.3)] | Grade 3 or 4a |
|
| Other Adverse Reactions
[see Adverse Reactions (6.1)] | Grade 3a |
|
| Grade 4a |
|
|
2.4 Preparation and Administration
Preparation
- A 2-step dilution process is required for preparation of the final KIMMTRAK dose for infusion.
- Use aseptic technique for dilution and preparation of intravenous infusion solutions.
- Visually inspect parenteral drug products and infusion bags for particulate matter and discoloration prior to administration, whenever solution and container permit.
Step 1: Preparation of the Infusion Bag
To prevent adsorption of tebentafusp-tebn to the infusion bag and other components of the drug delivery system, prepare an Albumin (Human) in 0.9% Sodium Chloride Injection, USP solution as follows:
- Using a 1 mL syringe with graduations of 2 decimal places and a sterile needle, withdraw the calculated volume of Albumin (Human) into the syringe (see Table 2 below) and add to the 100 mL 0.9% Sodium Chloride Injection, USP bag constructed of polyolefins (PO) [such as polyethylene (PE) and polypropylene (PP)] or polyvinyl chloride (PVC) to make a final Albumin (Human) concentration of 250 mcg/mL.
Table 2: Examples of Albumin (Human) Concentration and Volumes
*Albumin (Human); use concentration as per local availability. Examples include but are not restricted to the following strengths: 5%, 20%, or 25%. Albumin (Human) concentration* Albumin (Human) volume for addition to a 100 mL 0.9% Sodium Chloride Injection, USP Infusion Bag to prepare a concentration of 250 mcg/mL Albumin (Human) in 0.9% Sodium Chloride Injection, USP 5% (50 g/L) 0.5 mL 20% (200 g/L) 0.13 mL 25% (250 g/L) 0.1 mL - Gently homogenize the prepared solution by completing the following steps:
- Invert the infusion bag so that the bag is upside down with the entry port positioned on top. Then tap the side of the port tubing to ensure that any residual solution is released into the bulk solution.
- Mix the prepared solution by gently rotating the bag lengthwise 360 degrees from the inverted position at least 5 times. Do not shake the infusion bag.
- Repeat (i) and (ii) an additional three times.
Step 2- Preparation of KIMMTRAK Solution for Infusion
- Do not shake the KIMMTRAK vial.
- Using a 1 mL syringe with graduations of 2 decimal places and a sterile needle, withdraw the required volume of KIMMTRAK 100 mcg/ 0.5 mL as per the dose required (shown in Table 3 below) and add to the prepared 100 mL infusion bag containing 0.9% Sodium Chloride Injection, USP plus Albumin (Human).
- Discard the single-dose vial containing the unused portion of KIMMTRAK in accordance with local requirements. Do not prepare more than one dose from the vial.
Table 3: KIMMTRAK Volumes Required for Addition to the Infusion Bag
Day of treatment Dose (mcg) of KIMMTRAK Volume (mL) of KIMMTRAK Day 1 20 0.1 Day 8 30 0.15 Day 15 and weekly thereafter 68 0.34 - Mix the infusion bag by following the same procedure outlined in Step 1b.
Administration
- Immediately administer the diluted solution via intravenous infusion over 15-20 minutes through a dedicated intravenous line. A sterile, non-pyrogenic, low protein binding 0.2 micron in-line filter infusion set should be used. Administer the entire contents of the KIMMTRAK infusion bag.
- Administer the prepared infusion bag within 4 hours from the time of preparation including the duration of infusion. During the 4-hour window, the KIMMTRAK infusion bag should remain at room temperature.
- If not used immediately, store the KIMMTRAK infusion bag in a refrigerator at 2°C to 8°C (36°F to 46°F) and infuse within 24 hours from the time of preparation, which includes the storage time in the refrigerator, the time allowed for equilibration of the infusion bag to room temperature, and the duration of the infusion.
- Once removed from the refrigerator, do not refrigerate KIMMTRAK infusion bag again. Do not freeze. Discard unused KIMMTRAK solution beyond the recommended storage time.
- Do not mix KIMMTRAK with other drugs or administer other drugs through the same intravenous line.
- Upon completion of KIMMTRAK infusion, flush the infusion line with adequate volume of sterile 0.9% Sodium Chloride Injection, USP to ensure that the entire contents of the infusion bag are administered.
3. Dosage Forms and Strengths
Injection: 100 mcg/0.5 mL clear, colorless to slightly yellowish solution in a single-dose vial
5. Warnings and Precautions
5.1 Cytokine Release Syndrome
Cytokine release syndrome (CRS), which may be life threatening, occurred in patients receiving KIMMTRAK. Manifestations of CRS may include fever, hypotension, hypoxia, chills, nausea, vomiting, rash, elevated transaminases, fatigue, and headache. CRS (≥ Grade 2) occurred in 77% of patients in Study IMCgp100-202 who received KIMMTRAK [see Adverse Reactions (6.1)]. Among patients who received KIMMTRAK, 23% received systemic corticosteroids for at least 1 infusion, 8% received supplemental oxygen during at least 1 infusion, and 0.8% received a vasopressor for at least 1 infusion. CRS led to permanent discontinuation in 1.2% of patients.
In Study IMCgp100-202, 60% of patients experienced ≥ Grade 2 CRS with more than 1 infusion, with the median number of events being 2 (range 1 - 12). The majority (84%) of episodes of CRS started the day of infusion. Among cases that resolved, the median time to resolution of CRS was 2 days.
Ensure that healthcare providers administering KIMMTRAK have immediate access to medications and resuscitative equipment to manage CRS. Ensure patients are euvolemic prior to initiating the infusions. Closely monitor patients for signs or symptoms of CRS following infusions of KIMMTRAK [see Dosage and Administration (2.2)].
Monitor fluid status, vital signs, and oxygenation level and provide appropriate therapy. Withhold or discontinue KIMMTRAK depending on persistence and severity of CRS [see Dosage and Administration (2.3)].
5.2 Skin Reactions
Skin reactions, including rash, pruritus, and cutaneous edema occurred in patients treated with KIMMTRAK. In study IMCgp100-202, skin reactions occurred in 91% of patients treated with KIMMTRAK, including Grade 2 (44%) and Grade 3 (21%) events. Skin reactions included rash (83%), pruritus (69%), erythema (25%), and cutaneous edema (27%) [see Adverse Reactions (6.1)].
The median time to onset of skin reactions was 1 day (range: 1 – 55 days). The median time to improvement to ≤ Grade 1 was approximately 6 days.
Monitor patients for skin reactions. If skin reactions occur, treat with antihistamine and topical or systemic steroids based on persistence and severity of symptoms. Withhold or permanently discontinue KIMMTRAK depending on the severity of skin reactions [see Dosage and Administration (2.3)].
5.3 Elevated Liver Enzymes
In Study IMCgp100-202, increases in alanine aminotransferase or aspartate aminotransferase were observed in 65% of patients treated with KIMMTRAK.
In patients experiencing ALT/AST elevations, 73% initially occurred within the first 3 infusions with KIMMTRAK. Most patients experiencing Grade 3 or 4 ALT/AST elevations had improvement to ≤ Grade 1 within 7 days. For events that were observed outside the setting of CRS, the median time to onset was 129 days. Grade 3 or greater elevations in liver enzymes outside the setting of CRS occurred in approximately 8% of patients.
Elevations in liver enzymes led to permanent discontinuation in 0.4% of patients receiving KIMMTRAK.
Monitor alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total blood bilirubin prior to the start of and during treatment with KIMMTRAK. Withhold KIMMTRAK according to severity [see Dosage and Administration (2.3)].
5.4 Embryo-Fetal Toxicity
Based on the mechanism of action, KIMMTRAK may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with KIMMTRAK and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Cytokine Release Syndrome [see Boxed Warning, Warnings and Precautions (5.1)]
- Skin Reactions [see Warnings and Precautions (5.2)]
- Elevated Liver Enzymes [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
First line metastatic uveal melanoma
The safety of KIMMTRAK was evaluated in study IMCgp100-202, a randomized (2:1), open-label, active-controlled trial in patients who had not received prior systemic therapy for metastatic or advanced uveal melanoma [see Clinical Studies (14)]. Patients received either KIMMTRAK administered at 20 mcg intravenously on Day 1, 30 mcg intravenously on Day 8, 68 mcg intravenously on Day 15, and 68 mcg intravenously once every week thereafter (N=245) or investigator’s choice treatment (N=111). The median duration of exposure was 5.3 months (range: 0.3 to 33 months) in patients treated with KIMMTRAK.
Serious adverse reactions occurred in 28% of patients who received KIMMTRAK. Serious adverse reactions occurring in ≥ 2% of patients were cytokine release syndrome (10%), rashes (4.5%), pyrexia (2.4%), and hypotension (2%). One patient (0.4%) experienced a fatal adverse reaction (pulmonary embolism).
Adverse reactions led to permanent discontinuation in 3.3% of patients who received KIMMTRAK. Adverse reactions that led to permanent discontinuation of KIMMTRAK were anaphylactic reaction, brain edema, cytokine release syndrome, fatigue, hepatotoxicity, hypotension, and nausea (each 0.4%).
Adverse reactions resulting in dosage interruption occurred in 25% of patients who received KIMMTRAK. Adverse reactions which required dosage interruption in ≥ 2% of patients included fatigue (3.7%), lipase increased (2.9%), pyrexia (2.4%), alanine aminotransferase increase (2%), and aspartate aminotransferase increase (2%).
Adverse reactions leading to dose reduction occurred in 5% of patients who received KIMMTRAK. Adverse reactions which required dosage reduction in ≥ 2% of patients were cytokine release syndrome (2.4%), and rashes (2%).
The most common adverse reactions (≥30%) in patients who received KIMMTRAK were cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting. The most common (≥50%) laboratory abnormalities in patient who received KIMMTRAK were decreased lymphocyte count, increased creatinine, increased glucose, increased AST, increased ALT, decreased hemoglobin, and decreased phosphate.
Table 4 summarizes the adverse reactions observed in study IMCgp100-202.
| a Represents algorithmic identification of CRS cases based on ASTCT grading criteria (Lee et al. 2019). b Represents a composite of multiple related terms. |
||||
| Adverse Reactions | KIMMTRAK (N=245) | Investigator’s Choice (pembrolizumab, or ipilimumab, or dacarbazine) (N=111) |
||
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
| Immune system disorders | ||||
| Cytokine release syndromea | 89 | 0.8 | 2.7 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Rashb | 83 | 18 | 28 | 0 |
| Pruritus | 69 | 4.5 | 23 | 0 |
| Dry skin | 31 | 0 | 3.6 | 0 |
| Skin Hypopigmentationb | 28 | NA | 5 | NA |
| Erythema | 24 | 0 | 0.9 | 0 |
| Hair color changesb | 20 | NA | 0 | NA |
| General disorders and administration site conditions | ||||
| Pyrexia | 76 | 3.7 | 7 | 0.9 |
| Fatigueb | 64 | 6 | 42 | 0.9 |
| Chills | 48 | 0.4 | 3.6 | 0 |
| Edemab | 45 | 0 | 10 | 0 |
| Gastrointestinal disorders | ||||
| Nausea | 49 | 2 | 26 | 0.9 |
| Abdominal painb | 45 | 2.9 | 33 | 3.6 |
| Vomiting | 30 | 1.2 | 9 | 0 |
| Diarrhea | 25 | 1.2 | 20 | 2.7 |
| Vascular disorders | ||||
| Hypotension | 39 | 3.3 | 2.7 | 0 |
| Nervous system disorders | ||||
| Headache | 31 | 0.4 | 10 | 0.9 |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia | 22 | 0.8 | 16 | 0 |
Clinically relevant adverse reactions occurring in < 20% of patients who received KIMMTRAK included back pain, decreased appetite, constipation, hypertension, tachycardia or sinus tachycardia, dyspnea, paresthesia, dizziness, flushing, muscle spasms, myalgia, pain in extremity, alopecia, skin hyperpigmentation, influenza-like illness, oropharyngeal pain and night sweats.
Table 5 summarizes the selected laboratory abnormalities observed in study IMCgp100-202.
| Alk Phos = Alkaline Phosphatase; AST=aspartate aminotransferase; ALT=alanine aminotransferase a The denominator used to calculate the rate varied from 242 to 245 for KIMMTRAK and 105 to 109 for IC based on the number of patients with a baseline value and at least one post-treatment value for the laboratory assessment. |
||||
| KIMMTRAKa
(N=245) | Investigator’s Choicea(pembrolizumab, or ipilimumab, or dacarbazine) (N=111) |
|||
| Grades 1-4 (%) | Grades 3-4 (%) | Grades 1-4 (%) | Grades 3-4 (%) |
|
| HEMATOLOGY | ||||
| Lymphocyte count decreased | 91 | 56 | 26 | 1.8 |
| Hemoglobin decreased | 51 | 0.8 | 20 | 0.9 |
| Platelet count decreased | 16 | 0 | 15 | 0.9 |
| Neutrophil count decreased | 14 | 2 | 8 | 1.8 |
| CHEMISTRY | ||||
| Creatinine increased | 87 | 0.4 | 73 | 0 |
| Glucose increased | 66 | 3.3 | 39 | 4.6 |
| AST increased | 55 | 13 | 39 | 1.9 |
| ALT increased | 52 | 9 | 29 | 1.8 |
| Phosphate decreased | 51 | 11 | 20 | 2 |
| Albumin decreased | 47 | 2.1 | 14 | 0.9 |
| Calcium decreased | 45 | 1.6 | 15 | 1.9 |
| Lipase increased | 37 | 15 | 28 | 6 |
| Magnesium decreased | 34 | 0 | 8 | 0 |
| Alk phos increased | 34 | 2.9 | 36 | 1.8 |
| Sodium decreased | 30 | 2.9 | 15 | 0.9 |
| Potassium increased | 29 | 1.6 | 15 | 0.9 |
| Bilirubin increased | 27 | 4.1 | 14 | 7 |
| Amylase increased | 23 | 4.1 | 18 | 1 |
| Glucose decreased | 18 | 0.4 | 4.6 | 0 |
| Potassium decreased | 17 | 0.8 | 8 | 0.9 |
| Calcium increased | 13 | 0 | 3.7 | 0 |
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Treatment-emergent anti-drug antibodies (ADA) against tebentafusp-tebn were detected in 33% and 29% of patients receiving tebentafusp-tebn across all doses in study IMCgp100-102 and study IMCgp100-202, respectively. The median onset time to ADA formation was 6-9 weeks after tebentafusp-tebn treatment. The ability of these binding ADAs to neutralize tebentafusp-tebn is unknown. The tebentafusp-tebn clearance increased in patients with high titer ADAs [see Clinical Pharmacology (12.3)]. Exploratory analyses with limited data suggest that formation of ADA does not appear to have clinically significant effect on frequency or severity of hypersensitivity related adverse reactions and no observed sign of decreased overall survival.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on the mechanism of action, KIMMTRAK may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data with KIMMTRAK in pregnant woman. No animal reproductive and developmental toxicity studies have been conducted with KIMMTRAK. Molecules of similar molecular weight can cross the placenta resulting in fetal exposure. Advise women of the potential risk to the fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of tebentafusp-tebn in human milk, the effect on the breastfed child, or the effects on milk production. Because tebentafusp-tebn may be excreted in human milk and because of the potential for serious adverse reactions in a breastfed child, advise patients not to breastfeed during treatment with KIMMTRAK and for at least 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
KIMMTRAK may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating KIMMTRAK treatment.
Contraception
Females
Advise female of reproductive potential to use effective contraception during treatment and for 1 week following the last dose of KIMMTRAK [see Use in Specific Populations (8.1)].
8.5 Geriatric Use
Of the 245 patients with metastatic uveal melanoma treated with KIMMTRAK on IMCgp100-202, 47% were 65 years of age and older and 9% were 75 years of age and older. No overall differences in safety or efficacy were observed between patients ≥ 65 years of age compared to younger adult patients.
11. Kimmtrak Description
Tebentafusp-tebn is a bispecific gp100 peptide-HLA-directed T cell receptor CD3 T cell engager. Tebentafusp-tebn has an approximate molecular weight of 77 kDa. Tebentafusp-tebn is produced by recombinant DNA technology in Escherichia coli cells.
KIMMTRAK (tebentafusp-tebn) injection is supplied in a single-dose vial as a sterile, preservative-free, clear, colorless or slightly yellowish solution for intravenous administration by infusion.
Each single-dose vial contains tebentafusp-tebn (100 mcg), citric acid monohydrate (0.95 mg), di-sodium hydrogen phosphate (2.91 mg), mannitol (5 mg), polysorbate 20 (0.1 mg) trehalose (25 mg), and water for injection, with a pH of 6.5.
12. Kimmtrak - Clinical Pharmacology
12.1 Mechanism of Action
Tebentafusp-tebn is a bispecific gp100 peptide-HLA-A*02:01 directed T cell receptor CD3 T cell engager. The TCR arm binds to a gp100 peptide presented by human leukocyte antigen-A*02:01 (HLA-A*02:01) on the cell surface of uveal melanoma tumor cells.
In vitro, tebentafusp-tebn bound to HLA-A*02:01-positive uveal melanoma cells and activated polyclonal T cells to release inflammatory cytokines and cytolytic proteins, which results in direct lysis of uveal melanoma tumor cells.
12.2 Pharmacodynamics
Lymphocyte counts declined the day after the first 3 doses and returned to baseline prior to subsequent doses.
Serum levels of cytokines (IFN-γ, TNFα, IL-2, IL-6, IL-10 and IL-1RA) and chemokines (CXCL9, CXCL10, CXCL11, hepatocyte growth factor, and monocyte chemoattractant protein-1) were increased during the first three doses of KIMMTRAK with peak levels between 8 to 24 hours after treatment with KIMMTRAK and levels returned to baseline prior to subsequent doses. In subsequent treatment cycles, cytokine elevation occurred in fewer patients with lesser intensity compared to the first 3 doses.
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of KIMMTRAK have not been fully characterized.
12.3 Pharmacokinetics
After a single dose administration, tebentafusp-tebn Cmax and AUC0-7d increased in an approximately dose proportional manner from 20 to 68 mcg (0.3 to 1 times the approved recommended dose). Following administration of the approved recommended dosage in patients with metastatic uveal melanoma, the steady-state geometric mean (% CV) Cmax of tebentafusp-tebn is 13 ng/mL (34.6%) and AUC0-7d is 4.6 ng.day/mL (23%) with no accumulation.
Distribution
Tebentafusp-tebn geometric mean (% CV) steady-state volume of distribution is 7.56 L (24%).
Elimination
The geometric mean clearance of tebentafusp-tebn is 16.4 L/d (CV: 24.5%) and median terminal half-life is 7.5 hours (range: 6.8-7.5 hours).
Metabolism
Tebentafusp-tebn is expected to be catabolized into small peptides and amino acids.
Specific Populations
No clinically significant difference in the pharmacokinetics of tebentafusp-tebn were identified based on weight (43 to 163 kg), sex (48% female), age (23 to 91 years), or mild to moderate renal impairment based on creatinine clearance (CLcr) estimated by C-G formula (CLcr 30 to 89 mL/min) or mild hepatic impairment as measured by total bilirubin (TB) and aspartate aminotransferase (AST) (TB ≤ upper limit of normal (ULN) and AST > ULN or TB > 1 to 1.5x ULN and any AST).
Tebentafusp-tebn has not been studied in patients with severe (CLcr < 30 mL/min) renal impairment or in patients with moderate (TB >1.5 to 3x ULN, any AST) to severe (TB > 3 to 10x ULN, any AST) hepatic impairment.
Immunogenicity
Median titer in the ADA-positive subgroup was 8192 across the 67 treatment cycles. The exposure (AUC0-7 days) of tebentafusp-tebn decreased by 97% and terminal half-life decreased to 10-14 minutes in patients with ADA titers greater than 8192.
Drug Interaction
Elevation of certain proinflammatory cytokines may suppress CYP450 enzyme activities.
14. Clinical Studies
Study IMCgp100-202: First line metastatic uveal melanoma
KIMMTRAK was evaluated in IMCgp100-202, a randomized, open-label, multicenter trial (NCT03070392) that enrolled patients with metastatic uveal melanoma (N=378). Patients were required to be HLA-A*02:01 genotype positive identified by a central assay. Patients were excluded if they received prior systemic therapy for metastatic or advanced uveal melanoma or localized liver-directed therapy. Prior surgical resection of oligometastatic disease was permitted. Patients with clinically significant cardiac disease or the presence of symptomatic or untreated brain metastasis were excluded.
Patients were randomized (2:1) to receive KIMMTRAK weekly by intravenous infusion administered at 20 mcg intravenously on Day 1, 30 mcg intravenously on Day 8, 68 mcg intravenously on Day 15, and 68 mcg intravenously once every week thereafter (N=252) or Investigator’s choice (N=126) of pembrolizumab, ipilimumab, or dacarbazine. Randomization was stratified by lactate dehydrogenase (LDH) level at study entry. Across both arms, patients stopped treatment for disease progression, unless the patient was otherwise deriving benefit, or for unacceptable toxicity.
The major efficacy outcome was overall survival (OS). Additional efficacy outcomes were investigator-assessed progression free survival (PFS) and objective response rate (ORR) per RECIST 1.1.
The median age was 64 years (range 23 to 92 years); 50% were female; 87% were White, and 12% were unreported or unknown race. The reported ethnicity was Hispanic or Latino in 2.4% of patients. Baseline ECOG performance status was 0 (73%), 1 (21%), or 2 (0.3%); 36% had elevated LDH level; and 94% had liver metastasis.
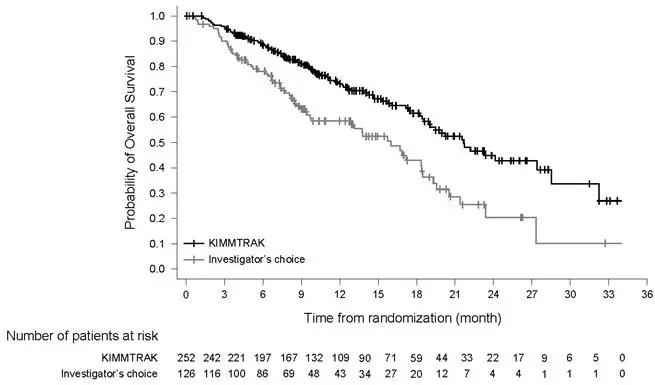
The efficacy results are summarized in Table 6 and Figure 1.
Table 6: Efficacy Results in Study IMCgp100-202
| CI= Confidence Interval, HR= Hazard Ratio
1 Based on prespecified interim analysis 2 Hazard ratio is from a cox proportional hazards model stratified by LDH status 3 Two-sided p-value based on log rank test stratified by LDH 4 Compared to the interim efficacy boundary of 0.006 5 Final PFS analysis 6 Compared to the efficacy boundary of 0.05. 7 Not formally tested |
||
| KIMMTRAK (N=252) | Investigator’s Choice (pembrolizumab, or ipilimumab, or dacarbazine) (N=126) |
|
| Overall Survival (OS)1 | ||
| Number of deaths | 87 (34.5%) | 63 (50%) |
| Median in months (95% CI) | 21.7 (18.6, 28.6) | 16 (9.7, 18.4) |
| HR (95% CI)2 | 0.51 (0.37, 0.71) | |
| p-value3, 4 | <0.0001 | |
| Progression-free Survival5 | ||
| Number (%) of patients with event | 198 (78.6%) | 97 (77%) |
| Median in months (95% CI) | 3.3 (3, 5) | 2.9 (2.8, 3) |
| HR (95% CI)2 | 0.73 (0.58, 0.94) | |
| p-value3, 6 | 0.0139 | |
| Objective Response Rate (95% CI)7 | 9.1% (5.9, 13.4) | 4.8% (1.8, 10.1) |
| Complete Response | 1 (0.4%) | 0 |
| Partial Response | 22 (8.7%) | 6 (4.8%) |
Figure 1: Kaplan-Meier Curves of Overall Survival in Study IMCgp100-202
16. How is Kimmtrak supplied
How Supplied
Each KIMMTRAK (tebentafusp-tebn) injection carton (NDC 80446-401-01) contains:
- One single-dose vial containing 100 mcg of tebentafusp-tebn in 0.5 mL of sterile, preservative-free, clear, colorless or slightly yellowish solution.
The vial stopper is not made with natural rubber latex.
Storage and Handling
- Store KIMMTRAK vials in the original carton refrigerated at 2°C to 8°C (36°F to 46°F) and protect from light until time of use. Do not freeze. Do not shake.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Cytokine Release Syndrome (CRS)
Inform patients of the risk of CRS, and to immediately contact their healthcare provider for signs and symptoms associated with CRS (e.g., pyrexia, hypotension, hypoxia, chills, nausea, vomiting, fatigue, or headache) [see Warnings and Precautions (5.1)].
Skin Reactions
Inform patients that rashes and skin reactions have occurred in patients who have received KIMMTRAK. Advise patients to contact their healthcare provider for signs and symptoms of progressive or intolerable skin reactions [see Warnings and Precautions (5.2)].
Elevated Liver Enzymes
Inform patients that elevations in liver enzymes have occurred in patients who have received KIMMTRAK. Advise patients to contact their healthcare provider for signs and symptoms of liver toxicity (e.g., right sided abdominal pain, jaundice, scleral icterus) [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
- Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform females of the risk to a fetus [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception while on KIMMTRAK and for 1 week after the last dose [see Use in Specific Populations (8.1) and (8.3)].
Lactation
- Advise patients not to breastfeed during treatment with KIMMTRAK and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
Immunocore Limited
92 Park Drive, Milton Park
Abingdon, Oxfordshire
United Kingdom, OX144RY
License no: 2239
At:
Baxter Oncology GmbH
Kantstraβe 2
33790 Halle/Westfalen
Germany
For:
Immunocore Commercial LLC
181 Washington Street,
Conshohocken, PA, US
| This Patient Information has been approved by the U.S. Food and Drug Administration Issued: January 2022 | |
|
PATIENT INFORMATION
KIMMTRAK®(KIM-track)
|
|
|
What is the most important information I should know about KIMMTRAK? KIMMTRAK can cause serious side effects that can be severe or, life threatening, and usually happens within the first three infusions. These side effects include:
Your healthcare provider will check for these problems during treatment with KIMMTRAK. Your healthcare provider may temporarily stop or completely stop your treatment with KIMMTRAK, if you have severe side effects. See “What are the possible side effects of KIMMTRAK?” for more information about side effects. |
|
|
What is KIMMTRAK? KIMMTRAK is a prescription medicine used to treat HLA-A*02:01-positive adults with uveal melanoma that cannot be removed by surgery or has spread. Your healthcare provider will test you for a presence of HLA-A*02:01 gene to make sure KIMMTRAK is right for you. It is not known if KIMMTRAK is safe and effective in children. |
|
|
Before you receive KIMMTRAK, tell your healthcare provider about all of your medical conditions, including if you:
For females who are able to become pregnant:
Tell your healthcare provider about all medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
|
|
How will I receive KIMMTRAK?
|
|
|
What are the possible side effects of KIMMTRAK? KIMMTRAK can cause serious side effects, including:
The most common side effects of KIMMTRAK include:
These are not all the possible side effects of KIMMTRAK. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
General information about safe and effective use of KIMMTRAK. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. If you would like more information about KIMMTRAK, talk with your healthcare provider. You can ask your healthcare provider for more information about KIMMTRAK that is written for healthcare professionals. |
|
|
What are the ingredients in KIMMTRAK? Active ingredient: tebentafusp Inactive ingredients: citric acid monohydrate, di-sodium hydrogen phosphate, mannitol, polysorbate 20, trehalose, and Water for injection.
Manufactured by:
at: Baxter Oncology GmbH, Kantstraβe 2, 33790 Halle/Westfalen Germany. For: Immunocore Commercial LLC 181 Washington Street Conshohocken, PA, US KIMMTRAK is a trademark of the Immunocore Limited For more information, go to www.KIMMTRAK.com or call 1-844-IMMUNO1 (1-844-466-8661). |
|

| KIMMTRAK
tebentafusp injection, solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Immunocore Commercial LLC (117793852) |
| Registrant - Immunocore Limited (211024107) |
