Drug Detail:Kynamro (Mipomersen [ mye-poe-mer-sen ])
Drug Class: Miscellaneous antihyperlipidemic agents
Highlights of Prescribing Information
KYNAMRO (mipomersen sodium) Injection
Solution for Subcutaneous Injection
Initial U.S. Approval: 2013
WARNING: RISK OF HEPATOTOXICITY
See full prescribing information for complete boxed warning.
KYNAMRO can cause elevations in transaminases (5.1).
- •
- Measure alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and total bilirubin before initiating treatment and then ALT and AST regularly as recommended (2.3, 5.1).
- •
- During treatment, withhold the dose of KYNAMRO if the ALT or AST is ≥ 3 times the upper limit of normal (ULN) (2.3, 5.1).
- •
- Discontinue KYNAMRO for clinically significant liver toxicity (2.3, 5.1).
KYNAMRO increases hepatic fat (hepatic steatosis) with or without concomitant increases in transaminases (5.1).
- •
- Hepatic steatosis associated with KYNAMRO may be a risk factor for progressive liver disease, including steatohepatitis and cirrhosis (5.1).
Because of the risk of hepatotoxicity, KYNAMRO is available only through a restricted program called the KYNAMRO REMS (5.2).
Prescribe KYNAMRO only to patients with a clinical or laboratory diagnosis consistent with HoFH. The safety and effectiveness of KYNAMRO have not been established in patients with hypercholesterolemia who do not have HoFH (1).
Recent Major Changes
Boxed Warning 05/2016
Indications and Usage (1) 05/2016
Dosage and Administration (2.1) 05/2016
Warnings and Precautions (5.1) 05/2016
Indications and Usage for Kynamro
KYNAMRO® is an oligonucleotide inhibitor of apolipoprotein B-100 synthesis indicated as an adjunct to lipid-lowering medications and diet to reduce low density lipoprotein-cholesterol (LDL-C), apolipoprotein B (apo B), total cholesterol (TC), and non-high density lipoprotein-cholesterol (non HDL-C) in patients with homozygous familial hypercholesterolemia (HoFH) (1).
Limitations of Use:
- •
- The safety and effectiveness of KYNAMRO have not been established in patients with hypercholesterolemia who do not have HoFH, including those with heterozygous familial hypercholesterolemia (HeFH) (1).
- •
- The effect of KYNAMRO on cardiovascular morbidity and mortality has not been determined (1).
- •
- The use of KYNAMRO as an adjunct to LDL apheresis is not recommended (1).
Kynamro Dosage and Administration
- •
- 200 mg once weekly as a subcutaneous injection (2.1)
- •
- Before treatment, measure ALT, AST, alkaline phosphatase, and total bilirubin (2.1)
Dosage Forms and Strengths
- •
- Single-use pre-filled syringe containing 1 mL of a 200 mg/mL solution (3)
Contraindications
- •
- Moderate or severe hepatic impairment, or active liver disease, including unexplained persistent elevations of serum transaminases (4)
- •
- Known sensitivity to product components (4)
Warnings and Precautions
- •
- Injection site reactions occur in 84% of patients and typically consist of one or more of the following: erythema, pain, tenderness, pruritus and local swelling (5.3)
- •
- Flu-like symptoms, which typically occur within 2 days after an injection, occur in 30% of patients and include one or more of the following: influenza-like illness, pyrexia, chills, myalgia, arthralgia, malaise or fatigue (5.4)
Adverse Reactions/Side Effects
The most commonly reported adverse reactions (incidence ≥ 10% and greater than placebo) are injection site reactions, flu-like symptoms, nausea, headache and elevations in serum transaminases, specifically ALT (5.4, 6)
To report SUSPECTED ADVERSE REACTIONS, contact Kastle Therapeutics at 1-877-279-2308 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Use In Specific Populations
- •
- Nursing mothers: Discontinue drug or nursing (8.3).
- •
- Pediatric Patients: Safety and effectiveness not established (8.4).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2016
Full Prescribing Information
WARNING: RISK OF HEPATOTOXICITY
KYNAMRO can cause elevations in transaminases. In the KYNAMRO clinical trial in patients with HoFH, 4 (12%) of the 34 patients treated with KYNAMRO compared with 0% of the 17 patients treated with placebo had at least one elevation in alanine aminotransferase (ALT) ≥ 3x upper limit of normal (ULN). There were no concomitant clinically meaningful elevations of total bilirubin, international normalized ratio (INR) or partial thromboplastin time (PTT) [see Warnings and Precautions (5.1)].
KYNAMRO also increases hepatic fat, with or without concomitant increases in transaminases. In the trials in patients with heterozygous familial hypercholesterolemia (HeFH) and hyperlipidemia, the median absolute increase in hepatic fat was 10% after 26 weeks of treatment, from 0% at baseline, measured by magnetic resonance imaging (MRI). Hepatic steatosis is a risk factor for advanced liver disease; including steatohepatitis and cirrhosis [see Warnings and Precautions (5.1)].
Measure ALT, AST, alkaline phosphatase, and total bilirubin before initiating treatment and then ALT, AST regularly as recommended. During treatment, withhold the dose of KYNAMRO if the ALT or AST are ≥ 3x ULN. Discontinue KYNAMRO for clinically significant liver toxicity [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
Because of the risk of hepatotoxicity, KYNAMRO is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the KYNAMRO REMS [see Warnings and Precautions (5.2)]. Prescribe KYNAMRO only to patients with a clinical or laboratory diagnosis consistent with HoFH. The safety and effectiveness of KYNAMRO have not been established in patients with hypercholesterolemia who do not have HoFH (1).
1. Indications and Usage for Kynamro
KYNAMRO® is indicated as an adjunct to lipid-lowering medications and diet to reduce low density lipoprotein-cholesterol (LDL-C), apolipoprotein B (apo B), total cholesterol (TC), and non-high density lipoprotein-cholesterol (non-HDL-C) in patients with homozygous familial hypercholesterolemia (HoFH).
Limitations of Use
- •
- The safety and effectiveness of KYNAMRO have not been established in patients with hypercholesterolemia who do not have HoFH, including those with heterozygous familial hypercholesterolemia (HeFH).
- •
- The effect of KYNAMRO on cardiovascular morbidity and mortality has not been determined.
- •
- The safety and effectiveness of KYNAMRO as an adjunct to LDL apheresis have not been established; therefore, the use of KYNAMRO as an adjunct to LDL apheresis is not recommended.
2. Kynamro Dosage and Administration
2.1 General Dosing Information
Before beginning treatment with KYNAMRO, measure transaminases (ALT, AST), alkaline phosphatase, and total bilirubin [see Warnings and Precautions (5.1)].
The recommended dose of KYNAMRO is 200 milligrams (mg) once weekly as a subcutaneous injection.
KYNAMRO is intended for subcutaneous use only. Do not administer intramuscularly or intravenously.
The injection should be given on the same day every week, but if a dose is missed, the injection should be given at least 3 days from the next weekly dose.
After initiation of KYNAMRO therapy lipid levels should be monitored at least every 3 months for the first year. Maximal reduction of LDL-C may be seen with KYNAMRO therapy after approximately 6 months (based on the time to steady state seen in clinical studies). Health care providers should assess the patient’s LDL-C level after 6 months to determine if the LDL-C reduction achieved with KYNAMRO is sufficiently robust to warrant the potential risk of liver toxicity. Monitor transaminases during treatment with KYNAMRO as described in Warnings and Precautions (5.1), and withhold the dose for patients who develop transaminase values >3x the upper limit of normal (ULN) during treatment with KYNAMRO [see Dosage and Administration (2.3)].
2.2 Administration
Each pre-filled syringe of KYNAMRO provides 200 mg of mipomersen sodium in a deliverable volume of 1 milliliter (mL) of solution and is intended for single-use only.
The KYNAMRO pre-filled syringe should be removed from 2-8°C (36-46°F) refrigerated storage and allowed to reach room temperature for at least 30 minutes prior to administration.
Parenteral drug products should be inspected visually prior to administration. If the solution is cloudy or contains visible particulate matter, the contents must not be injected and the product should be returned to the pharmacy.
The first injection administered by the patient or caregiver should be performed under the guidance and supervision of an appropriately qualified health care professional.
KYNAMRO should be injected into the abdomen, thigh region, or outer area of the upper arm. KYNAMRO should not be injected in areas of active skin disease or injury such as sunburns, skin rashes, inflammation, skin infections, active areas of psoriasis, etc. Areas of tattooed skin and scarring should also be avoided.
2.3 Adjustments for Patients Developing Transaminase Elevations
Table 1 summarizes recommendations for monitoring for patients who develop elevated transaminases during therapy with KYNAMRO [see Warnings and Precautions (5.1)].
|
|
|
ALT or AST |
Treatment and monitoring recommendations* |
|
≥ 3x and < 5x ULN |
|
|
≥ 5x ULN |
|
If transaminase elevations are accompanied by clinical symptoms of liver injury (e.g., nausea, vomiting, abdominal pain, fever, jaundice, lethargy, flu-like symptoms), increases in bilirubin ≥ 2x ULN, or active liver disease, discontinue treatment with KYNAMRO and investigate to identify the probable cause [see Warnings and Precautions (5.1)].
3. Dosage Forms and Strengths
- •
- Single-use pre-filled syringe containing 1 mL of a 200 mg/mL clear, colorless to slightly yellow solution.
4. Contraindications
KYNAMRO is contraindicated in the following conditions:
- •
- Moderate or severe hepatic impairment (Child-Pugh B or C) or active liver disease, including unexplained persistent elevations of serum transaminases [see Warnings and Precautions (5.1) and Use in Specific Populations (8.8)]
- •
- Patients with a known hypersensitivity to any component of this product [see Adverse Reactions (6.1)].
5. Warnings and Precautions
5.1 Risk of Hepatotoxicity
KYNAMRO can cause elevations in transaminases and hepatic steatosis, as described below. To what extent KYNAMRO-associated hepatic steatosis promotes the elevations in transaminases is unknown. There is concern that KYNAMRO could induce steatohepatitis, which can progress to cirrhosis over several years. The clinical studies supporting the safety and efficacy of KYNAMRO in HoFH would have been unlikely to detect this adverse outcome given their size and duration [see Clinical Studies (14)].
Elevation of Transaminases
KYNAMRO can cause increases in serum transaminases (alanine aminotransferase [ALT] and/or aspartate aminotransferase [AST]). In the clinical trial, 4 (12%) of the 34 subjects with HoFH treated with KYNAMRO compared to 0% of the 17 subjects treated with placebo had an elevation in ALT ≥ 3x ULN, and 3 (9%) of those treated with KYNAMRO compared to 0% treated with placebo had at least one elevation in ALT ≥ 5x ULN.
Monitoring of Transaminases
Before initiating KYNAMRO and during treatment, monitor transaminases as recommended in Table 2.
|
TIME |
RECOMMENDATIONS |
|
Before initiating treatment |
|
|
During the first year |
|
|
After the first year |
|
|
At any time during treatment |
|
Hepatic Steatosis
KYNAMRO increases hepatic fat (steatosis) with or without concomitant increases in transaminases [see Adverse Reactions (6.1)]. Hepatic steatosis is a risk factor for advanced liver disease, including steatohepatitis and cirrhosis. The long-term consequences of hepatic steatosis associated with KYNAMRO therapy are unknown. During the clinical trials in patients with heterozygous familial hypercholesterolemia (HeFH) and hyperlipidemia, the median absolute increase in hepatic fat was 10% after 26 weeks of treatment, from 0% at baseline, measured by magnetic resonance imaging (MRI).
Alcohol may increase levels of hepatic fat and induce or exacerbate liver injury. It is recommended that patients taking KYNAMRO should consume no more than one alcoholic drink per day.
Caution should be exercised when KYNAMRO is used with other medications known to have potential for hepatotoxicity, for example isotretinoin, amiodarone, acetaminophen (>4 g/day for ≥ 3 days/week), methotrexate, tetracyclines, and tamoxifen. The effect of concomitant administration of KYNAMRO with other hepatotoxic medications is unknown. More frequent monitoring of liver-related tests may be warranted.
Mipomersen has not been studied concomitantly with other LDL-lowering agents that can also increase hepatic fat. Therefore, the combined use of such agents is not recommended.
5.2 KYNAMRO REMS
Because of the risk of hepatotoxicity, KYNAMRO is available only through a limited program under the REMS. Under the KYNAMRO REMS, only certified healthcare providers and pharmacies may prescribe and distribute KYNAMRO. Further information is available at www.KynamroREMS.com or by telephone at 1-877-KYNAMRO (1-877-596-2676).
5.3 Injection Site Reactions
Injection site reactions have been reported in 84% of patients receiving KYNAMRO therapy. These local reactions typically consist of one or more of the following: erythema, pain, tenderness, pruritus and local swelling. Injection site reactions do not occur with all injections but resulted in discontinuation of therapy in 5% of patients in pooled Phase 3 trials. [see Adverse Reactions (6.1)] To minimize the potential for injection site reactions, proper technique for subcutaneous administration should be followed. [see Patient Counseling Information (17)]
5.4 Flu-Like Symptoms
Flu-like symptoms have been reported in 30% of patients receiving KYNAMRO therapy and include one or more of the following: influenza-like illness, pyrexia, chills, myalgia, arthralgia, malaise or fatigue. Flu-like symptoms, which typically occur within 2 days after an injection, do not occur with all injections but resulted in discontinuation of therapy in 3% of patients in pooled Phase 3 trials. [see Adverse Reactions (6.1)]
6. Adverse Reactions/Side Effects
The following important adverse reactions have been observed and are discussed in detail in other sections of the label:
- •
- Risk of hepatotoxicity [see Warnings and Precautions (5.1)]
6.1 Clinical Trials
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trial of another drug and may not reflect the rates observed in patients in clinical practice.
Safety data are based on pooled results from four Phase 3, randomized, double-blind, placebo-controlled trials with a total of 390 patients of which 261 patients received weekly subcutaneous injections of 200 mg of KYNAMRO and 129 patients received placebo for a median treatment duration of 25 weeks (age range 12-81 years, 47% women, 84% Caucasian, 10% Blacks, 3% Asian, 3% other). For the 141 participants who subsequently were treated in the open-label extension trial, the mean length of study treatment, including exposure to KYNAMRO in the index study, was 19.8 months and the median was 18.2 months. A total of 41 individuals with HoFH were exposed to KYNAMRO for at least 6 months and 25 were exposed for at least 12 months.
Eighteen percent of patients on KYNAMRO and 2% of patients on placebo discontinued treatment due to adverse reactions. The five most common adverse reactions in patients treated with KYNAMRO that led to treatment discontinuation and occurred at a rate greater than placebo were: injection site reactions (5.0%), alanine aminotransferase increased (3.4%), flu-like symptoms (2.7%), aspartate aminotransferase increased (2.3%), and liver function test abnormal (1.5%).
Common Adverse Reactions
enumerates adverse reactions that occurred among pooled Phase 3 patients treated with KYNAMRO at an incidence that was at least 2% more than that observed in the placebo-treated patients, listed by system organ class and frequency (MedDRA v.13.0). Similar types and severities of adverse reactions were observed across all populations in this pooled table including the subset of patients with HoFH.
|
||
|
System Organ Class Preferred Term |
Treatment Group |
|
|
KYNAMRO (%) (N=261) |
Placebo (%) (N=129) |
|
|
Total Patients with Events |
95% |
85% |
|
Cardiac disorders |
9% |
6% |
|
Angina pectoris |
4% |
2% |
|
Palpitations |
3% |
0% |
|
Gastrointestinal disorders |
30% |
29% |
|
Nausea |
14% |
8% |
|
Vomiting |
4% |
2% |
|
Abdominal pain |
3% |
1% |
|
General disorders and administration site conditions |
87% |
47% |
|
Injection site reactions* |
84% |
33% |
|
Fatigue |
15% |
8% |
|
Influenza like illness |
13% |
3% |
|
Pyrexia |
8% |
3% |
|
Chills |
6% |
1% |
|
Edema peripheral |
5% |
2% |
|
Hepatobiliary disorders |
9% |
5% |
|
Hepatic steatosis |
7% |
2% |
|
Investigations |
30% |
15% |
|
Alanine aminotransferase increased |
10% |
1% |
|
Aspartate aminotransferase increased |
6% |
2% |
|
Liver function test abnormal |
5% |
1% |
|
Hepatic enzyme increased |
3% |
1% |
|
Musculoskeletal and connective tissue disorders |
26% |
26% |
|
Pain in extremity |
7% |
3% |
|
Musculoskeletal pain |
4% |
2% |
|
Nervous system disorders |
25% |
17% |
|
Headache |
12% |
9% |
|
Psychiatric disorders |
10% |
3% |
|
Insomnia |
3% |
1% |
|
Vascular disorders |
11% |
5% |
|
Hypertension |
7% |
3% |
In the pooled Phase 3 trials, neoplasms (benign and malignant) were reported in 4% of patients receiving KYNAMRO and 0% of patients receiving placebo. In addition, 9% of patients receiving KYNAMRO and 3% of patients receiving placebo developed 1+ or greater proteinuria by dipstick measurement by the end of the trial.
In the open-label extension trial, one case of hypersensitivity reaction with angioedema and one case of glomerular nephritis were reported.
Platelets
In the phase 3 trial in patients with HoFH, the mean change in platelet count from baseline to Week 28/Early Termination was -30.6 × 103/µL in the mipomersen group and +8.1 × 103/µL in the placebo group. In the pooled Phase 3 trials the mean change in platelet count from baseline to Week 28/Early Termination was -23.8 × 103/µL in the mipomersen group and -3.5 × 103/µL in the placebo group.
Transaminase Elevations
In the pooled, placebo-controlled clinical trials with KYNAMRO, elevated serum transaminase levels, mainly ALT, have been observed as presented in Table 4. Elevated ALT levels ≥ 3x ULN have been reported on two consecutive occasions at least 7 days apart in 8.4% of patients receiving KYNAMRO therapy (versus 0% of placebo patients) with 16.5% of patients receiving KYNAMRO therapy having at least 1 result that was ≥ 3X ULN (versus 0.8% for placebo patients). The ALT elevations observed in the pooled, placebo-controlled trials were generally accompanied by lesser AST elevations and were not associated with increased total bilirubin, changes in INR or PTT, nor by decreased albumin levels. After stopping therapy, in the patients in whom an elevation was observed, transaminase elevations trended toward baseline over a period of weeks to months.
|
Parameter |
Statistic |
Kynamro
|
Placebo
|
|
ALT maximum |
Incidence rate, % |
||
|
≥ 3 × ULN and < 5 × ULN |
12% |
1% |
|
|
≥ 5 × ULN and < 10 × ULN |
3% |
0% |
|
|
≥ 10 x ULN |
1% |
0% |
|
|
ALT |
≥ 3 × ULN, two consecutive results (at least 7 days apart), % |
8% |
0% |
|
AST maximum |
Incidence rate, % |
||
|
≥ 3 × ULN and < 5 × ULN |
7% |
1% |
|
|
≥ 5 × ULN and < 10 × ULN |
3% |
0% |
|
|
≥ 10 x ULN |
0% |
0% |
|
|
AST |
≥ 3 × ULN, two consecutive results (at least 7 days apart), % |
4% |
0% |
Adults: ALT ULN= 41 U/L; AST ULN = 34 U/L
Hepatic Steatosis
Increases in liver fat as measured by MRI were greater in patients receiving KYNAMRO therapy than in patients receiving placebo. Data from Phase 3 supportive trials in patients with heterozygous familial hypercholesterolemia and coronary artery disease and in patients with high risk hypercholesterolemia demonstrated after 26 weeks of treatment, a median nominal increase in fat fraction of 9.6% relative to baseline following KYNAMRO therapy versus a nominal 0.02% change in the placebo group (mean increases were 12.2% mipomersen vs 0.4% placebo). The maximum change in fat fraction was 46% for the KYNAMRO group and 28% for the placebo group. Sixty-two percent of patients receiving KYNAMRO developed a 5% or greater increase in hepatic fat versus 8% of patients receiving placebo. In general, these elevations in fat fraction decreased when assessed by MRI performed 24 weeks after cessation of KYNAMRO in the Phase 3 trial of patients with high-risk hypercholesterolemia. In the open-label extension trial, among individuals with a measurement at baseline and at 12 months or longer on KYNAMRO, 25% had an average liver fat fraction > 20% on at least one occasion.
Injection Site Reactions
The most commonly-reported adverse reactions were injection site reactions occurring in 84% of patients receiving KYNAMRO versus 33% of placebo treated patients. The most common injection site reactions were erythema (59%), pain (56%), hematoma (32%), pruritus (29%), swelling (18%) and discoloration (17%). Injection site reactions did not occur with every injection. Injection site reactions resulted in discontinuation of KYNAMRO in 5% of patients. Recall reactions, consisting of local erythema, tenderness and/or pruritus at previous injection sites when subsequent injections were administered, were observed in 8% of patients, all of whom were receiving KYNAMRO.
Flu-like Symptoms
Flu-like symptoms, defined as any one of the following: influenza-like illness, pyrexia, chills, myalgia, arthralgia, malaise or fatigue and occurring within 2 days of injection, have been reported more frequently in patients receiving KYNAMRO (29.9%) versus placebo (16.3%) in the pooled Phase 3 studies. Flu-like symptoms did not occur with all injections. Flu-like symptoms resulted in discontinuation of KYNAMRO in 2.7% of patients. In the open-label extension trial, in which all patients received KYNAMRO therapy, 66% reported flu-like symptoms, 25% discontinued treatment due to flu-like symptoms and 9% experienced severe flu-like symptoms.
Immunogenicity
In the pooled Phase 3 trials, 38% of KYNAMRO-treated patients tested positive for anti-KYNAMRO antibodies during the 6-month trials. Efficacy results in the Phase 3 trials in patients who tested positive for anti-KYNAMRO antibodies were similar to patients who remained negative for antibodies (mean LDL-C percent change from baseline was -32% for antibody-positive and -34% for antibody-negative participants). In the open-label extension trial, approximately 72% of patients receiving KYNAMRO therapy tested positive for anti-KYNAMRO antibodies (35% with titers >3200). The incidence of flu-like symptoms and the incidence of discontinuation of KYNAMRO were higher in antibody-positive patients. Antibodies to KYNAMRO were associated with higher trough levels for the drug. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to KYNAMRO with the incidence of antibodies to other products may be misleading.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of KYNAMRO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- •
- Idiopathic thrombocytopenic purpura
- •
- Allergic Reactions: Hypersensitivity reactions (e.g., urticaria, rash, angioedema)
7. Drug Interactions
No clinically relevant pharmacokinetic interactions were reported between KYNAMRO and warfarin, or between KYNAMRO and simvastatin or ezetimibe [see Clinical Pharmacology (12.3)]. Additionally, coadministration of KYNAMRO with warfarin did not result in a pharmacodynamic interaction as determined by INR, aPTT and PT.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Category B
There are no adequate and well-controlled studies in pregnant women. Reproduction and embryofetal development studies performed in mice at doses up to 87.5 mg/kg/wk given by subcutaneous administration from mating through organogenesis and in pregnant rabbits given 52.5 mg/kg/wk, show no evidence of impaired fertility or harm to the fetus at 2 (mice) to 5 (rabbits) times clinical exposure at a 200 mg/wk therapeutic dose.
Because animal reproduction studies are not always predictive of the human response, this drug should be used during pregnancy only if clearly needed.
Pregnant rats given subcutaneous doses of 7, 35, 70 mg/kg/wk mipomersen sodium from gestation day 6 through weaning on lactation day 20, resulted in decreased rat pup survival at 70 mg/kg/wk, 3-times clinical exposure at a 200 mg/wk therapeutic dose based on body surface area comparisons across species. Dose related decreases in pup body weights, impaired reflexes and grip strength were observed at 35 mg/kg/wk (2-times the anticipated human dose. Levels of mipomersen in rat milk were very low (≤0.92 µg/mL at subcutaneous doses up to 70 mg/kg/wk). Due to the poor oral bioavailability of mipomersen sodium, it was considered unlikely that these low milk exposure levels adversely affected the pups during lactation.
8.3 Nursing Mothers
It is not known whether KYNAMRO is excreted in human milk. Because many drugs are excreted in human milk a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Levels of mipomersen present in rat milk were low (≤ 0.92 µg/mL) given subcutaneous doses up to 70 mg/kg/wk. Oral bioavailability is expected to be less than 10%. However a risk to newborns/infants cannot be excluded, therefore caution should be used when KYNAMRO is administered to a nursing woman.
Lactating rats administered mipomersen sodium at doses up to 70 mg/kg/wk (3-times the anticipated systemic exposure from a 200 mg/wk dose, based on body surface area comparison) consumed less food while nursing. This correlated with reduced weight gain in the rat pups, and decreased pup survival in litters of dams given 70 mg/kg/wk.
8.4 Pediatric Use
Safety and effectiveness have not been established in pediatric patients.
A juvenile toxicity study was conducted in rats at doses up to 50 mg/kg/wk (2-times the systemic exposure from a 200 mg/wk clinical dose based on body surface area comparisons). Doses > 10 mg/kg/wk were associated with reduced body weight gain in young rats, but had no effect on long bone growth or sexual development.
8.5 Geriatric Use
Clinical studies of KYNAMRO did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Of the 51 patients enrolled in the Phase 3 trial in HoFH, the mean age was 31 years and the oldest patient in the trial was 53 years. Of the 261 patients who received KYNAMRO in the pooled Phase 3 trials, 59 (22.6%) were ≥ 65 years old and 10 (3.8%) were ≥ 75 years old. In the pooled Phase 3 trials, patients ≥ 65 years of age treated with KYNAMRO had a higher incidence of hypertension and peripheral edema compared to placebo patients in this age group, as well as compared to the younger KYNAMRO-treated age group. Hepatic steatosis was also reported with greater frequency in the ≥ 65 group (13.6%) compared to the <65 group (10.4%).
8.6 Females of Reproductive Potential
KYNAMRO may cause fetal harm [see Use in Specific Populations (8.1)]. Females who become pregnant during KYNAMRO therapy should notify their healthcare provider.
Contraception
Females of reproductive potential should use effective contraception during KYNAMRO therapy.
8.7 Renal Impairment
The safety and efficacy of KYNAMRO treatment in patients with known renal impairment or in patients undergoing renal dialysis have not been established. Due to the lack of clinical data and KYNAMRO’s renal safety profile, KYNAMRO is not recommended in patients with severe renal impairment, clinically significant proteinuria, or on renal dialysis.
8.8 Hepatic Impairment
The safety and efficacy of KYNAMRO treatment in patients with known hepatic impairment have not been established. KYNAMRO is contraindicated in patients with clinically significant hepatic dysfunction, which may include persistent elevations of transaminases. [seeContraindications (4) and Warnings and Precautions (5.1)]
10. Overdosage
There have been no reports of overdose with KYNAMRO treatment. In clinical trials, patients receiving higher doses of KYNAMRO (300 mg and 400 mg once weekly for 13 weeks) experienced adverse reactions similar to the adverse reactions experienced by patients receiving treatment with 200 mg once weekly but at slightly higher rates and greater severity. Liver-related tests should be monitored. Although there is no information on the effect of hemodialysis in treating an overdose with mipomersen, hemodialysis is unlikely to be useful in overdose management since mipomersen is highly bound to plasma proteins.
11. Kynamro Description
KYNAMRO (mipomersen sodium) Injection is a sterile, preservative-free, clear, colorless to slightly yellow, aqueous solution for subcutaneous injection. KYNAMRO is supplied in single-use, 1 mL, clear glass pre-filled syringes filled to deliver 1 mL of solution containing 200 mg of mipomersen sodium (200 mg per 1 mL). KYNAMRO is formulated in water for injection and may include hydrochloric acid and/or sodium hydroxide for pH adjustment to 7.5 – 8.5.
Mipomersen sodium is an oligonucleotide inhibitor of apo B-100 synthesis. ApoB is the principal apolipoprotein of LDL and its metabolic precursor, very low density lipoprotein (VLDL). Mipomersen inhibits synthesis of apoB by sequence-specific binding to its messenger ribonucleic acid (mRNA) resulting in degradation of the mRNA through enzyme-mediated pathways or disruption of mRNA function through binding alone.
Mipomersen sodium is a synthetic phosphorothioate oligonucleotide sodium salt, 20 nucleotides in length, with the following sequence:
5'-GMeCMeCMeUMeCAGTMeCTGMeCTTMeC GMeCAMeCMeC- 3'
where the underlined residues are 2'-O-(2-methoxyethyl) nucleosides; all other residues are 2'-deoxynucleosides. Substitution at the 5-position of the cytosine (C) and uracil (U) bases with a methyl group is indicated by Me.
Mipomersen sodium is represented by the following structural formula:
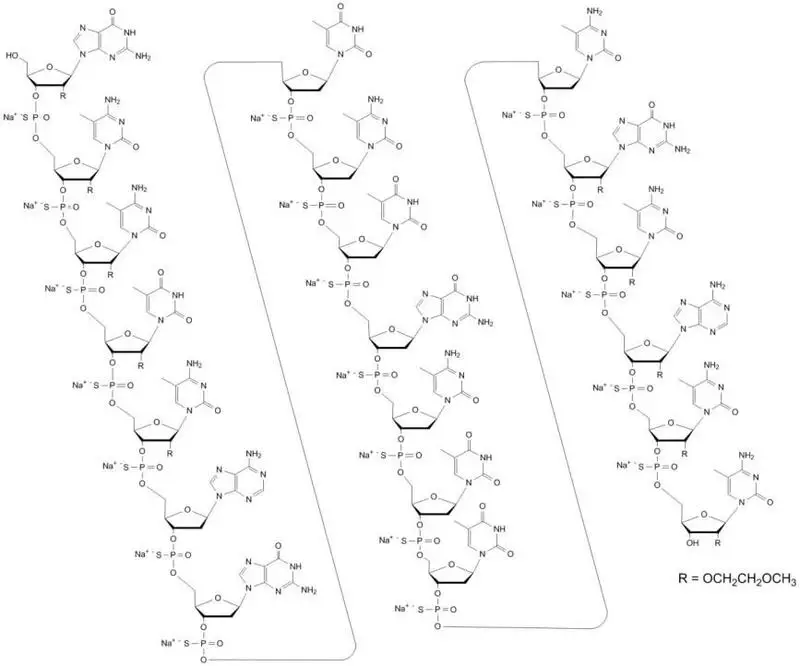
The molecular formula of mipomersen sodium is C‑230‑H305N67O122P19S19Na19 and the molecular weight is 7594.9 g/mol.
12. Kynamro - Clinical Pharmacology
12.1 Mechanism of Action
Mipomersen is an antisense oligonucleotide targeted to human messenger ribonucleic acid (mRNA) for apo B-100, the principal apolipoprotein of LDL and its metabolic precursor, VLDL. Mipomersen is complementary to the coding region of the mRNA for apo B-100, and binds by Watson and Crick base pairing. The hybridization of mipomersen to the cognate mRNA results in RNase H-mediated degradation of the cognate mRNA thus inhibiting translation of the apo B-100 protein.
The in vitro pharmacologic activity of mipomersen was characterized in human hepatoma cell lines (HepG2, Hep3B) and in human and cynomolgus monkey primary hepatocytes. In these experiments, mipomersen selectively reduced apo B mRNA, protein and secreted protein in a concentration- and time-dependent manner. The effects of mipomersen were shown to be highly sequence-specific. The binding site for mipomersen lies within the coding region of the apo B mRNA at the position 3249-3268 relative to the published sequence GenBank accession number NM_000384.1.
12.2 Pharmacodynamics
Cardiac ECG Effects
At a concentration of 3.8 times the Cmax of the maximum recommended dose (200 mg subcutaneous injection), mipomersen does not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
Single- and multiple-dose pharmacokinetics of mipomersen in healthy volunteers and in patients with FH and non-FH has shown that mipomersen plasma exposure increases with increasing dose in the range of 30 mg to 400 mg.
Absorption
Following subcutaneous injection, peak concentrations of mipomersen are typically reached in 3 to 4 hours. The estimated plasma bioavailability of mipomersen following subcutaneous administration over a dose range of 50 mg to 400 mg, relative to intravenous administration, ranged from 54% to 78%.
Distribution
Mipomersen is highly bound to human plasma proteins (≥ 90%) at clinically relevant concentrations (1-8 µg/mL). Mipomersen has a distribution plasma half-life of approximately 2 to 5 hours.
With once weekly dosing, plasma trough levels increase over time and approach steady-state, typically within 6 months.
Metabolism
Mipomersen is not a substrate for CYP450 metabolism, and is metabolized in tissues by endonucleases to form shorter oligonucleotides that are then substrates for additional metabolism by exonucleases.
Excretion
The elimination of mipomersen involves both metabolism in tissues and excretion, primarily in urine. Both mipomersen and putative shorter oligonucleotide metabolites were identified in human urine. Urinary recovery was limited in humans with less than 4% within the 24 hours post dose. Following subcutaneous administration, elimination half-life for mipomersen is approximately 1 to 2 months.
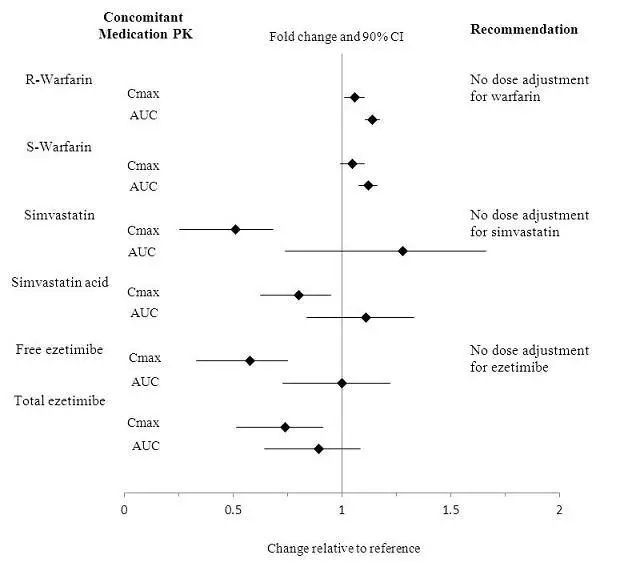
Drug Interactions
No clinically relevant pharmacokinetic interactions were reported between mipomersen and warfarin, or between mipomersen and simvastatin or ezetimibe. The results of these studies are summarized in Figures 1 and 2.
Figure 1: Impact of Other Drugs on Mipomersen Pharmacokinetics

Figure 2: Impact of Mipomersen on the Pharmacokinetics of Other Drugs
Specific Populations
Renal Impairment
Pharmacokinetics of KYNAMRO in patients with renal impairment has not been established [see Use in Specific Populations (8.7)].
Hepatic Impairment
Pharmacokinetics of KYNAMRO in patients with hepatic impairment has not been established [see Use in Specific Populations (8.8)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a subcutaneous carcinogenicity study in mice, mipomersen sodium was administered for up to 104 weeks at doses of 5, 20, 60 mg/kg/week. There were statistically significant increases in the incidences of hepatocellular adenoma and combined adenoma and carcinoma in female mice at 60 mg/kg/wk (2-times the systemic clinical exposure at 200 mg/wk, based on a body surface area comparison) for both mipomersen sodium and the mouse-specific analog. This dose also resulted in statistically significant increases in the incidence of hemangiosarcomas in female mice and fibrosarcomas of the skin/subcutis in male mice.
In a subcutaneous carcinogenicity study in rats, mipomersen sodium was administered for up to 104 weeks at doses of 3, 10, 20 mg/kg/wk. The incidence of fibrosarcomas of the skin/subcutis and the combination of fibroma, fibrosarcomas and malignant fibrous histiocytoma of the skin/subcutis was statistically significantly increased in female rats at 10 mg/kg/wk, at less than clinical exposure at the 200 mg/wk dose based on body surface area comparisons. Both sexes of rats also had statistically significant increases in the incidence of malignant fibrous histiocytoma of the skin/subcutis at 20 mg/kg/wk (at clinical exposure at the 200 mg/wk dose based on body surface area comparisons.
Mipomersen did not exhibit genotoxic potential in a battery of studies, including the in vitro Bacterial Reverse Mutation (Ames) assay, an in vitro cytogenetics assay using a mouse lymphoma cell line, and an in vivo micronucleus assay in mice.
Mipomersen sodium had no effect on fertility in mice at doses up to 87.5 mg/kg/wk (2-times clinical exposure at the 200 mg/wk dose based on body surface area comparisons).
13.2 Animal Pharmacology and/or Toxicology
The principal target organs for mipomersen pathology are the kidneys and liver. These organs represent the highest distribution of compound, and exhibit microscopic changes reflective of cellular uptake in macrophages. The most widespread toxicological effect of mipomersen was a spectrum of inflammatory changes in numerous organs, including lymphohistiocytic cell infiltrates and increases in lymphoid organ weights, associated with increases in plasma cytokines, chemokines and total serum IgG. In a chronic monkey study, multi-focal intimal hyperplasia with mixed inflammatory infiltrates was evident in vascular beds in 2 of 6 monkeys treated for 12 months with 30 mg/kg/week with a no-observed-adverse-effect-level (NOAEL) of 10 mg/kg/week (approximately equal to clinical exposures anticipated from a 200 mg/wk dose based on body surface area comparisons across species).
14. Clinical Studies
The safety and effectiveness of KYNAMRO, given as 200 mg weekly subcutaneous injections, as an adjunct to lipid-lowering medications in individuals with HoFH were evaluated in a multinational, randomized (34 KYNAMRO; 17 placebo), placebo-controlled, 26-week trial in 51 patients with HoFH. A diagnosis of functional HoFH was defined by the presence of at least one of the following clinical or laboratory criteria: (1) history of genetic testing confirming 2 mutated alleles at the LDLr gene locus, or (2) documented history of untreated LDL-C > 500 mg/dL and at least one of the criteria (a) tendinous and/or cutaneous xanthoma prior to age 10 years or (b) documentation of elevated LDL-C > 190 mg/dL prior to lipid-lowering therapy consistent with HeFH in both parents. In case a parent was not available, a history of coronary artery disease in a first degree male relative of the parent younger than 55 years or first degree female relative of the parent younger than 60 years was acceptable.
The baseline demographic characteristics were well-matched between the KYNAMRO and placebo patients. The mean age was 32 years (range, 12 to 53 years), the mean body mass index (BMI) was 26 kg/m2, 43% were men, and the majority (75%) were Caucasian. In 50 of 51 (98%) patients, the background therapy of maximally tolerated lipid-lowering medication included statins. In total, 44 of the 50 (88%) patients were on maximum-dose statin therapy with or without other lipid-lowering medications. Thirty-eight of the 50 (76%) patients were also taking at least one other lipid-lowering medication, most commonly ezetimibe in 37 of 50 (74%) patients; patients were not on LDL apheresis. Eighty-two percent of the KYNAMRO group and 100% of the placebo group completed the efficacy endpoint at week 28. Adverse events contributed to premature discontinuation for four patients, all in the KYNAMRO group [see Adverse Reactions (6)].
The primary efficacy endpoint was percent change in LDL-C from baseline to Week 28. At Week 28, the mean and median percent changes in LDL-C from baseline were -25% (p<0.001) and -19%, respectively, for the KYNAMRO group. The mean and median treatment difference from placebo was -21% (95% confidence interval [CI]: -33, -10) and -19%, respectively. Changes in lipids and lipoproteins through the efficacy endpoint at Week 28 are presented in Table 5.
|
|||
|
|
KYNAMRO n=34 |
Placebo n=17 | |
|
Mean Baseline LDL-C(mg/dL) (range) |
439 (190, 704) |
400 (172, 639) |
|
|
Parameter (mg/dL) |
Mean or Median Percent Change from Baseline to End of Treatment* |
Mean (95% CI) or Median Treatment Difference from Placebo (%) |
|
|
LDL-C† |
-25 |
-3 |
-21 (-33, -10) |
|
Apo B† |
-27 |
-3 |
-24 (-34, -15) |
|
TC† |
-21 |
-2 |
-19 (-29, -9) |
|
Non-HDL-C† |
-25 |
-3 |
-22 (-33, -11) |
|
TG‡ |
-18 |
1 |
-18 |
|
HDL-C§ ‡ |
15 |
4 |
11 |
LDL-C percent changes from baseline with KYNAMRO were variable among individuals with HoFH ranging from a 2% increase to an 82% reduction. The LDL-C percent changes from baseline in the placebo group range from a 43% increase to a 33% reduction. Mean LDL-C percent changes over time are presented in Figure 3.
Figure 3: Mean Percent Change in LDL-C in Patients with HoFH (Completers Population)
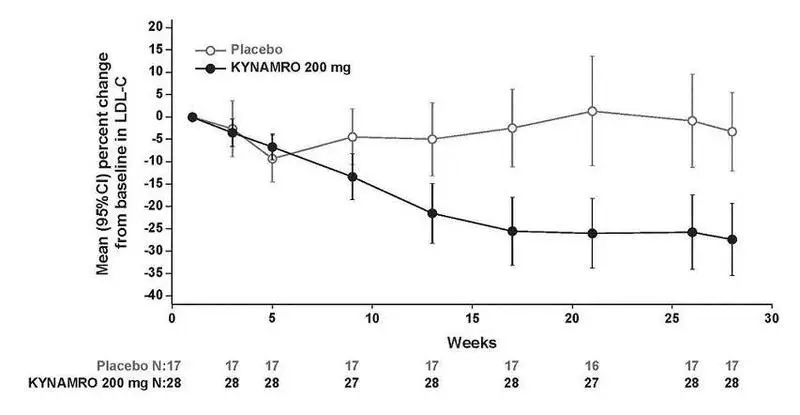
16. How is Kynamro supplied
KYNAMRO is supplied in single-use, 1 mL, clear pre-filled syringes with staked needles. Each single-use pre-filled syringe of KYNAMRO is filled to deliver 1 mL of 200 mg/mL solution containing 200 mg of mipomersen sodium.
KYNAMRO is available in cartons containing 1 or 4 pre-filled syringes.
Pack of 1 pre-filled syringe: NDC 70688-0502-1
Pack of 4 pre-filled syringe: NDC 70688-0502-2
Store refrigerated KYNAMRO at 2-8 °C (36-46 °F). KYNAMRO should be protected from light and kept in the original carton until time of use. When refrigeration is not available KYNAMRO may be stored at or below 30 °C (86 °F), away from heat sources, for up to 14 days. Do not use KYNAMRO after the expiration date on the label.
17. Patient Counseling Information
See FDA-approved labeling (Medication Guide)
Advise patients of the following:
Risk of hepatotoxicity [see Warnings and Precautions (5.1)]
- •
- KYNAMRO can cause elevations in transaminases and hepatic steatosis. Discuss with the patient the importance of monitoring liver-related laboratory tests before taking KYNAMRO and periodically thereafter.
- •
- Patients should be advised of the potential for increased risk of liver injury if alcohol is consumed while taking KYNAMRO. It is recommended that patients taking KYNAMRO should consume no more than one alcoholic drink per day.
- •
- Advise patients to promptly report symptoms of possible liver injury, such as nausea, vomiting, fever, anorexia, fatigue, jaundice, dark urine, pruritus, or abdominal pain.
KYNAMRO REMS [see Warnings and Precautions (5.2)]
- •
- KYNAMRO is only available through a restricted program called KYNAMRO REMS and therefore, KYNAMRO is only available from certified pharmacies that are enrolled in the program. Additional information may be obtained at 1-877-KYNAMRO (1-877-596-2676).
Injection Site Reactions [see Warnings and Precautions (5.3)]
- •
- Injection site reactions have been reported frequently in patients receiving KYNAMRO.
- •
- These local reactions typically consist of one or more of the following: erythema, pain, tenderness, pruritus and local swelling.
Flu-like symptoms [see Warnings and Precautions (5.4)]
- •
- Flu-like symptoms have been reported in patients receiving KYNAMRO.
- •
- Flu-like symptoms typically occur within 2 days after an injection and include one or more of the following: influenza-like illness, pyrexia, chills, myalgia, arthralgia, malaise or fatigue.
Dosing [see Dosage and Administration (2)]
- •
- The patient or caregiver should be instructed to review the KYNAMRO Medication Guide and Instructions for Use carefully.
- •
- KYNAMRO is administered as a subcutaneous injection given once a week.
- •
- Do not remove the needle cover from the pre-filled syringe while allowing the syringe to reach room temperature.
- •
- The patient or caregiver should be instructed by a physician or an appropriately qualified healthcare professional in the proper technique for administering subcutaneous injections, including the use of aseptic technique.
- •
- The patient and caregiver should be cautioned that needles or syringes must not be re-used and instructed in safe disposal procedures. A puncture-resistant container for disposal of used needles and syringes should be supplied to the patient along with instructions for safe disposal of the full container.
- •
- KYNAMRO should be injected into the abdomen, thigh region, or outer area of the upper arm. Patients and caregivers should be advised to alternate sites for subcutaneous injections. KYNAMRO should not be injected in areas of active skin disease or injury such as sunburns, skin rashes, inflammation, skin infections, active areas of psoriasis, or areas of tattooed skin and scarring.
- •
- Patients and caregivers should be advised to alternate sites for subcutaneous injection. The injection should be performed slowly and steadily and the needle should not be withdrawn until the injection is complete.
- •
- Protect from light. Do not mix or co-administer KYNAMRO with other products.
KYNAMRO is manufactured for:
Kastle Therapeutics
Chicago, IL
1-877-596-2676 (phone)
KYNAMRO is a registered trademark of Kastle Therapeutics.
|
Medication Guide |
|
KYNAMRO® (kye-NAM-roe) |
|
(mipomersen sodium) |
|
injection |
What is the most important information I should know about KYNAMRO?
- •
- KYNAMRO is available only through certified pharmacies that are enrolled in the KYNAMRO REMS Program. Your doctor must be enrolled in the program in order for you to be prescribed KYNAMRO.
KYNAMRO may cause serious side effects, including liver problems. KYNAMRO can cause liver problems such as increased liver enzymes or increased fat in the liver.
- •
- Your doctor should do blood tests to check your liver before you start KYNAMRO and during your treatment. If your tests show some liver problems, your doctor may stop KYNAMRO.
- •
- Tell your doctor if you have had liver problems, including liver problems while taking other medicines.
- •
- Tell your doctor right away if you have any of these symptoms of liver problems while taking KYNAMRO:
|
|
|
|
|
|
|
|
|
- •
- Drinking alcohol may increase your chance of having liver problems or make your liver problems worse. You should not have more than 1 alcoholic drink each day while using KYNAMRO.
What is KYNAMRO?
KYNAMRO is a prescription medicine used along with diet and other lipid-lowering treatments in people with homozygous familial hypercholesterolemia (HoFH) to reduce:
- •
- LDL (“bad”) cholesterol
- •
- total cholesterol
- •
- a protein that carries “bad” cholesterol in the blood (apolipoprotein B)
- •
- non-high-density lipoprotein cholesterol (non-HDL-C)
It is not known if KYNAMRO can decrease problems from high cholesterol, such as heart attack, stroke, death or other health problems.
It is not known if KYNAMRO is safe and effective in people with high cholesterol but who do not have HoFH, including those with heterozygous familial hypercholesterolemia (HeFH).
It is not known if KYNAMRO is safe and effective as an additional treatment to LDL-apheresis.
It is not known if KYNAMRO is safe and effective in people with kidney and liver problems, including people who are on kidney dialysis.
It is not known if KYNAMRO is safe and effective when used in children under the age of 18.
Who should not take KYNAMRO?
Do not take KYNAMRO if you:
- •
- have moderate or severe liver problems or active liver disease, including people who have unexplained abnormal liver tests.
- •
- are allergic to mipomersen or any of the ingredients in KYNAMRO. See the end of this leaflet for a complete list of ingredients in KYNAMRO.
What should I tell my doctor before taking KYNAMRO?
Before you take KYNAMRO, tell your doctor if you:
- •
- have liver problems
- •
- have kidney problems
- •
- drink alcohol
- •
- are pregnant or plan to become pregnant. KYNAMRO may cause harm to your unborn baby. If you are a female who can get pregnant, you should use effective birth control while using KYNAMRO. Talk with your doctor to find the best method of birth control for you. If you become pregnant while taking KYNAMRO, stop taking KYNAMRO and call your doctor right away.
- •
- are breastfeeding or plan to breastfeed. It is not known if KYNAMRO passes into your breast milk. You and your doctor should decide if you will use KYNAMRO or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Before starting a new medicine while taking KYNAMRO, even if you will only be taking it for a short time, ask your doctor or pharmacist if it is safe to take while you are using KYNAMRO.
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
How should I take KYNAMRO?
- •
- See the Instructions for Use that comes with this Medication Guide for complete information on how to use KYNAMRO.
- •
- KYNAMRO is given by injection under your skin (subcutaneous) 1 time each week. KYNAMRO is available in single-use (1 time) pre-filled syringe.
- •
- Take KYNAMRO exactly as your doctor tells you to take it.
- •
- Make sure that you are or your caregiver is trained by your doctor or other healthcare provider on how to inject KYNAMRO the right way.
- •
- Do not try to give yourself or have another person give you injections at home until you or both of you understand and are comfortable with how to prepare for your dose and give the injection.
- •
- Take KYNAMRO on the same day of the week at the same time of day.
- •
- If you miss a dose or forget to take your dose of KYNAMRO at your usual weekly time, you can take it when you remember, unless it is less than 3 days until your next weekly dose. If it is less than 3 days until your next weekly dose, wait and take your next weekly dose at your regularly scheduled time. Do not take a double dose at the same time to make up for a forgotten or missed dose.
- •
- It is important that KYNAMRO is at room temperature when it is injected.
- •
- Do not mix KYNAMRO with other injectable medicines.
- •
- Do not use KYNAMRO at the same time as other injectable medicines.
- •
- If you use too much KYNAMRO, call your doctor right away.
- •
- Do not stop taking KYNAMRO without talking to your doctor.
What are the possible side effects of KYNAMRO?
KYNAMRO can cause serious side effects, including:
- •
- See “What is the most important information I should know about KYNAMRO?”
- •
- injection site problems. Skin reactions can happen in some people including redness or discoloration of the skin, pain, tenderness, itching, and swelling around the injection site. You may also get a reaction at a former site of injection, when injecting at a different site, or after an injury to an injection site.
- •
- flu-like symptoms, including fever, chills, aches, and tiredness. These symptoms usually happen within 2 days of an injection.
Call your doctor right away if you have any of the serious side effects of KYNAMRO.
The most common side effects of KYNAMRO include:
|
|
|
|
Tell your doctor about any side effect that bothers you or that does not go away.
These are not all the possible side effects of KYNAMRO. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store KYNAMRO?
- •
- Store KYNAMRO in a refrigerator between 36°F to 46°F (2°C to 8°C). If a refrigerator is not available, KYNAMRO may be stored at room temperature between 68°F to 77°F (20°C to 25°C) for up to 14 days if it is kept away from heat.
- •
- Protect KYNAMRO from light and store in the original carton.
- •
- Safely throw away medicine that is out of date or no longer needed.
Keep KYNAMRO and all medicines out of the reach of children.
General information about the safe and effective use of KYNAMRO
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use KYNAMRO for a condition for which it was not prescribed. Do not give KYNAMRO to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about KYNAMRO. You can ask your pharmacist or doctor for information about KYNAMRO that is written for healthcare professionals.
For more information, go to www.KYNAMRO.com or call 1-877-KYNAMRO (1-877-596-2676).
What are the ingredients in KYNAMRO?
Active ingredient: mipomersen sodium
Inactive ingredients: sterile water, hydrochloric acid, and sodium hydroxide
This Medication Guide has been approved by the U.S. Food and Drug Administration.
KYNAMRO is manufactured for:
Kastle Therapeutics
Chicago, IL
KYNAMRO is a registered trademark of Kastle Therapeutics.
Issued: June 2016
|
INSTRUCTIONS FOR USE |
|
KYNAMRO® (kye-NAM-roe) |
|
(mipomersen sodium) injection |
|
Solution for Subcutaneous Injection |
|
Single-use Pre-filled Syringe |
Read the Instructions for Use that come with your KYNAMRO before you start using it and each time you get a refill. There may be new information. This leaflet does not take the place of talking to your doctor about your medical condition or your treatment. Before you use KYNAMRO for the first time, make sure your doctor shows you the right way to use it.
This Instructions for Use is only to be used for KYNAMRO in pre-filled syringes.
Do not use the KYNAMRO pre-filled syringe if:
- •
- the expiration date on the barrel of the container has passed.
- •
- the packaging or seals are torn or broken, or the syringe looks cracked or damaged when you receive your KYNAMRO.
- •
- the medicine in the pre-filled syringe is discolored (it should be colorless to slightly yellow), or if it is cloudy or has any visible particles in it (it should be completely clear).
Supplies needed for your KYNAMRO injection:
- •
- KYNAMRO pre-filled syringe
- •
- NOTE: It is important that KYNAMRO be at room temperature prior to the injection. Allow KYNAMRO to come to room temperature for at least 30 minutes. When KYNAMRO is cold, it may cause redness or sensitivity after your injection. KYNAMRO should not be heated and should be kept in original packaging to protect from light.
- •
- alcohol wipe
- •
- cotton ball
- •
- puncture-proof container to dispose of the used syringes. See the detailed instructions for the “Dispose of used syringes” at the end of this Instructions for Use.
The figure below shows what the pre-filled syringe for KYNAMRO looks like before and after use.

PREPARE:
Step 1. Place Supplies and Wash Hands. Place the supplies you will need on a clean, flat surface in a well-lit area. Wash and dry your hands well.
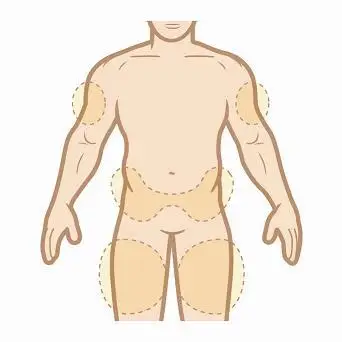
Step 2. Choose an injection site. KYNAMRO is injected under the skin and into the fat layer between the skin and the muscles (subcutaneous). KYNAMRO should be injected in the abdomen (belly), thigh, or back of the upper arm. If you choose your abdomen, do not use the area 2 inches around your belly button (navel). NOTE: Choose a different site each time you give yourself an injection to reduce the chance of redness or pain. Avoid injecting KYNAMRO into areas of skin that are damaged, such as scars, tattoos, active skin disease, sunburns, rashes, inflammation, skin infections, or active areas of psoriasis.
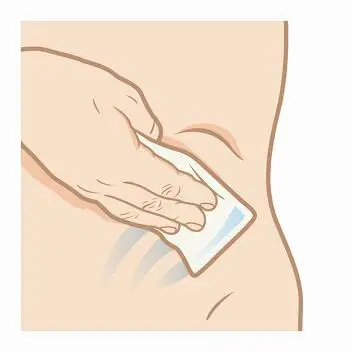
Step 3. Clean the injection site. Use an alcohol wipe and allow the site to dry.
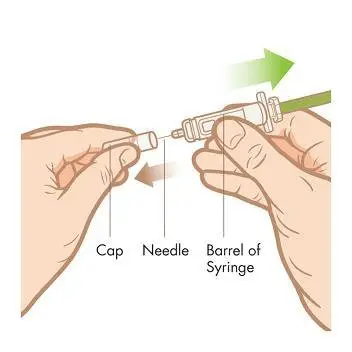
Step 4. Remove the syringe from the tray. Peel back the foil lid from the tray. Grab the syringe from the center and pull straight out of the tray. NOTE: Do not remove the syringe by pulling on the plunger or the cap as you may bend the needle or move the plunger.
INJECT:
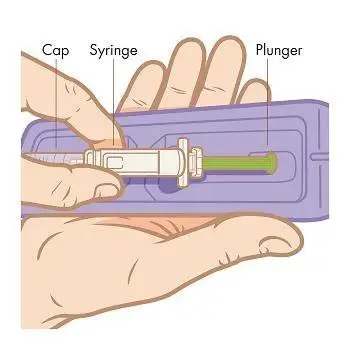
Step 5. Remove the cap. Pull the cap straight off the syringe to avoid bending the needle. NOTE: Hold the barrel of the syringe in 1 hand like a pencil or a dart. Do not touch the needle.
Step 6. Insert the needle. Gently pinch and lift the skin around the injection site. Stick the needle straight down into your skin with a quick, firm motion. Be careful not to stick the needle into the fingers of your other hand.
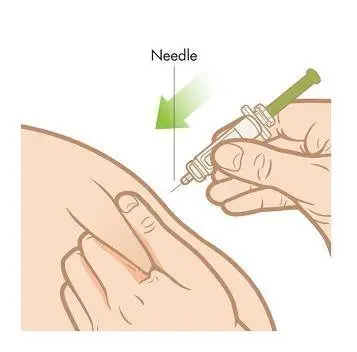
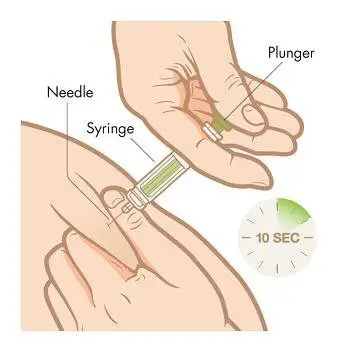
Step 7. Slowly, over a period of at least 10 seconds, push down the plunger with your thumb until the syringe is empty. Once the syringe is empty, pull the needle straight out, release the skin, and hold a clean cotton ball at the injection site.Do not rub the area because rubbing may cause reddening or pain at your injection site.
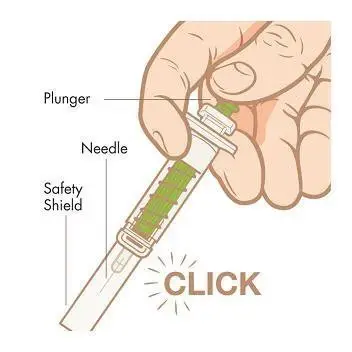
Step 8. Activate safety shield. Point the needle down away from yourself and others, and then fully push down on the plunger to activate the safety shield. Do not try to re-cap the needle with the cap.
DISPOSE:
Step 9. Dispose of used syringes.
- •
- Put your used syringes in a FDA-cleared sharps disposal container right away after use.
- •
- Do not throw away (dispose of) loose syringes in your household trash.
- •
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- •
- made of a heavy-duty plastic
- •
- can be closed with a tight-fitting, puncture-proof lid, without sharps being able to come out
- •
- upright and stable during use
- •
- leak resistant
- •
- properly labeled to warn of hazardous waste inside the container.
- •
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal
- •
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- •
- Do not recycle your used sharps disposal container.
How should I store KYNAMRO?
- •
- Store KYNAMRO in a refrigerator between 36°F to 46°F (2°C to 8°C). If a refrigerator is not available, KYNAMRO can be stored at or below 86°F (30°C) for up to 14 days if it is kept away from heat.
- •
- Protect KYNAMRO from light and store in the original carton.
- •
- Safely throw away medicine that is out of date or no longer needed.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
KYNAMRO is manufactured for:
Kastle Therapeutics
Chicago, IL
1-877-596-2676 (phone)
KYNAMRO is a registered trademark of Kastle Therapeutics
Issued: June 2016

| KYNAMRO
mipomersen sodium injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Kastle Therapeutics Llc (080134022) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Biosurgery | 098066215 | ANALYSIS(70688-0502) , MANUFACTURE(70688-0502) , PACK(70688-0502) , LABEL(70688-0502) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Isis Pharmaceuticals | 361949092 | ANALYSIS(70688-0502) , API MANUFACTURE(70688-0502) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Limited | 229522842 | ANALYSIS(70688-0502) , LABEL(70688-0502) , PACK(70688-0502) | |
