Drug Detail:Kyprolis (Carfilzomib [ kar-filz-oh-mib ])
Drug Class: Proteasome inhibitors
Highlights of Prescribing Information
KYPROLIS® (carfilzomib) for injection, for intravenous use
Initial U.S. Approval: 2012
Recent Major Changes
| Indications and Usage (1.1) | 11/2021 |
| Indications and Usage (1.1) | 6/2022 |
| Dosage and Administration (2.2) | 6/2022 |
| Dosage and Administration (2.2) | 11/2021 |
Indications and Usage for Kyprolis
Kyprolis is a proteasome inhibitor that is indicated:
- for the treatment of adult patients with relapsed or refractory multiple myeloma who have received one to three lines of therapy in combination with
- Lenalidomide and dexamethasone; or
- Dexamethasone; or
- Daratumumab and dexamethasone; or
- Daratumumab and hyaluronidase-fihj and dexamethasone; or
- Isatuximab and dexamethasone. (1, 14)
- as a single agent for the treatment of patients with relapsed or refractory multiple myeloma who have received one or more lines of therapy. (1, 14)
Kyprolis Dosage and Administration
- Hydrate prior to and following Kyprolis as needed. (2.1)
- Premedicate prior to all Cycle 1 doses and if infusion-related reactions develop or reappear. (2.1)
- The recommended dosing regimens are as follows. See Full Prescribing Information for additional dosage information. (2.2)
| Regimen | Dosage | Infusion Time |
|---|---|---|
| Kyprolis and Dexamethasone (Kd) or Kyprolis, Daratumumab and Dexamethasone (DKd) or Kyprolis, Daratumumab and hyaluronidase-fihj and Dexamethasone (DKd) | 20/70 mg/m2 once weekly | 30 minutes |
| Kyprolis and Dexamethasone (Kd) or Kyprolis, Daratumumab and Dexamethasone (DKd) or Kyprolis, Daratumumab and hyaluronidase-fihj and Dexamethasone (DKd) or Kyprolis, Isatuximab and Dexamethasone (Isa-Kd) or Kyprolis Monotherapy | 20/56 mg/m2 twice weekly | 30 minutes |
| Kyprolis, Lenalidomide and Dexamethasone (KRd) or Kyprolis Monotherapy | 20/27 mg/m2 twice weekly | 10 minutes |
Dosage Forms and Strengths
For injection: 10 mg, 30 mg or 60 mg lyophilized powder in single-dose vial for reconstitution. (3)
Contraindications
None. (4)
Warnings and Precautions
- Cardiac Toxicities: Monitor for signs and symptoms of cardiac failure or ischemia. Withhold Kyprolis and evaluate promptly. (5.1)
- Acute Renal Failure: Monitor serum creatinine regularly. (5.2)
- Tumor Lysis Syndrome (TLS): Administer pre-treatment hydration. (2.1) Monitor for TLS, including uric acid levels and treat promptly. (5.3)
- Pulmonary Toxicity, including Acute Respiratory Distress Syndrome, Acute Respiratory Failure, and Acute Diffuse Infiltrative Pulmonary Disease: Withhold Kyprolis and evaluate promptly. (5.4)
- Pulmonary Hypertension: Withhold Kyprolis and evaluate. (5.5)
- Dyspnea: For severe or life-threatening dyspnea, withhold Kyprolis and evaluate. (5.6)
- Hypertension, including Hypertensive Crisis: Monitor blood pressure regularly. If hypertension cannot be controlled, interrupt treatment with Kyprolis. (5.7)
- Venous Thrombosis: Thromboprophylaxis is recommended. (5.8)
- Infusion-related Reactions: Premedicate with dexamethasone. (2.1, 5.9)
- Hemorrhage: Fatal or serious cases of hemorrhage may occur, including gastrointestinal, pulmonary, and intracranial hemorrhage. Promptly evaluate signs and symptoms of blood loss. (5.10)
- Thrombocytopenia: Monitor platelet counts; interrupt or reduce Kyprolis dosing as clinically indicated. (2.3, 5.11)
- Hepatic Toxicity and Hepatic Failure: Monitor liver enzymes regularly. Withhold Kyprolis if suspected. (5.12)
- Thrombotic Microangiopathy: Monitor for signs and symptoms. Discontinue Kyprolis if suspected. (5.13)
- Posterior Reversible Encephalopathy Syndrome (PRES): Consider neuro-radiological imaging (MRI) for onset of visual or neurological symptoms; discontinue Kyprolis if suspected. (5.14)
- Progressive Multifocal Leukoencephalopathy: Consider PML if new or worsening neurologic manifestations. Discontinue Kyprolis in patients who develop PML. (5.15)
- Increased Fatal and Serious Toxicities in Combination with Melphalan and Prednisone in Newly Diagnosed Transplant-Ineligible Patients (5.16)
- Embryo-Fetal Toxicity: Kyprolis can cause fetal harm. Advise females of reproductive potential and males with female partners of reproductive potential of potential risk to a fetus and to use effective contraception. (5.17, 8.1)
Adverse Reactions/Side Effects
- The most common adverse reactions occurring in at least 20% of patients treated with Kyprolis in monotherapy trials: anemia, fatigue, thrombocytopenia, nausea, pyrexia, dyspnea, diarrhea, headache, cough, edema peripheral. (6)
- The most common adverse reactions occurring in at least 20% of patients treated with Kyprolis in the combination therapy trials: anemia, diarrhea, hypertension, fatigue, upper respiratory tract infection, thrombocytopenia, pyrexia, cough, dyspnea, and insomnia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Medical Information at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Geriatric Use: In the Kyprolis clinical trials, the incidence of adverse reactions was greater in patients ≥ 75 years of age. (8.5)
- Hepatic Impairment: Reduce the dose of Kyprolis by 25% in patients with mild or moderate hepatic impairment. (2.4)
- Patients on Hemodialysis: Administer Kyprolis after the hemodialysis procedure. (2.1)
- Lactation: Advise women not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2022
Full Prescribing Information
1. Indications and Usage for Kyprolis
1.1 Relapsed or Refractory Multiple Myeloma
-
Kyprolis is indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received one to three lines of therapy in combination with:
- Lenalidomide and dexamethasone; or
- Dexamethasone; or
- Daratumumab and dexamethasone; or
- Daratumumab and hyaluronidase-fihj and dexamethasone; or
- Isatuximab and dexamethasone.
- Kyprolis is indicated as a single agent for the treatment of adult patients with relapsed or refractory multiple myeloma who have received one or more lines of therapy.
2. Kyprolis Dosage and Administration
2.2 Recommended Dosage
Once Weekly 20/70 mg/m2 (30-minute infusion)
Kyprolis once weekly 20/70 mg/m2 administered in combination with
- dexamethasone (Kd),
- daratumumab plus dexamethasone (DKd), or
- daratumumab and hyaluronidase-fihj plus dexamethasone (DKd).
The recommended starting dosage of Kyprolis is 20 mg/m2 on Cycle 1, Day 1. If tolerated, escalate the dose to 70 mg/m2 on Cycle 1, Day 8. Administer Kyprolis intravenously as a 30-minute infusion on Days 1, 8, and 15 of each 28-day cycle until disease progression or unacceptable toxicity as shown in Table 1 [see Clinical Studies (14.2)]. Administer dexamethasone 30 minutes to 4 hours before Kyprolis and 1 to 3 hours before daratumumab or daratumumab and hyaluronidase-fihj. For dosage instructions of combination agents with Kyprolis, see Clinical Studies sections 14.2 (Kd) and 14.3 (DKd). Refer to the Prescribing Information for dexamethasone, intravenous daratumumab, and subcutaneous daratumumab and hyaluronidase-fihj for additional dosage information.
| Cycle 1 | ||||||||||||
| Week 1 | Week 2 | Week 3 | Week 4 | |||||||||
| Day 1 | Day 2 | Days 3-7 | Day 8 | Day 9 | Days 10-14 | Day 15 | Day 16 | Days 17-21 | Day 22 | Day 23 | Days 24-28 | |
| Kyprolis (mg/m2) | 20 | - | - | 70 | - | - | 70 | - | - | - | - | - |
| Cycles 2 and later | ||||||||||||
| Week 1 | Week 2 | Week 3 | Week 4 | |||||||||
| Day 1 | Day 2 | Days 3-7 | Day 8 | Day 9 | Days 10-14 | Day 15 | Day 16 | Days 17-21 | Day 22 | Day 23 | Days 24-28 | |
| Kyprolis (mg/m2) | 70 | - | - | 70 | - | - | 70 | - | - | - | - | - |
Twice Weekly 20/56 mg/m2 (30-minute infusion)
Kyprolis twice weekly 20/56 mg/m2 administered as monotherapy or in combination with
- dexamethasone (Kd),
- daratumumab plus dexamethasone (DKd),
- daratumumab and hyaluronidase-fihj plus dexamethasone (DKd), or
- isatuximab plus dexamethasone (Isa-Kd).
The recommended starting dosage of Kyprolis is 20 mg/m2 on Cycle 1, Days 1 and 2. If tolerated, escalate the dose to 56 mg/m2 on Cycle 1, Day 8. Administer Kyprolis intravenously as a 30-minute infusion on Days 1, 2, 8, 9, 15, and 16 of each 28-day cycle as shown in Table 2 until disease progression or unacceptable toxicity [see Clinical Studies (Section 14)]. If given as monotherapy, administer 8 mg dexamethasone orally or intravenously 30 minutes to 4 hours before Kyprolis then as needed to minimize infusion-related reactions [see Dosage and Administration (2.1)]. Kyprolis given as monotherapy may be omitted on Days 8 and 9 of cycle 13 onward. For dosage instructions of combination agents administered with Kyprolis, see Clinical Studies sections 14.2 (Kd), 14.3 (DKd), 14.4 (Isa-Kd), and 14.5 (Monotherapy). Refer to the Prescribing Information for dexamethasone, intravenous daratumumab, subcutaneous daratumumab and hyaluronidase-fihj, and isatuximab for additional dosage information.
|
||||||||||||
| Cycle 1 | ||||||||||||
| Week 1 | Week 2 | Week 3 | Week 4 | |||||||||
| Day 1 | Day 2 | Days 3-7 | Day 8 | Day 9 | Days 10-14 | Day 15 | Day 16 | Days 17-21 | Day 22 | Day 23 | Days 24-28 | |
| Kyprolis* (mg/m2) | 20 | 20 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
| Cycles 2 and later | ||||||||||||
| Week 1 | Week 2 | Week 3 | Week 4 | |||||||||
| Day 1 | Day 2 | Days 3-7 | Day 8 | Day 9 | Days 10-14 | Day 15 | Day 16 | Days 17-21 | Day 22 | Day 23 | Days 24-28 | |
| Kyprolis (mg/m2) | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Twice Weekly 20/27 mg/m2 (10-minute infusion)
Kyprolis twice weekly 20/27mg/m2 is administered as monotherapy or in combination with lenalidomide and dexamethasone (KRd).
The recommended starting dosage of Kyprolis is 20 mg/m2 in Cycle 1 on Days 1 and 2. If tolerated, escalate the dose to 27 mg/m2 on Day 8 of Cycle 1 and thereafter. Administer Kyprolis intravenously as a 10-minute infusion [see Clinical Studies (14.4)]. In Cycles 1 through 12, administer Kyprolis on Days 1, 2, 8, 9, 15 and 16 of each 28-day cycle as shown in Table 3. From Cycle 13, administer Kyprolis on Days 1, 2, 15 and 16 of each 28-day cycle. If given as monotherapy, premedicate with dexamethasone 4 mg orally or intravenously 30 minutes to 4 hours before each Kyprolis dose in Cycle 1, then as needed to minimize infusion-related reactions [see Dosage and Administration (2.1)]. Continue Kyprolis with the regimens shown in Table 3 until disease progression or unacceptable toxicity occurs. When combined with lenalidomide and dexamethasone, discontinue Kyprolis after Cycle 18 and continue lenalidomide and dexamethasone until disease progression or unacceptable toxicity occurs. For dosage instructions of combination agents with Kyprolis, see Clinical Studies sections 14.1 (KRd) and 14.5 (Monotherapy). Refer to the Prescribing Information for dexamethasone and lenalidomide for additional dosage information.
|
||||||||||
| Cycle 1 | ||||||||||
| Week 1 | Week 2 | Week 3 | Week 4 | |||||||
| Day 1 | Day 2 | Days 3-7 | Day 8 | Day 9 | Days 10-14 | Day 15 | Day 16 | Days 17-21 | Days 22-28 | |
| Kyprolis (mg/m2)* | 20 | 20 | - | 27 | 27 | - | 27 | 27 | - | - |
| Cycles 2 to 12 | ||||||||||
| Week 1 | Week 2 | Week 3 | Week 4 | |||||||
| Day 1 | Day 2 | Days 3-7 | Day 8 | Day 9 | Days 10-14 | Day 15 | Day 16 | Days 17-21 | Days 22-28 | |
| Kyprolis (mg/m2) | 27 | 27 | - | 27 | 27 | - | 27 | 27 | - | - |
| Cycles 13 and later† | ||||||||||
| Week 1 | Week 2 | Week 3 | Week 4 | |||||||
| Day 1 | Day 2 | Days 3-7 | Day 8 | Day 9 | Days 10-14 | Day 15 | Day 16 | Days 17-21 | Days 22-28 | |
| Kyprolis (mg/m2) | 27 | 27 | - | - | - | - | 27 | 27 | - | - |
2.3 Dosage Modifications for Adverse Reactions
Recommended actions and dosage modifications for Kyprolis are presented in Table 4. Dose level reductions are presented in Table 5. See the lenalidomide, intravenous daratumumab, subcutaneous daratumumab and hyaluronidase-fihj, isatuximab, and dexamethasone Prescribing Information respectively for recommended dosage modifications associated with each product.
| ANC = absolute neutrophil count | |
|
|
| Hematologic Toxicity
[see Warnings and Precautions (5.11), Adverse Reactions (6.1)] | Recommended Action |
|
|
|
|
|
|
| Renal Toxicity
[see Warnings and Precautions (5.2)] | Recommended Action |
|
|
| Other Non-hematologic Toxicity
[see Adverse Reactions (6.1)]. | Recommended Action |
|
|
| Regimen | Kyprolis Frequency | Dose | First Dose Reduction | Second Dose Reduction | Third Dose Reduction |
|---|---|---|---|---|---|
| Note: Infusion times remain unchanged during dose reduction(s). | |||||
|
|||||
| Kyprolis and Dexamethasone OR Kyprolis, Daratumumab, and Dexamethasone | Once weekly | 70 mg/m2 | 56 mg/m2 | 45 mg/m2 | 36 mg/m2* |
| Kyprolis and Dexamethasone OR Kyprolis, Daratumumab, and Dexamethasone OR Kyprolis, Isatuximab, and Dexamethasone OR Kyprolis Monotherapy | Twice weekly | 56 mg/m2 | 45 mg/m2 | 36 mg/m2 | 27 mg/m2* |
| Kyprolis, Lenalidomide, and Dexamethasone OR Kyprolis Monotherapy | Twice weekly | 27 mg/m2 | 20 mg/m2 | 15 mg/m2* | — |
2.4 Dosage Modifications for Hepatic Impairment
For patients with mild (total bilirubin 1 to 1.5 × ULN and any AST or total bilirubin ≤ ULN and AST > ULN) or moderate (total bilirubin > 1.5 to 3 × ULN and any AST) hepatic impairment, reduce the dose of Kyprolis by 25% [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.5 Recommended Dosage for End Stage Renal Disease
For patients with end stage renal disease who are on hemodialysis, administer Kyprolis after the hemodialysis procedure.
2.6 Preparation and Administration
Kyprolis vials contain no antimicrobial preservatives and are intended for single-dose only. The reconstituted solution contains carfilzomib at a concentration of 2 mg/mL.
Read the complete preparation instructions prior to reconstitution. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Reconstitution/Preparation Steps:
- Remove vial from refrigerator just prior to use.
- Calculate the dose (mg/m2) and number of vials of Kyprolis required using the patient's BSA at baseline.
- Aseptically reconstitute each Kyprolis vial only with Sterile Water for Injection, USP using the volumes described in Table 6. Use a 21-gauge or larger needle (0.8 mm or smaller external diameter needle) to reconstitute each vial by slowly injecting Sterile Water for Injection, USP through the stopper and directing the Sterile Water for Injection, USP onto the INSIDE WALL OF THE VIAL to minimize foaming. There is no data to support the use of closed system transfer devices with Kyprolis.
Table 6: Reconstitution Volumes Strength Amount of Sterile Water for Injection, USP required for reconstitution 10 mg vial 5 mL 30 mg vial 15 mL 60 mg vial 29 mL - Gently swirl and/or invert the vial slowly for about 1 minute, or until complete dissolution. DO NOT SHAKE to avoid foam generation. If foaming occurs, allow the solution to settle in the vial until foaming subsides (approximately 5 minutes) and the solution is clear.
- Visually inspect for particulate matter and discoloration prior to administration. The reconstituted product should be a clear, colorless solution and should not be administered if any discoloration or particulate matter is observed.
- Discard any unused portion left in the vial. DO NOT pool unused portions from the vials. DO NOT administer more than one dose from a vial.
- Administer Kyprolis directly by intravenous infusion or in a 50 mL to 100 mL intravenous bag containing 5% Dextrose Injection, USP. Do not administer as an intravenous push or bolus.
- When administering in an intravenous bag, use a 21-gauge or larger gauge needle (0.8 mm or smaller external diameter needle) to withdraw the calculated dose from the vial and dilute into 50 mL or 100 mL intravenous bag containing only 5% Dextrose Injection, USP (based on the calculated total dose and infusion time).
- Flush the intravenous administration line with normal saline or 5% Dextrose Injection, USP immediately before and after Kyprolis administration.
- Do not mix Kyprolis with or administer as an infusion with other medicinal products.
The stabilities of reconstituted Kyprolis under various temperature and container conditions are shown in Table 7.
| Storage Conditions of Reconstituted Kyprolis | Stability* per Container | ||
|---|---|---|---|
| Vial | Syringe | Intravenous Bag (D5W†) |
|
|
|||
| Refrigerated 2°C to 8°C (36°F to 46°F) | 24 hours | 24 hours | 24 hours |
| Room Temperature 15°C to 30°C (59°F to 86°F) | 4 hours | 4 hours | 4 hours |
3. Dosage Forms and Strengths
For injection: 10 mg, 30 mg and 60 mg as a lyophilized cake or powder in single-dose vial for reconstitution.
5. Warnings and Precautions
5.1 Cardiac Toxicities
New onset or worsening of pre-existing cardiac failure (e.g., congestive heart failure, pulmonary edema, decreased ejection fraction), cardiomyopathy, myocardial ischemia, and myocardial infarction including fatalities have occurred following administration of Kyprolis. Some events occurred in patients with normal baseline ventricular function. In clinical studies with Kyprolis, these events occurred throughout the course of Kyprolis therapy. Death due to cardiac arrest has occurred within one day of Kyprolis administration. In randomized, open-label, multicenter trials for combination therapies, the incidence of cardiac failure events was 8% and that of arrythmias was 8% (majority of which were atrial fibrillation and sinus tachycardia) [see Adverse Reactions (6.1)].
Monitor patients for clinical signs or symptoms of cardiac failure or cardiac ischemia. Evaluate promptly if cardiac toxicity is suspected. Withhold Kyprolis for Grade 3 or 4 cardiac adverse reactions until recovery and consider whether to restart Kyprolis at 1 dose level reduction based on a benefit/risk assessment [see Dosage and Administration (2.3)].
While adequate hydration is required prior to each dose in Cycle 1, monitor all patients for evidence of volume overload, especially patients at risk for cardiac failure. Adjust total fluid intake as clinically appropriate in patients with baseline cardiac failure or who are at risk for cardiac failure [see Dosage and Administration (2.1)].
In patients ≥ 75 years of age, the risk of cardiac failure is increased compared to younger patients. Patients with New York Heart Association Class III and IV heart failure, recent myocardial infarction, conduction abnormalities, angina, or arrhythmias uncontrolled by medications were not eligible for the clinical trials. These patients may be at greater risk for cardiac complications; for these patients, complete a comprehensive medical assessment (including blood pressure control and fluid management) prior to starting treatment with Kyprolis and remain under close follow-up [see Use in Specific Populations (8.5)].
5.2 Acute Renal Failure
Cases of acute renal failure have occurred in patients receiving Kyprolis. Some of these events have been fatal. Renal insufficiency (including renal failure) has occurred in approximately 9% of patients who received Kyprolis. Acute renal failure was reported more frequently in patients with advanced relapsed and refractory multiple myeloma who received Kyprolis monotherapy. The risk of fatal renal failure was greater in patients with a baseline reduced estimated creatinine clearance (calculated using Cockcroft-Gault equation).
Monitor renal function with regular measurement of the serum creatinine and/or estimated creatinine clearance. Reduce or withhold dose as appropriate [see Dosage and Administration (2.3)].
5.3 Tumor Lysis Syndrome
Cases of TLS, including fatal outcomes, have been reported in patients who received Kyprolis. Patients with multiple myeloma and a high tumor burden should be considered to be at greater risk for TLS.
Administer oral and intravenous fluids before administration of Kyprolis in Cycle 1 and in subsequent cycles as needed. Consider uric acid-lowering drugs in patients at risk for TLS. Monitor for TLS during treatment and manage promptly, including interruption of Kyprolis until TLS is resolved [see Dosage and Administration (2.1)].
5.4 Pulmonary Toxicity
Acute Respiratory Distress Syndrome (ARDS) and acute respiratory failure have occurred in approximately 2% of patients who received Kyprolis. In addition, acute diffuse infiltrative pulmonary disease, such as pneumonitis and interstitial lung disease, occurred in approximately 2% of patients who received Kyprolis. Some events were fatal.
In the event of drug-induced pulmonary toxicity, discontinue Kyprolis.
5.5 Pulmonary Hypertension
Pulmonary arterial hypertension was reported in approximately 2% of patients who received Kyprolis, with Grade 3 or greater in less than 1%.
Evaluate with cardiac imaging and/or other tests as indicated. Withhold Kyprolis for pulmonary hypertension until resolved or returned to baseline and consider whether to restart Kyprolis based on a benefit/risk assessment.
5.6 Dyspnea
Dyspnea was reported in 25% of patients treated with Kyprolis, with Grade 3 or greater in 4%.
Evaluate dyspnea to exclude cardiopulmonary conditions including cardiac failure and pulmonary syndromes. Stop Kyprolis for Grade 3 or 4 dyspnea until resolved or returned to baseline. Consider whether to restart Kyprolis based on a benefit/risk assessment [see Warnings and Precautions (5.1, 5.4) and Adverse Reactions (6.1)].
5.7 Hypertension
Hypertension, including hypertensive crisis and hypertensive emergency, has been observed with Kyprolis. In ASPIRE, the incidence of hypertension events was 17% in the KRd arm versus 9% in the Rd arm. In ENDEAVOR, the incidence of hypertension events was 34% in the Kd arm versus 11% in the Vd arm. In CANDOR, the incidence of hypertension events was 31% in the DKd arm versus 28% in the Kd arm. Some of these events have been fatal.
Optimize blood pressure prior to starting Kyprolis. Monitor blood pressure regularly in all patients while on Kyprolis. If hypertension cannot be adequately controlled, withhold Kyprolis and evaluate. Consider whether to restart Kyprolis based on a benefit/risk assessment.
5.8 Venous Thrombosis
Venous thromboembolic events (including deep venous thrombosis and pulmonary embolism) have been observed with Kyprolis. In ASPIRE, with thromboprophylaxis used in both arms, the incidence of venous thromboembolic events in the first 12 cycles was 13% in the KRd arm versus 6% in the Rd arm. In ENDEAVOR, the incidence of venous thromboembolic events in months 1–6 was 9% in the Kd arm versus 2% in the Vd arm. With Kyprolis monotherapy, the incidence of venous thromboembolic events was 2% [see Adverse Reactions (6.1)].
Provide thromboprophylaxis for patients being treated with Kyprolis in combination with lenalidomide and dexamethasone; with dexamethasone; or with intravenous daratumumab and dexamethasone. Select the thromboprophylaxis regimen based on the patient's underlying risks.
For patients using oral contraceptives or hormonal contraception associated with a risk of thrombosis, consider non-hormonal contraception during treatment when Kyprolis is administered in combination [see Use in Specific Populations (8.3)].
5.9 Infusion-Related Reactions
Infusion-related reactions, including life-threatening reactions, have occurred in patients receiving Kyprolis. Signs and symptoms include fever, chills, arthralgia, myalgia, facial flushing, facial edema, laryngeal edema, vomiting, weakness, shortness of breath, hypotension, syncope, chest tightness, or angina. These reactions can occur immediately following or up to 24 hours after administration of Kyprolis.
Administer dexamethasone prior to Kyprolis to reduce the incidence and severity of infusion-related reactions [see Dosage and Administration (2.1, 2.2), Adverse Reactions (6.1)].
5.10 Hemorrhage
Fatal or serious cases of hemorrhage have been reported in patients treated with Kyprolis [see Adverse Reactions (6.1)]. Hemorrhagic events have included gastrointestinal, pulmonary, and intracranial hemorrhage and epistaxis. The bleeding can be spontaneous and intracranial hemorrhage has occurred without trauma. Hemorrhage has been reported in patients having either low or normal platelet counts. Hemorrhage has also been reported in patients who were not on antiplatelet therapy or anticoagulation.
Promptly evaluate signs and symptoms of blood loss. Reduce or withhold dose as appropriate [see Dosage and Administration (2.3)].
5.11 Thrombocytopenia
Kyprolis causes thrombocytopenia with platelet nadirs observed between Day 8 and Day 15 of each 28-day cycle, with recovery to baseline platelet count usually by the start of the next cycle [see Adverse Reactions (6.1)]. Thrombocytopenia was reported in approximately 32% of patients in clinical trials with Kyprolis. Hemorrhage may occur [see Adverse Reactions (6.1), Warnings and Precautions (5.10)].
Monitor platelet counts frequently during treatment with Kyprolis. Reduce or withhold dose as appropriate [see Dosage and Administration (2.3)].
5.12 Hepatic Toxicity and Hepatic Failure
Cases of hepatic failure, including fatal cases, have been reported (2%) during treatment with Kyprolis. Kyprolis can cause increased serum transaminases [see Adverse Reactions (6.1)].
Monitor liver enzymes regularly, regardless of baseline values. Reduce or withhold dose as appropriate [see Dosage and Administration (2.3)].
5.13 Thrombotic Microangiopathy
Cases of thrombotic microangiopathy, including thrombotic thrombocytopenic purpura/ hemolytic uremic syndrome (TTP/HUS), have been reported in patients who received Kyprolis. Some of these events have been fatal.
Monitor for signs and symptoms of TTP/HUS. If the diagnosis is suspected, stop Kyprolis and evaluate. If the diagnosis of TTP/HUS is excluded, Kyprolis may be restarted. The safety of reinitiating Kyprolis therapy in patients previously experiencing TTP/HUS is not known.
5.14 Posterior Reversible Encephalopathy Syndrome
Cases of posterior reversible encephalopathy syndrome (PRES) have been reported in patients receiving Kyprolis. PRES, formerly termed Reversible Posterior Leukoencephalopathy Syndrome (RPLS), is a neurological disorder which can present with seizure, headache, lethargy, confusion, blindness, altered consciousness, and other visual and neurological disturbances, along with hypertension, and the diagnosis is confirmed by neuro-radiological imaging (MRI).
Discontinue Kyprolis if PRES is suspected and evaluate. The safety of reinitiating Kyprolis therapy in patients previously experiencing PRES is not known.
5.15 Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML), which can be fatal, has been reported with Kyprolis. In addition to Kyprolis, other possible contributory factors include prior or concurrent immunosuppressive therapy that may cause immunosuppression.
Consider PML in any patient with new onset of or changes in pre-existing neurological signs or symptoms. If PML is suspected, discontinue Kyprolis and initiate evaluation for PML including neurology consultation.
5.16 Increased Fatal and Serious Toxicities in Combination with Melphalan and Prednisone in Newly Diagnosed Transplant-Ineligible Patients
In CLARION, a clinical trial of 955 transplant-ineligible patients with newly diagnosed multiple myeloma randomized to Kyprolis (20/36 mg/m2 by 30-minute infusion twice weekly for four of each six-week cycle), melphalan and prednisone (KMP) or bortezomib, melphalan and prednisone (VMP), a higher incidence of fatal adverse reactions (7% versus 4%) and serious adverse reactions (50% versus 42%) were observed in the KMP arm compared to patients in the VMP arm, respectively. Patients in the KMP arm were observed to have a higher incidence of any grade adverse reactions involving cardiac failure (11% versus 4%), hypertension (25% versus 8%), acute renal failure (14% versus 6%), and dyspnea (18% versus 9%). This study did not meet its primary outcome measure of superiority in progression-free survival (PFS) for the KMP arm. Kyprolis in combination with melphalan and prednisone is not indicated for transplant-ineligible patients with newly diagnosed multiple myeloma.
5.17 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, Kyprolis can cause fetal harm when administered to a pregnant woman. Carfilzomib administered intravenously to pregnant rabbits during organogenesis at a dose approximately 40% of the clinical dose of 27 mg/m2 based on BSA caused post-implantation loss and a decrease in fetal weight.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with Kyprolis and for 6 months following the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with Kyprolis and for 3 months following the last dose [see Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Cardiac Toxicities [see Warnings and Precautions (5.1)]
- Acute Renal Failure [see Warnings and Precautions (5.2)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.3)]
- Pulmonary Toxicity [see Warnings and Precautions (5.4)]
- Pulmonary Hypertension [see Warnings and Precautions (5.5)]
- Dyspnea [see Warnings and Precautions (5.6)]
- Hypertension [see Warnings and Precautions (5.7)]
- Venous Thrombosis [see Warnings and Precautions (5.8)]
- Infusion-Related Reactions [see Warnings and Precautions (5.9)]
- Hemorrhage [see Warnings and Precautions (5.10)]
- Thrombocytopenia [see Warnings and Precautions (5.11)]
- Hepatic Toxicity and Hepatic Failure [see Warnings and Precautions (5.12)]
- Thrombotic Microangiopathy [see Warnings and Precautions (5.13)]
- Posterior Reversible Encephalopathy Syndrome [see Warnings and Precautions (5.14)]
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.15)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the Warnings and Precautions reflect exposure to Kyprolis in 2,239 patients administered in combination with other drugs in ASPIRE, ENDEAVOR, A.R.R.O.W., CANDOR, IKEMA, EQUULEUS, and PLEIADES. The most common adverse reactions occurring in at least 20% of patients who received Kyprolis in combination were anemia, diarrhea, hypertension, fatigue, upper respiratory tract infection, thrombocytopenia, pyrexia, cough, dyspnea, and insomnia.
Kyprolis in Combination with Lenalidomide and Dexamethasone
The safety of Kyprolis 20/27 mg/m2 twice weekly in combination with lenalidomide and dexamethasone (KRd) was evaluated in ASPIRE [see Clinical Studies (14.1)]. The median number of cycles initiated was 22 cycles for the KRd arm and 14 cycles for the Rd arm.
Deaths due to adverse reactions within 30 days of the last dose of any therapy in the KRd arm occurred in 45/392 (12%) patients compared with 42/389 (11%) patients who died due to adverse reactions within 30 days of the last dose of any Rd therapy. The most frequent cause of deaths occurring in patients (%) in the two arms (KRd versus Rd) included infection 12 (3%) versus 11 (3%), cardiac 10 (3%) versus 9 (2%), and other adverse reactions 23 (6%) versus 22 (6%).
Serious adverse reactions were reported in 65% of the patients in the KRd arm and 57% of the patients in the Rd arm. The most frequent serious adverse reactions reported in the KRd arm as compared with the Rd arm were pneumonia (17% versus 13%), respiratory tract infection (4% versus 2%), pyrexia (4% versus 3%), and pulmonary embolism (3% versus 2%).
Discontinuation due to any adverse reaction occurred in 33% in the KRd arm versus 30% in the Rd arm. Adverse reactions leading to discontinuation of Kyprolis occurred in 12% of patients and the most common reactions included pneumonia (1%), myocardial infarction (0.8%), and upper respiratory tract infection (0.8%). The incidence of cardiac failure events was 7% in the KRd arm versus 4% in the Rd arm.
Table 8 summarizes the adverse reactions in the first 12 cycles in ASPIRE.
| Adverse Reactions | KRd (N = 392) n (%) | Rd (N = 389) n (%) |
||
|---|---|---|---|---|
| Any Grade | ≥ Grade 3 | Any Grade | ≥ Grade 3 | |
| KRd = Kyprolis, lenalidomide, and dexamethasone; Rd = lenalidomide and dexamethasone | ||||
|
||||
| Blood and Lymphatic System Disorders | ||||
| Anemia | 138 (35) | 53 (14) | 127 (33) | 47 (12) |
| Neutropenia | 124 (32) | 104 (27) | 115 (30) | 89 (23) |
| Thrombocytopenia | 100 (26) | 58 (15) | 75 (19) | 39 (10) |
| Gastrointestinal Disorders | ||||
| Diarrhea | 119 (30) | 8 (2) | 106 (27) | 12 (3) |
| Constipation | 68 (17) | 0 (0) | 55 (14) | 1 (0) |
| Nausea | 63 (16) | 1 (0) | 43 (11) | 3 (1) |
| General Disorders and Administration Site Conditions | ||||
| Fatigue | 113 (29) | 23 (6) | 107 (28) | 20 (5) |
| Pyrexia | 93 (24) | 5 (1) | 64 (17) | 1 (0) |
| Edema peripheral | 59 (15) | 3 (1) | 48 (12) | 2 (1) |
| Asthenia | 54 (14) | 11 (3) | 49 (13) | 7 (2) |
| Infections | ||||
| Upper respiratory tract infection | 87 (22) | 7 (2) | 54 (14) | 4 (1) |
| Bronchitis | 55 (14) | 5 (1) | 40 (10) | 2 (1) |
| Viral upper respiratory tract infection | 55 (14) | 0 (0) | 44 (11) | 0 (0) |
| Pneumonia* | 54 (14) | 35 (9) | 43 (11) | 27 (7) |
| Metabolism and Nutrition Disorders | ||||
| Hypokalemia | 78 (20) | 22 (6) | 35 (9) | 12 (3) |
| Hypocalcemia | 55 (14) | 10 (3) | 39 (10) | 5 (1) |
| Hyperglycemia | 43 (11) | 18 (5) | 33 (9) | 15 (4) |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Muscle spasms | 92 (24) | 3 (1) | 75 (19) | 3 (1) |
| Back pain | 41 (11) | 4 (1) | 54 (14) | 6 (2) |
| Nervous System Disorders | ||||
| Peripheral neuropathies† | 43 (11) | 7 (2) | 39 (10) | 4 (1) |
| Psychiatric Disorders | ||||
| Insomnia | 64 (16) | 6 (2) | 51 (13) | 8 (2) |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Cough‡ | 93 (24) | 2 (1) | 54 (14) | 0 (0) |
| Dyspnea§ | 71 (18) | 8 (2) | 61 (16) | 6 (2) |
| Skin and Subcutaneous Tissue Disorders | ||||
| Rash | 45 (12) | 5 (1) | 54 (14) | 5 (1) |
| Vascular Disorders | ||||
| Embolic and thrombotic events¶ | 49 (13) | 16 (4) | 23 (6) | 9 (2) |
| Hypertension# | 41 (11) | 12 (3) | 15 (4) | 4 (1) |
There were 274 (70%) patients in the KRd arm who received treatment beyond Cycle 12. There were no new clinically relevant adverse reactions that emerged in the later treatment cycles.
Kyprolis in Combination with Dexamethasone
The safety of Kyprolis in combination with dexamethasone was evaluated in two open-label, randomized trials (ENDEAVOR and A.R.R.O.W.).
ENDEAVOR
The safety of Kyprolis 20/56 mg/m2 twice weekly in combination with dexamethasone (Kd) was evaluated in ENDEAVOR [see Clinical Studies (14.2)]. Patients received treatment for a median duration of 48 weeks in the Kd arm and 27 weeks in the bortezomib/dexamethasone (Vd) arm.
Deaths due to adverse reactions within 30 days of last study treatment occurred in 32/463 (7%) patients in the Kd arm and 21/456 (5%) patients in the Vd arm. The causes of death occurring in patients (%) in the two arms (Kd versus Vd) included cardiac 4 (1%) versus 5 (1%), infections 8 (2%) versus 8 (2%), disease progression 7 (2%) versus 4 (1%), pulmonary 3 (1%) versus 2 (< 1%), renal 1 (< 1%) versus 0 (0%), and other adverse reactions 9 (2%) versus 2 (< 1%).
Serious adverse reactions were reported in 59% of the patients in the Kd arm and 40% of the patients in the Vd arm. In both arms, pneumonia was the most frequently reported serious adverse reaction (8% versus 9%).
Discontinuation due to any adverse reaction occurred in 29% in the Kd arm versus 26% in the Vd arm. The most frequent adverse reaction leading to discontinuation was cardiac failure in the Kd arm (n = 8, 2%) and peripheral neuropathy in the Vd arm (n = 22, 5%). The incidence of cardiac failure events was 11% in the Kd arm versus 3% in the Vd arm.
Adverse reactions in the first 6 months of therapy that occurred at a rate of 10% or greater in the Kd arm are presented in Table 10.
| Adverse Reactions | Kd (N = 463) n (%) | Vd (N = 456) n (%) |
||
|---|---|---|---|---|
| Any Grade | Grade ≥ 3 | Any Grade | Grade ≥ 3 | |
| Kd = Kyprolis and dexamethasone; Vd = bortezomib and dexamethasone | ||||
|
||||
| Blood and Lymphatic System Disorders | ||||
| Anemia | 161 (35) | 57 (12) | 112 (25) | 43 (9) |
| Thrombocytopenia* | 125 (27) | 45 (10) | 112 (25) | 64 (14) |
| Gastrointestinal Disorders | ||||
| Diarrhea | 117 (25) | 14 (3) | 149 (33) | 27 (6) |
| Nausea | 70 (15) | 4 (1) | 68 (15) | 3 (1) |
| Constipation | 60 (13) | 1 (0) | 113 (25) | 6 (1) |
| Vomiting | 45 (10) | 5 (1) | 33 (7) | 3 (1) |
| General Disorders and Administration Site Conditions | ||||
| Fatigue | 116 (25) | 14 (3) | 126 (28) | 25 (6) |
| Pyrexia | 102 (22) | 9 (2) | 52 (11) | 3 (1) |
| Asthenia | 73 (16) | 9 (2) | 65 (14) | 13 (3) |
| Peripheral edema | 62 (13) | 3 (1) | 62 (14) | 3 (1) |
| Infections | ||||
| Upper respiratory tract infection | 67 (15) | 4 (1) | 55 (12) | 3 (1) |
| Bronchitis | 54 (12) | 5 (1) | 25 (6) | 2 (0) |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Muscle spasms | 70 (15) | 1 (0) | 23 (5) | 3 (1) |
| Back pain | 64 (14) | 8 (2) | 61 (13) | 10 (2) |
| Nervous System Disorders | ||||
| Headache | 67 (15) | 4 (1) | 39 (9) | 2 (0) |
| Peripheral neuropathies†,‡ | 56 (12) | 7 (2) | 170 (37) | 23 (5) |
| Psychiatric Disorders | ||||
| Insomnia | 105 (23) | 5 (1) | 116 (25) | 10 (2) |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Dyspnea§ | 128 (28) | 23 (5) | 69 (15) | 8 (2) |
| Cough¶ | 97 (21) | 0 (0) | 61 (13) | 2 (0) |
| Vascular Disorders | ||||
| Hypertension# | 83 (18) | 30 (7) | 33 (7) | 12 (3) |
The event rate of ≥ Grade 2 peripheral neuropathy in the Kd arm was 7% (95% CI: 5, 9) versus 35% (95% CI: 31, 39) in the Vd arm.
Adverse Reactions Occurring at a Frequency of < 10%
- Blood and lymphatic system disorders: febrile neutropenia, leukopenia, lymphopenia, neutropenia, thrombotic microangiopathy, thrombotic thrombocytopenic purpura
- Cardiac disorders: atrial fibrillation, cardiac arrest, cardiac failure, cardiac failure congestive, myocardial infarction, myocardial ischemia, palpitations, tachycardia
- Ear and labyrinth disorders: tinnitus
- Eye disorders: cataract, vision blurred
- Gastrointestinal disorders: abdominal pain, abdominal pain upper, dyspepsia, gastrointestinal hemorrhage, toothache
- General disorders and administration site conditions: chest pain, chills, influenza like illness, infusion site reactions (including inflammation, pain, and erythema), malaise, pain
- Hepatobiliary disorders: cholestasis, hepatic failure, hyperbilirubinemia
- Immune system disorders: drug hypersensitivity
- Infections: bronchopneumonia, gastroenteritis, influenza, lung infection, nasopharyngitis, pneumonia, rhinitis, sepsis, urinary tract infection, viral infection
- Metabolism and nutrition disorders: decreased appetite, dehydration, hypercalcemia, hyperkalemia, hyperuricemia, hypoalbuminemia, hypocalcemia, hypomagnesemia, hyponatremia, hypophosphatemia, tumor lysis syndrome
- Musculoskeletal and connective tissue disorders: muscular weakness, musculoskeletal chest pain, musculoskeletal pain, myalgia
- Nervous system disorders: cerebrovascular accident, dizziness, hypoesthesia, paresthesia, posterior reversible encephalopathy syndrome
- Psychiatric disorders: anxiety
- Renal and urinary disorders: renal failure, renal failure acute, renal impairment
- Respiratory, thoracic and mediastinal disorders: acute respiratory distress syndrome, dysphonia, epistaxis, interstitial lung disease, oropharyngeal pain, pneumonitis, pulmonary embolism, pulmonary edema, pulmonary hypertension, wheezing
- Skin and subcutaneous tissue disorders: erythema, hyperhidrosis, pruritus, rash
- Vascular disorders: deep vein thrombosis, flushing, hypotension
Table 11 describes Grade 3–4 laboratory abnormalities reported at a rate of ≥ 10% in the Kd arm.
| Laboratory Abnormality | Kd (N = 463) n (%) | Vd (N = 456) n (%) |
|---|---|---|
| Kd = Kyprolis and dexamethasone; Vd = bortezomib and dexamethasone | ||
|
||
| Decreased lymphocytes | 249 (54) | 180 (40) |
| Increased uric acid | 244 (53) | 198 (43) |
| Decreased hemoglobin | 79 (17) | 68 (15) |
| Decreased platelets | 85 (18) | 77 (17) |
| Decreased phosphorus | 74 (16) | 61 (13) |
| Decreased creatinine clearance* | 65 (14) | 49 (11) |
| Increased potassium | 55 (12) | 21 (5) |
A.R.R.O.W.
The safety of Kyprolis in combination with dexamethasone was evaluated in A.R.R.O.W. [see Clinical Studies (14.2)]. Patients received treatment for a median duration of 38 weeks in the Kd 20/70 mg/m2 arm once weekly and 29.1 weeks in the Kd 20/27 mg/m2 twice weekly arm. The safety profile for the once weekly Kd 20/70 mg/m2 regimen was similar to the twice weekly Kd 20/27 mg/m2 regimen.
Deaths due to adverse reactions within 30 days of last study treatment occurred in 22/238 (9%) patients in the Kd 20/70 mg/m2 arm and 18/235 (8%) patients in the Kd 20/27 mg/m2 arm. The most frequent fatal adverse reactions occurring in patients (%) in the two arms (once weekly Kd 20/70 mg/m2 versus twice weekly Kd 20/27 mg/m2) were sepsis 2 (< 1%) versus 2 (< 1%), septic shock 2 (< 1%) versus 1 (< 1%), and infection 2 (< 1%) versus 0 (0%).
Serious adverse reactions were reported in 43% of the patients in the Kd 20/70 mg/m2 arm and 41% of the patients in the Kd 20/27 mg/m2 arm. In both arms, pneumonia was the most frequently reported serious adverse reaction (8% versus 7%).
Discontinuation due to any adverse reaction occurred in 13% in the Kd 20/70 mg/m2 arm versus 12% in the Kd 20/27 mg/m2 arm. The most frequent adverse reaction leading to discontinuation was acute kidney injury (2% versus 2%). The incidence of cardiac failure events was 3.8% in the once weekly Kd 20/70 mg/m2 arm versus 5.1% in the twice weekly Kd 20/27 mg/m2 arm.
Adverse reactions that occurred at a rate of 10% or greater in either Kd arm are presented in Table 12.
| Adverse Reactions | Once weekly Kd 20/70 mg/m2 (N = 238) n (%) | Twice weekly Kd 20/27 mg/m2 (N = 235) n (%) |
||
|---|---|---|---|---|
| Any Grade | Grade ≥ 3 | Any Grade | Grade ≥ 3 | |
| Kd = Kyprolis and dexamethasone | ||||
|
||||
| Blood and Lymphatic System Disorders | ||||
| Anemia* | 64 (27) | 42 (18) | 76 (32) | 42 (18) |
| Thrombocytopenia† | 53 (22) | 26 (11) | 41 (17) | 27 (12) |
| Neutropenia‡ | 30 (13) | 21 (9) | 27 (12) | 17 (7) |
| Gastrointestinal Disorders | ||||
| Diarrhea | 44 (19) | 2 (1) | 47 (20) | 3 (1) |
| Nausea | 34 (14) | 1 (< 1) | 26 (11) | 2 (1) |
| General Disorders and Administration Site Conditions | ||||
| Pyrexia | 55 (23) | 2 (1) | 38 (16) | 4 (2) |
| Fatigue | 48 (20) | 11 (5) | 47 (20) | 5 (2) |
| Asthenia | 24 (10) | 3 (1) | 25 (11) | 2 (1) |
| Peripheral edema | 18 (8) | 0 (0) | 25 (11) | 2 (1) |
| Infections | ||||
| Respiratory tract infection§ | 70 (29) | 7 (3) | 79 (34) | 7 (3) |
| Pneumonia | 28 (12) | 24 (10) | 20 (9) | 16 (7) |
| Bronchitis | 27 (11) | 2 (1) | 25 (11) | 5 (2) |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Back pain | 28 (12) | 2 (1) | 28 (12) | 4 (2) |
| Nervous System Disorders | ||||
| Headache | 25 (11) | 1 (< 1) | 23 (10) | 1 (< 1) |
| Psychiatric Disorders | ||||
| Insomnia | 35 (15) | 2 (1) | 47 (20) | 0 (0) |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Cough¶ | 37 (16) | 2 (1) | 31 (13) | 0 (0) |
| Dyspnea# | 28 (12) | 1 (< 1) | 26 (11) | 2 (1) |
| Vascular Disorders | ||||
| HypertensionÞ | 51 (21) | 13 (6) | 48 (20) | 12 (5) |
Kyprolis in Combination with Intravenous Daratumumab and Dexamethasone
The safety of Kyprolis in combination with intravenous daratumumab and dexamethasone was evaluated in two trials (CANDOR and EQUULEUS).
CANDOR
The safety of Kyprolis 20/56 mg/m2 twice weekly in combination with intravenous daratumumab and dexamethasone (DKd) was evaluated in CANDOR [see Clinical Studies (14.3)]. Patients received Kyprolis for a median duration of 58 weeks in the DKd arm and 40 weeks in the Kd arm.
Serious adverse reactions were reported in 56% of the patients in the DKd arm and 46% of the patients in the Kd arm. The most frequent serious adverse reactions reported in the DKd arm as compared with the Kd arm were pneumonia (14% versus 9%), pyrexia (4.2% versus 2.0%), influenza (3.9% versus 1.3%), sepsis (3.9% versus 1.3%), anemia (2.3% versus 0.7%), bronchitis (1.9% versus 0%) and diarrhea (1.6% versus 0%). Fatal adverse reactions within 30 days of the last dose of any study treatment occurred in 10% of 308 patients in the DKd arm compared with 5% of 153 patients in the Kd arm. The most frequent fatal adverse reaction (DKd versus Kd) was infection 4.5% versus 2.6%.
Permanent discontinuation due to an adverse reaction in patients who received Kyprolis occurred in 21% of patients in the DKd arm versus 22% in the Kd arm. The most frequent adverse reactions leading to discontinuation of Kyprolis were cardiac failure (1.9%) and fatigue (1.9%) in the DKd arm and cardiac failure (2.0%), hypertension (2.0%) and acute kidney injury (2.0%) in the Kd arm. Interruption of Kyprolis due to adverse reactions occurred in 71% of patients in DKd arm versus 63% in the Kd arm. Dose reduction of Kyprolis due to adverse reactions occurred in 25% of patients in DKd arm versus 20% in the Kd arm.
Infusion-related reactions that occurred following the first Kyprolis dose was 13% in the DKd arm versus 1% in the Kd arm.
Table 13 summarizes the adverse reactions in CANDOR.
| Adverse Reactions | Twice weekly DKd (N = 308) | Twice weekly Kd (N = 153) |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
| DKd = Kyprolis, daratumumab, and dexamethasone; Kd = Kyprolis and dexamethasone | ||||
|
||||
| General Disorders and Administration Site Conditions | ||||
| Infusion-related reaction* | 41 | 12 | 28 | 5 |
| Fatigue† | 32 | 11 | 28 | 8 |
| Pyrexia | 20 | 1.9 | 15 | 0.7 |
| Infections | ||||
| Respiratory tract infection‡ | 40§ | 7 | 29 | 3.3 |
| Pneumonia | 18§ | 13 | 12 | 9 |
| Bronchitis | 17 | 2.6 | 12 | 1.3 |
| Blood and Lymphatic System Disorders | ||||
| Thrombocytopenia¶ | 37 | 25 | 30 | 16 |
| Anemia# | 33 | 17 | 31 | 14 |
| Gastrointestinal Disorders | ||||
| Diarrhea | 32 | 3.9 | 14 | 0.7 |
| Nausea | 18 | 0 | 13 | 0.7 |
| Vascular Disorders | ||||
| Hypertension | 31 | 18 | 28 | 13 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| CoughÞ | 21 | 0 | 21 | 0 |
| Dyspnea | 20 | 3.9 | 22 | 2.6 |
| Psychiatric Disorders | ||||
| Insomnia | 18 | 3.9 | 11 | 2.0 |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Back pain | 16 | 1.9 | 10 | 1.3 |
EQUULEUS
The safety of Kyprolis 20/70 mg/m2 once weekly in combination with intravenous daratumumab and dexamethasone (DKd) was evaluated in EQUULEUS [see Clinical Studies (14.3)]. Patients received Kyprolis for a median duration of 66 weeks.
Serious adverse reactions were reported in 48% of patients. The most frequent serious adverse reactions reported were pneumonia (4.7%), upper respiratory tract infection (4.7%), basal cell carcinoma (4.7%), influenza (3.5%), general physical health deterioration (3.5%) and hypercalcemia (3.5%). Fatal adverse reactions within 30 days of the last dose of any study treatment occurred in 3.5% of patients who died of general physical health deterioration, multi-organ failure secondary to pulmonary aspergillosis, and disease progression.
Discontinuation of Kyprolis occurred in 19% of patients. The most frequent adverse reaction leading to discontinuation was asthenia (2%). Interruption of Kyprolis due to adverse reactions occurred in 77% of patients. Dose reduction of Kyprolis due to adverse reactions occurred in 31% of patients in DKd.
Infusion-related reactions that occurred following the first Kyprolis dose was 11%. Pulmonary hypertension adverse reactions were reported in 4.7% of patients in EQUULEUS.
Table 14 summarizes the adverse reactions in EQUULEUS.
| Adverse Reactions | Once weekly DKd (N = 85) |
|
|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) |
|
| DKd = Kyprolis, daratumumab, and dexamethasone; Kd = Kyprolis and dexamethasone | ||
|
||
| Blood and Lymphatic System Disorders | ||
| Thrombocytopenia* | 68 | 32 |
| Anemia† | 52 | 21 |
| Neutropenia‡ | 31 | 21 |
| Lymphopenia§ | 29 | 25 |
| General Disorders and Administration Site Conditions | ||
| Fatigue¶ | 54 | 18 |
| Infusion-related reaction# | 53 | 12 |
| Pyrexia | 37 | 1.2 |
| Infections | ||
| Respiratory tract infectionÞ | 53 | 3.5 |
| Bronchitis | 19 | 0 |
| Nasopharyngitis | 18 | 0 |
| Influenza | 17 | 3.5 |
| Gastrointestinal Disorders | ||
| Nausea | 42 | 1.2 |
| Vomiting | 40 | 1.2 |
| Diarrhea | 38 | 2.4 |
| Constipation | 17 | 0 |
| Respiratory, Thoracic and Mediastinal Disorders | ||
| Dyspnea | 35 | 3.5 |
| Coughß | 33 | 0 |
| Vascular Disorders | ||
| Hypertension | 33 | 20 |
| Psychiatric Disorders | ||
| Insomnia | 33 | 4.7 |
| Nervous System Disorders | ||
| Headache | 27 | 1.2 |
| Musculoskeletal and Connective Tissue Disorders | ||
| Back pain | 25 | 0 |
| Pain in extremity | 15 | 0 |
Kyprolis in Combination with Subcutaneous Daratumumab and Dexamethasone
The safety of Kyprolis in combination with daratumumab and hyaluronidase-fihj and dexamethasone was evaluated in PLEIADES [see Clinical Studies (14.3)].
PLEIADES
The safety of Kyprolis in combination with daratumumab and hyaluronidase-fihj and dexamethasone (DKd) was evaluated in a single-arm cohort of PLEIADES. Patients received Kyprolis as a 30-minute IV infusion once weekly for three weeks (Days 1, 8, and 15), followed by a 13-day rest period (Days 16 to 28) and continued until disease progression or unacceptable toxicity (N=66) in combination with daratumumab and hyaluronidase-fihj and dexamethasone. Among these patients, 77% were exposed for 6 months or longer and 27% were exposed for greater than one year.
Serious adverse reactions occurred in 27% of patients who received Kyprolis in combination with daratumumab and hyaluronidase-fihj and dexamethasone. Fatal adverse reactions occurred in 3% of patients who received Kyprolis in combination with daratumumab and hyaluronidase-fihj and dexamethasone.
Permanent discontinuation of Kyprolis due to an adverse reaction occurred in 6% of patients who received Kyprolis.
Dosage interruptions due to an adverse reaction occurred in 46% of patients who received Kyprolis.
The most common adverse reactions (≥20%) were upper respiratory tract infection, fatigue, insomnia, hypertension, diarrhea, cough, dyspnea, headache, pyrexia, nausea and edema peripheral.
Table 15 summarizes the adverse reactions in patients who received Kyprolis with subcutaneous daratumumab and dexamethasone (DKd) in PLEIADES.
| Adverse Reaction | DKd (N=66) |
|
|---|---|---|
| All Grades (%) | Grade ≥3 (%) |
|
|
||
| Infections and infestations | ||
| Upper respiratory tract infection* | 52 | 0 |
| Bronchitis† | 12 | 2‡ |
| General disorders and administration site conditions | ||
| Fatigue§ | 39 | 2‡ |
| Pyrexia | 21 | 2‡ |
| Edema peripheral¶ | 20 | 0 |
| Psychiatric disorders | ||
| Insomnia | 33 | 6‡ |
| Vascular disorders | ||
| Hypertension# | 32 | 21‡ |
| Gastrointestinal disorders | ||
| Diarrhea | 29 | 0 |
| Nausea | 21 | 0 |
| Vomiting | 15 | 0 |
| Respiratory, thoracic and mediastinal disorders | ||
| CoughÞ | 24 | 0 |
| Dyspneaß | 23 | 2‡ |
| Nervous system disorders | ||
| Headache | 23 | 0 |
| Peripheral sensory neuropathy | 11 | 0 |
| Musculoskeletal and connective tissue disorders | ||
| Back pain | 17 | 2‡ |
| Musculoskeletal chest pain | 11 | 0 |
Clinically relevant adverse reactions in <10% of patients who received Kyprolis with subcutaneous daratumumab and dexamethasone include:
- Gastrointestinal disorders: abdominal pain, constipation, pancreatitis
- Infection and infestations: pneumonia, influenza, urinary tract infection, herpes zoster, sepsis
- Metabolism and nutrition disorders: hyperglycemia, decreased appetite, hypocalcemia
- Musculoskeletal and connective tissue disorders: muscle spasms, arthralgia
- Nervous system disorders: paresthesia, dizziness, syncope
- General disorders and administration site conditions: injection site reaction, infusion reactions, chills
- Skin and subcutaneous tissue disorders: rash, pruritus
- Cardiac disorders: cardiac failure
- Vascular disorders: hypotension
Table 16 summarizes the laboratory abnormalities in patients who received Kyprolis with subcutaneous daratumumab and dexamethasone in PLEIADES.
| Laboratory Abnormality | DKd* | |
|---|---|---|
| All Grades (%) | Grades 3-4 (%) | |
|
||
| Decreased platelets | 88 | 18 |
| Decreased lymphocytes | 83 | 50 |
| Decreased leukocytes | 68 | 18 |
| Decreased neutrophils | 55 | 15 |
| Decreased hemoglobin | 47 | 6 |
| Decreased corrected calcium | 45 | 2 |
| Increased alanine aminotransferase (ALT) | 35 | 5 |
Kyprolis in Combination with Isatuximab and Dexamethasone
The safety of Kyprolis in combination with isatuximab and dexamethasone was evaluated in IKEMA, a randomized, open-label clinical trial in patients with previously treated multiple myeloma [see Clinical Studies (14.4)]. Patients received Kyprolis 20/56 mg/m2 twice weekly in combination with isatuximab and dexamethasone (Isa-Kd) (n=177) or Kyprolis and dexamethasone (Kd) (n=122). Among patients receiving Isa-Kd, the median exposure to Kyprolis was 65 weeks.
Serious adverse reactions occurred in 59% of patients receiving Isa-Kd. The most frequent serious adverse reactions in >5% of patients who received Isa-Kd were pneumonia (25%) and upper respiratory tract infections (9%). Adverse reactions with a fatal outcome during treatment were reported in 3.4% of patients in the Isa-Kd group (those occurring in more than 1% of patients were pneumonia occurring in 1.7% and cardiac failure in 1.1% of patients).
Permanent treatment discontinuation due to an adverse reaction (grades 1-4) occurred in 8% of patients who received Isa-Kd. The most frequent adverse reactions requiring permanent discontinuation in patients who received Isa-Kd were infections (2.8%).
Dosage interruptions due to an adverse reaction occurred in 4% of patients who received Kyprolis in the Isa-Kd group. The most frequent adverse reactions requiring dosage interruption in patients who received Kyprolis in the Isa-Kd group were administration site extravasation (1.1%) and infusion site extravasation (1.1%).
Dose reductions or omissions of Kyprolis due to an adverse reaction in the Isa-Kd group occurred in 67% of patients. Adverse reactions which required dose reductions or omissions in >10% of patients who received Kyprolis in the Isa-Kd group were upper respiratory tract infection (12.4%) and hypertension (11.9%).
The most common adverse reactions (≥20%) were upper respiratory tract infection, infusion-related reactions, fatigue, hypertension, diarrhea, pneumonia, dyspnea, insomnia, bronchitis, cough and back pain.
Table 17 summarizes the adverse reactions in IKEMA.
| Adverse Reactions | Isa-Kd (N = 177) | Kd (N = 122) |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
| Isa-Kd = Kyprolis, isatuximab, and dexamethasone | ||||
|
||||
| Infections | ||||
| Upper respiratory tract infection* | 67 | 9 | 57 | 7 |
| Pneumonia† | 36 | 22 | 30 | 18 |
| Bronchitis‡ | 24 | 2.3 | 13 | 0.8 |
| General Disorders and Administration Site Conditions | ||||
| Infusion-related reaction§ | 46 | 0.6 | 3.3 | 0 |
| Fatigue¶ | 42 | 5 | 32 | 3.3 |
| Vascular Disorders | ||||
| Hypertension# | 37 | 21 | 32 | 20 |
| Gastrointestinal Disorders | ||||
| Diarrhea | 36 | 2.8 | 29 | 2.5 |
| Vomiting | 15 | 1.1 | 9 | 0.8 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| DyspneaÞ | 29 | 5 | 24 | 0.8 |
| Coughß | 23 | 0 | 15 | 0 |
Table 18 summarizes the laboratory abnormalities in patients who received Kyprolis with isatuximab and dexamethasone versus Kyprolis with dexamethasone in IKEMA.
| Laboratory Abnormality | Isa-Kd*
(N=177) | Kd (N=122) |
|||
|---|---|---|---|---|---|
| All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
||
|
|||||
| Hemoglobin decreased | 99 | 22 | 99 | 20 | |
| Lymphocytes decreased | 94 | 69 | 95 | 57 | |
| Platelets decreased | 94 | 30 | 88 | 24 | |
| Neutrophils decreased | 55 | 20 | 43 | 8 | |
Kyprolis in Patients who Received Monotherapy
The safety of Kyprolis 20/27 mg/m2 as a 10-minute infusion was evaluated in clinical trials consisting of 598 patients with relapsed and/or refractory myeloma [see Clinical Studies (14.5)]. Premedication with dexamethasone 4 mg was required before each dose in Cycle 1 and was optional for subsequent cycles. The median age was 64 years (range 32–87), and approximately 57% were male. The patients received a median of 5 (range 1–20) prior regimens. The median number of cycles initiated was 4 (range 1–35).
Deaths due to adverse reactions within 30 days of the last dose of Kyprolis occurred in 30/598 (5%) patients receiving Kyprolis monotherapy. These adverse reactions were related to cardiac disorders in 10 (2%) patients, infections in 8 (1%) patients, renal disorders in 4 (< 1%) patients, and other adverse reactions in 8 (1%) patients.
Serious adverse reactions were reported in 50% of patients in the pooled Kyprolis monotherapy studies (N = 598). The most frequent serious adverse reactions were: pneumonia (8%), acute renal failure (5%), disease progression (4%), pyrexia (3%), hypercalcemia (3%), congestive heart failure (3%), multiple myeloma (3%), anemia (2%), and dyspnea (2%).
In FOCUS, a randomized trial comparing Kyprolis as a single agent versus corticosteroids with optional oral cyclophosphamide for patients with relapsed and refractory multiple myeloma, mortality was higher in the patients treated with Kyprolis in comparison to the control arm in the subgroup of 48 patients ≥ 75 years of age. The most common cause of discontinuation due to an adverse reaction was acute renal failure (2%).
Safety of Kyprolis monotherapy dosed at 20/56 mg/m2 by 30-minute infusion was evaluated in a multicenter, open-label study in patients with relapsed and/or refractory multiple myeloma [see Clinical Studies (14.5)]. The patients received a median of 4 (range 1–10) prior regimens.
Adverse reactions occurring with Kyprolis monotherapy are presented in Table 19.
| Adverse Reactions | 20/56 mg/m2 by 30-minute infusion (N = 24) | 20/27 mg/m2 by 2- to 10-minute infusion (N = 598) |
||
|---|---|---|---|---|
| All Grades n (%) | Grades 3-5 n (%) | All Grades n (%) | Grades 3-5 n (%) |
|
|
||||
| Fatigue | 14 (58) | 2 (8) | 238 (40) | 25 (4) |
| Dyspnea* | 14 (58) | 2 (8) | 202 (34) | 21 (4) |
| Pyrexia | 14 (58) | 0 | 177 (30) | 11 (2) |
| Thrombocytopenia | 13 (54) | 13 (54) | 220 (37) | 152 (25) |
| Nausea | 13 (54) | 0 | 211 (35) | 7 (1) |
| Anemia | 10 (42) | 7 (29) | 291 (49) | 141 (24) |
| Hypertension† | 10 (42) | 3 (13) | 90 (15) | 22 (4) |
| Chills | 9 (38) | 0 | 73 (12) | 1 (< 1) |
| Headache | 8 (33) | 0 | 141 (24) | 7 (1) |
| Cough‡ | 8 (33) | 0 | 134 (22) | 2 (< 1) |
| Vomiting | 8 (33) | 0 | 104 (17) | 4 (1) |
| Lymphopenia | 8 (33) | 8 (33) | 85 (14) | 73 (12) |
| Insomnia | 7 (29) | 0 | 75 (13) | 0 |
| Dizziness | 7 (29) | 0 | 64 (11) | 5 (1) |
| Diarrhea | 6 (25) | 1 (4) | 160 (27) | 8 (1) |
| Blood creatinine increased | 6 (25) | 1 (4) | 103 (17) | 15 (3) |
| Peripheral edema | 5 (21) | 0 | 118 (20) | 1 (< 1) |
| Back pain | 5 (21) | 1 (4) | 115 (19) | 19 (3) |
| Upper respiratory tract infection | 5 (21) | 1 (4) | 112 (19) | 15 (3) |
| Decreased appetite | 5 (21) | 0 | 89 (15) | 2 (< 1) |
| Muscle spasms | 5 (21) | 0 | 62 (10) | 2 (< 1) |
| Chest pain | 5 (21) | 0 | 20 (3) | 1 (< 1) |
Adverse Reactions Occurring at a Frequency of < 20%
- Blood and lymphatic system disorders: febrile neutropenia, leukopenia, neutropenia
- Cardiac disorders: cardiac arrest, cardiac failure, cardiac failure congestive, myocardial infarction, myocardial ischemia
- Ear and labyrinth disorders: tinnitus
- Eye disorders: cataract, blurred vision
- Gastrointestinal disorders: abdominal pain, abdominal pain upper, constipation, dyspepsia, gastrointestinal hemorrhage, toothache
- General disorders and administration site conditions: asthenia, infusion site reaction, multi-organ failure, pain
- Hepatobiliary disorders: hepatic failure
- Infections: bronchitis, bronchopneumonia, influenza, lung infection, pneumonia, nasopharyngitis, respiratory tract infection, rhinitis, sepsis, urinary tract infection
- Metabolism and nutrition disorders: hypercalcemia, hyperglycemia, hyperkalemia, hyperuricemia, hypoalbuminemia, hypocalcemia, hypokalemia, hypomagnesemia, hyponatremia, hypophosphatemia, tumor lysis syndrome
- Musculoskeletal and connective tissue disorders: arthralgia, musculoskeletal pain, musculoskeletal chest pain, myalgia, pain in extremity
- Nervous system disorders: hypoesthesia, intracranial hemorrhage, paresthesia, peripheral motor neuropathy, peripheral neuropathy, peripheral sensory neuropathy
- Psychiatric disorders: anxiety
- Renal and urinary disorders: acute renal failure, renal failure, renal impairment
- Respiratory, thoracic and mediastinal disorders: dysphonia, epistaxis, oropharyngeal pain, pulmonary edema, pulmonary hemorrhage
- Skin and subcutaneous tissue disorders: erythema, hyperhidrosis, pruritus, rash
- Vascular disorders: embolic and thrombotic events, venous (including deep vein thrombosis and pulmonary embolism), hemorrhage, hypotension
Grade 3 and higher adverse reactions occurring at an incidence of > 1% include febrile neutropenia, cardiac arrest, cardiac failure congestive, pain, sepsis, urinary tract infection, hyperglycemia, hyperkalemia, hyperuricemia, hypoalbuminemia, hypocalcemia, hyponatremia, hypophosphatemia, renal failure, renal failure acute, renal impairment, pulmonary edema, and hypotension.
Table 20 describes Grade 3–4 laboratory abnormalities reported at a rate of > 10% for patients who received Kyprolis monotherapy.
| Laboratory Abnormality | Kyprolis 20/56 mg/m2 (N = 24) | Kyprolis 20/27 mg/m2 (N = 598) |
|---|---|---|
| Decreased lymphocytes | 15 (63) | 151 (25) |
| Decreased platelets | 11 (46) | 184 (31) |
| Decreased hemoglobin | 7 (29) | 132 (22) |
| Decreased total white blood cell count | 3 (13) | 71 (12) |
| Decreased sodium | 2 (8) | 69 (12) |
| Decreased absolute neutrophil count | 2 (8) | 67 (11) |
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Kyprolis. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure: hemolytic uremic syndrome (HUS), hepatitis B virus reactivation, gastrointestinal perforation, pericarditis, and cytomegalovirus infection, including chorioretinitis, pneumonitis, enterocolitis, viremia, intestinal obstruction, and acute pancreatitis.
8. Use In Specific Populations
8.3 Females and Males of Reproductive Potential
Based on its mechanism of action and findings in animals, Kyprolis can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of Kyprolis in pediatric patients have not been established.
8.5 Geriatric Use
Of the 2,837 patients with relapsed or refractory multiple myeloma exposed to Kyprolis in monotherapy and combination therapy studies [see Clinical Studies (14.1, 14.2, 14.3, 14.4, 14.5)], 50% were 65 years and older, while 13% were 75 years and older. The incidence of serious adverse reactions was 50% in patients < 65 years of age, 60% in patients 65 to 74 years of age, and 63% in patients ≥ 75 years of age. Of the 308 patients in CANDOR who received DKd, 47% of patients were 65 years and older, while 9% were 75 years and older. Fatal adverse reactions in the DKd arm of CANDOR occurred in 6% of patients <65 years of age, 14% of patients between 65 to 74 years of age, and 14% of patients ≥ 75 years of age [see Adverse Reactions (6.1)]. No overall differences in effectiveness were observed between older and younger patients.
8.6 Hepatic Impairment
Reduce the dose of Kyprolis by 25% in patients with mild (total bilirubin 1 to 1.5 × ULN and any AST or total bilirubin ≤ ULN and AST > ULN) or moderate (total bilirubin > 1.5 to 3 × ULN and any AST) hepatic impairment. A recommended dosage of Kyprolis has not been established for patients with severe hepatic impairment (total bilirubin > 3 × ULN and any AST) [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
The incidence of serious adverse reactions was higher in patients with mild, moderate, and severe hepatic impairment combined (22/35 or 63%) than in patients with normal hepatic function (3/11 or 27%) [see Warnings and Precautions (5.12), Clinical Pharmacology (12.3)].
10. Overdosage
Acute onset of chills, hypotension, renal insufficiency, thrombocytopenia, and lymphopenia has been reported following a dose of 200 mg of Kyprolis administered in error.
There is no known specific antidote for Kyprolis overdosage. In the event of overdose, monitor patients for adverse reactions and provide supportive care as appropriate.
11. Kyprolis Description
Carfilzomib is a proteasome inhibitor. The chemical name for carfilzomib is (2S)-N-((S)-1-((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-ylcarbamoyl)-2-phenylethyl)-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)-4-methylpentanamide. Carfilzomib has the following structure:
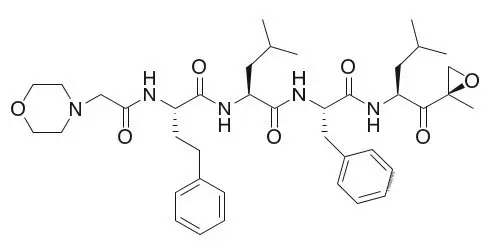
Carfilzomib is a crystalline substance with a molecular weight of 719.9. The molecular formula is C40H57N5O7. Carfilzomib is practically insoluble in water and very slightly soluble in acidic conditions.
Kyprolis for injection, for intravenous use is a sterile, white to off-white lyophilized powder in a single-dose vial. Each 10 mg vial contains 10 mg of carfilzomib, 500 mg sulfobutylether beta-cyclodextrin, and 9.6 mg anhydrous citric acid and sodium hydroxide for pH adjustment (target pH 3.5). Each 30 mg vial contains 30 mg of carfilzomib, 1500 mg sulfobutylether beta-cyclodextrin, and 28.8 mg anhydrous citric acid and sodium hydroxide for pH adjustment (target pH 3.5). Each 60 mg vial contains 60 mg of carfilzomib, 3000 mg sulfobutylether beta-cyclodextrin, 57.7 mg citric acid, and sodium hydroxide for pH adjustment (target pH 3.5).
12. Kyprolis - Clinical Pharmacology
12.1 Mechanism of Action
Carfilzomib is a tetrapeptide epoxyketone proteasome inhibitor that irreversibly binds to the N-terminal threonine-containing active sites of the 20S proteasome, the proteolytic core particle within the 26S proteasome. Carfilzomib had antiproliferative and proapoptotic activities in vitro in solid and hematologic tumor cells. In animals, carfilzomib inhibited proteasome activity in blood and tissue and delayed tumor growth in models of multiple myeloma, hematologic, and solid tumors.
12.2 Pharmacodynamics
Intravenous carfilzomib administration resulted in suppression of proteasome chymotrypsin-like (CT-L) activity when measured in blood 1 hour after the first dose. Doses of carfilzomib ≥ 15 mg/m2 with or without lenalidomide and dexamethasone induced a ≥ 80% inhibition of the CT-L activity of the proteasome. In addition, carfilzomib, 20 mg/m2 intravenously as a single agent, resulted in a mean inhibition of the low molecular mass polypeptide 2 (LMP2) and multicatalytic endopeptidase complex-like 1 (MECL1) subunits of the proteasome ranging from 26% to 32% and 41% to 49%, respectively. Proteasome inhibition was maintained for ≥ 48 hours following the first dose of carfilzomib for each week of dosing.
12.3 Pharmacokinetics
Carfilzomib at doses between 20 mg/m2 and 70 mg/m2 administered as a 30-minute infusion resulted in dose-dependent increases in maximum plasma concentrations (Cmax) and area under the curve over time to infinity (AUC0-INF) in patients with multiple myeloma. A dose-dependent increase in Cmax and AUC0-INF was also observed between carfilzomib 20 mg/m2 and 56 mg/m2 as a 2- to 10-minute infusion in patients with relapsed or refractory multiple myeloma. A 30-minute infusion resulted in a similar AUC0-INF, but 2- to 3-fold lower Cmax than that observed with a 2- to 10-minute infusion at the same dose. There was no evidence of carfilzomib accumulation following repeated administration of carfilzomib 70 mg/m2 as a 30-minute once weekly infusion or 15 and 20 mg/m2 as a 2- to 10-minute twice weekly infusion.
Table 21 lists the estimated mean average daily area under the curve in the first cycle (AUCC1,avg), average daily area under the curve at steady-state (AUCss) and Cmax at the highest dose in the first cycle (Cmax,C1) for the different dosing regimens.
| Estimated Parameters (%CV) | 20/27 mg/m2 twice weekly with 2- to 10-minute infusion | 20/56 mg/m2 twice weekly with 30-minute infusion | 20/70 mg/m2 once weekly with 30-minute infusion |
|---|---|---|---|
| CV = Coefficient of variation | |||
| AUCC1,avg (ng∙hr/mL) | 95 (40) | 170 (35) | 114 (36) |
| AUCss (ng∙hr/mL) | 111 (34) | 228 (28) | 150 (35) |
| Cmax,C1 (ng/mL) | 1282 (17) | 1166 (29) | 1595 (36) |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with carfilzomib.
Carfilzomib was clastogenic in the in vitro chromosomal aberration test in peripheral blood lymphocytes. Carfilzomib was not mutagenic in the in vitro bacterial reverse mutation (Ames) test and was not clastogenic in the in vivo mouse bone marrow micronucleus assay.
Fertility studies with carfilzomib have not been conducted. No effects on reproductive tissues were noted during 28-day repeat-dose rat and monkey toxicity studies or in 6-month rat and 9-month monkey chronic toxicity studies.
14. Clinical Studies
14.1 In Combination with Lenalidomide and Dexamethasone for Relapsed or Refractory Multiple Myeloma
ASPIRE (NCT01080391)
ASPIRE was a randomized, open-label, multicenter trial which evaluated the combination of Kyprolis with lenalidomide and dexamethasone (KRd) versus lenalidomide and dexamethasone alone (Rd) in patients with relapsed or refractory multiple myeloma who had received 1 to 3 lines of therapy (A line of therapy is a planned course of treatment [including sequential induction, transplantation, consolidation, and/or maintenance] without an interruption for lack of efficacy, such as for relapse or progressive disease). Patients who had the following were excluded from the trial: refractory to bortezomib in the most recent regimen, refractory to lenalidomide and dexamethasone in the most recent regimen, not responding to any prior regimen, creatinine clearance < 50 mL/min, ALT/AST > 3.5 × ULN and bilirubin > 2 × ULN, New York Heart Association Class III to IV congestive heart failure, or myocardial infarction within the last 4 months.
In the KRd arm, Kyprolis was evaluated at a starting dose of 20 mg/m2, which was increased to 27 mg/m2 on Cycle 1, Day 8 onward. Kyprolis was administered as a 10-minute infusion on Days 1, 2, 8, 9, 15, and 16 of each 28-day cycle for Cycle 1 through 12. Kyprolis was dosed on Days 1, 2, 15, and 16 of each 28-day cycle from Cycle 13 through 18. Dexamethasone 40 mg was administered orally or intravenously on Days 1, 8, 15 and 22 of each cycle. Lenalidomide was given 25 mg orally on Days 1 to 21 of each 28-day cycle. The Rd treatment arm had the same regimen for lenalidomide and dexamethasone as the KRd treatment arm. Kyprolis was administered for a maximum of 18 cycles unless discontinued early for disease progression or unacceptable toxicity. Lenalidomide and dexamethasone administration could continue until progression or unacceptable toxicity. Concurrent use of thromboprophylaxis and a proton pump inhibitor were required for both arms and antiviral prophylaxis was required for the KRd arm.
The 792 patients in ASPIRE were randomized 1:1 to the KRd or Rd arm. The demographics and baseline characteristics were well-balanced between the two arms (see Table 22). Only 53% of the patients had testing for genetic mutations; a high-risk genetic mutation was identified for 12% of patients in the KRd arm and in 13% in the Rd arm.
| Characteristics | KRd (N = 396) | Rd (N = 396) |
|---|---|---|
| ECOG = Eastern Cooperative Oncology Group; IgG = immunoglobulin G; ISS = International Staging System; KRd = Kyprolis, lenalidomide, and dexamethasone; Rd = lenalidomide and dexamethasone | ||
|
||
| Age, Median, Years (min, max) | 64 (38, 87) | 65 (31, 91) |
| Age ≥ 75 Years, n (%) | 43 (11) | 53 (13) |
| Males, n (%) | 215 (54) | 232 (59) |
| Race, n (%) | ||
| White | 377 (95) | 377 (95) |
| Black | 12 (3) | 11 (3) |
| Other or Not Reported | 7 (2) | 8 (2) |
| Number of Prior Regimens, n (%) | ||
| 1 | 184 (46) | 157 (40) |
| 2 | 120 (30) | 139 (35) |
| 3* | 92 (23) | 100 (25) |
| Prior Transplantation, n (%) | 217 (55) | 229 (58) |
| ECOG Performance Status, n (%) | ||
| 0 | 165 (42) | 175 (44) |
| 1 | 191 (48) | 186 (47) |
| 2 | 40 (10) | 35 (9) |
| ISS Stage at Study Baseline, n (%) | ||
| I | 167 (42) | 154 (39) |
| II | 148 (37) | 153 (39) |
| III | 73 (18) | 82 (21) |
| Unknown | 8 (2) | 7 (2) |
| Creatinine Clearance mL/min, Median (min, max) | 79 (39, 212) | 79 (30, 208) |
| 30 to < 50, n (%) | 19 (5) | 32 (8) |
| 50 to < 80, n (%) | 185 (47) | 170 (43) |
| Refractory to Last Therapy, n (%) | 110 (28) | 119 (30) |
| Refractory at Any Time to, n (%): | ||
| Bortezomib | 60 (15) | 58 (15) |
| Lenalidomide | 29 (7) | 28 (7) |
| Bortezomib + immunomodulatory agent | 24 (6) | 27 (7) |
Patients in the KRd arm demonstrated improved PFS compared with those in the Rd arm (HR = 0.69, with 2-sided P-value = 0.0001) as determined using standard International Myeloma Working Group (IMWG)/European Blood and Marrow Transplantation (EBMT) response criteria by an Independent Review Committee (IRC). The median PFS was 26.3 months in the KRd arm versus 17.6 months in the Rd arm (see Table 23 and Figure 1).
A pre-planned overall survival (OS) analysis was performed after 246 deaths in the KRd arm and 267 deaths in the Rd arm. The median follow-up was approximately 67 months. A statistically significant advantage in OS was observed in patients in the KRd arm compared to patients in the Rd arm (see Table 23 and Figure 2).
| Combination Therapy | ||
|---|---|---|
| KRd (N = 396) | Rd (N = 396) |
|
| CI = confidence interval; CR = complete response; HR = hazard ratio; KRd = Kyprolis, lenalidomide, and dexamethasone; ORR = overall response rate; PFS = progression-free survival; PR = partial response; Rd = lenalidomide and dexamethasone; sCR = stringent CR; VGPR = very good partial response | ||
|
||
| PFS† | ||
| Median‡, Months (95% CI) | 26.3 (23.3, 30.5) | 17.6 (15.0, 20.6) |
| HR (95% CI)§ | 0.69 (0.57, 0.83) | |
| P-value (2-sided)¶ | 0.0001 | |
| Overall Survival | ||
| Median‡, Months (95% CI) | 48.3 (42.4, 52.8) | 40.4 (33.6, 44.4) |
| HR (95% CI)§ | 0.79 (0.67, 0.95) | |
| P-value (2-sided)¶ | 0.0091 | |
| Overall Response† | ||
| N with response | 345 | 264 |
| ORR (%) (95% CI)# | 87 (83, 90) | 67 (62, 71) |
| P-value (2-sided)Þ | < 0.0001 | |
| Response Category, n (%) | ||
| sCR | 56 (14) | 17 (4) |
| CR | 70 (18) | 20 (5) |
| VGPR | 151 (38) | 123 (31) |
| PR | 68 (17) | 104 (26) |
The median duration of response (DOR) was 28.6 months (95% CI: 24.9, 31.3) for the 345 patients achieving a response in the KRd arm and 21.2 months (95% CI: 16.7, 25.8) for the 264 patients achieving a response in the Rd arm. The median time to response was 1 month (range 1 to 14 months) in the KRd arm and 1 month (range 1 to 16 months) in the Rd arm.
| CI = confidence interval; EBMT = European Blood and Marrow Transplantation; HR = hazard ratio; IMWG = International Myeloma Working Group; KRd = Kyprolis, lenalidomide, and dexamethasone; mo = months; PFS = progression-free survival; Rd = lenalidomide and dexamethasone arm Note: The response and PD outcomes were determined using standard objective IMWG/EBMT response criteria. |
|
Figure 1: Kaplan-Meier Curve of Progression-Free Survival in ASPIRE |
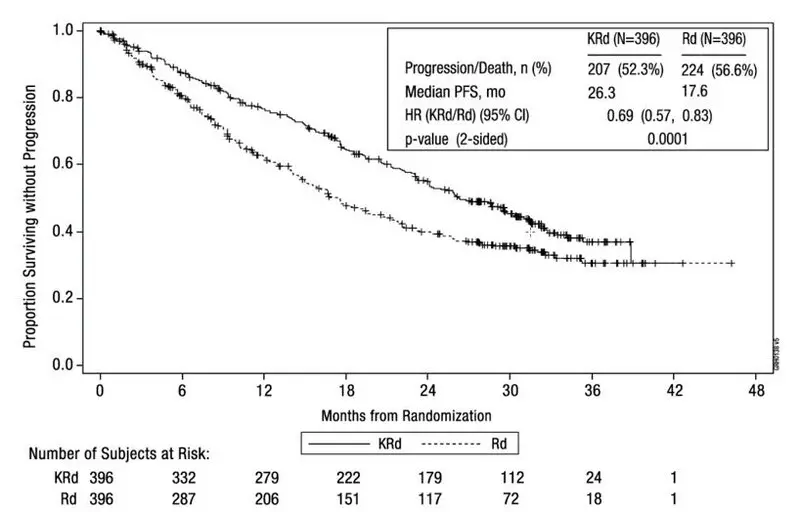 |
| CI = confidence interval; HR = hazard ratio; KRd = Kyprolis, lenalidomide, and dexamethasone; mo = months; OS = overall survival; Rd = lenalidomide and dexamethasone arm |
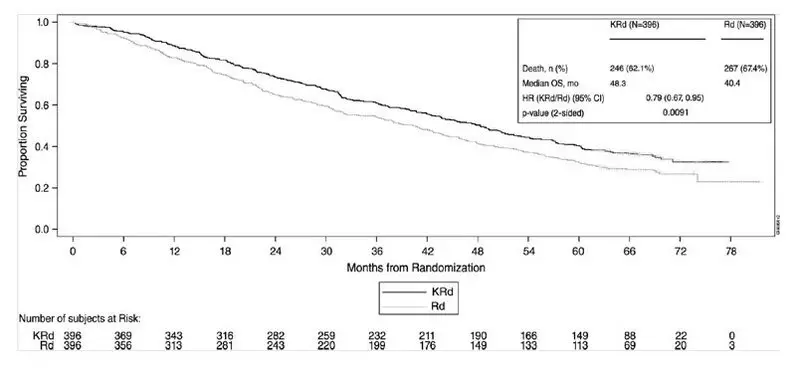 |
14.2 In Combination with Dexamethasone for Relapsed or Refractory Multiple Myeloma
The efficacy of Kyprolis in combination with dexamethasone was evaluated in two open-label randomized trials (ENDEAVOR and A.R.R.O.W.).
ENDEAVOR (NCT01568866)
ENDEAVOR was a randomized, open-label, multicenter trial of Kyprolis and dexamethasone (Kd) versus bortezomib and dexamethasone (Vd) in patients with relapsed or refractory multiple myeloma who had received 1 to 3 lines of therapy. A total of 929 patients were enrolled and randomized (464 in the Kd arm; 465 in the Vd arm). Randomization was stratified by prior proteasome inhibitor therapy (yes versus no), prior lines of therapy (1 versus 2 or 3), current International Staging System stage (1 versus 2 or 3), and planned route of bortezomib administration. Patients were excluded if they had less than PR to all prior regimens; creatinine clearance < 15 mL/min; hepatic transaminases ≥ 3 × ULN; or left-ventricular ejection fraction < 40% or other significant cardiac conditions.
This trial evaluated Kyprolis at a starting dose of 20 mg/m2, which was increased to 56 mg/m2 on Cycle 1, Day 8 onward. Kyprolis was administered twice weekly as a 30-minute infusion on Days 1, 2, 8, 9, 15, and 16 of each 28-day cycle. Dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 8, 9, 15, 16, 22, and 23 of each cycle. In the Vd arm, bortezomib was dosed at 1.3 mg/m2 intravenously or subcutaneously on Days 1, 4, 8, and 11 of a 21-day cycle, and dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 4, 5, 8, 9, 11, and 12 of each cycle. Concurrent use of thromboprophylaxis was optional, and prophylaxis with an antiviral agent and proton pump inhibitor was required. Of the 465 patients in the Vd arm, 381 received bortezomib subcutaneously. Treatment continued until disease progression or unacceptable toxicity.
The demographics and baseline characteristics are summarized in Table 24.
| Characteristics | Kd (N = 464) | Vd (N = 465) |
|---|---|---|
| ECOG = Eastern Cooperative Oncology Group; FISH = Fluorescence in situ hybridization; ISS = International Staging System; Kd = Kyprolis and dexamethasone; Vd = bortezomib and dexamethasone | ||
|
||
| Age, Years | ||
| Median (min, max) | 65 (35, 89) | 65 (30, 88) |
| < 65, n (%) | 223 (48) | 210 (45) |
| 65 – 74, n (%) | 164 (35) | 189 (41) |
| ≥ 75, n (%) | 77 (17) | 66 (14) |
| Sex, n (%) | ||
| Female | 224 (48) | 236 (51) |
| Male | 240 (52) | 229 (49) |
| Race, n (%) | ||
| White | 353 (76) | 361 (78) |
| Black | 7 (2) | 9 (2) |
| Asian | 56 (12) | 57 (12) |
| Other or Not Reported | 48 (10) | 38 (8) |
| ECOG Performance Status, n (%) | ||
| 0 | 221 (48) | 232 (50) |
| 1 | 210 (45) | 203 (44) |
| 2 | 33 (7) | 30 (6) |
| Creatinine Clearance (mL/min) | ||
| Median (min, max) | 73 (14, 185) | 72 (12, 208) |
| < 30, n (%) | 28 (6) | 28 (6) |
| 30 – < 50, n (%) | 57 (12) | 71 (15) |
| 50 – < 80, n (%) | 186 (40) | 177 (38) |
| ≥ 80, n (%) | 193 (42) | 189 (41) |
| FISH, n (%) | ||
| High-risk | 97 (21) | 113 (24) |
| Standard-risk | 284 (61) | 291 (63) |
| Unknown-risk | 83 (18) | 61 (13) |
| ISS Stage at Study Baseline, n (%) | ||
| ISS I | 219 (47) | 212 (46) |
| ISS II | 138 (30) | 153 (33) |
| ISS III | 107 (23) | 100 (22) |
| Number of Prior Regimens, n (%) | ||
| 1 | 232 (50) | 231 (50) |
| 2 | 158 (34) | 144 (31) |
| 3 | 74 (16) | 88 (19) |
| 4 | 0 (0) | 2 (0.4) |
| Prior Therapies, n (%) | 464 (100) | 465 (100) |
| Bortezomib | 250 (54) | 252 (54) |
| Transplant for Multiple Myeloma | 266 (57) | 272 (59) |
| Thalidomide | 212 (46) | 249 (54) |
| Lenalidomide | 177 (38) | 178 (38) |
| Bortezomib + immunomodulatory agent | 159 (34) | 168 (36) |
| Refractory to last prior therapy, n (%)* | 184 (40) | 189 (41) |
The efficacy of Kyprolis was evaluated by PFS as determined by an IRC using IMWG response criteria. The trial showed a median PFS of 18.7 months in the Kd arm versus 9.4 months in the Vd arm (see Table 25 and Figure 3).
Figure 3: Kaplan-Meier Plot of Progression-Free Survival in ENDEAVOR
| CI = confidence interval; HR = hazard ratio; Kd = Kyprolis and dexamethasone; mo = months; PFS = progression-free survival; Vd = bortezomib and dexamethasone |
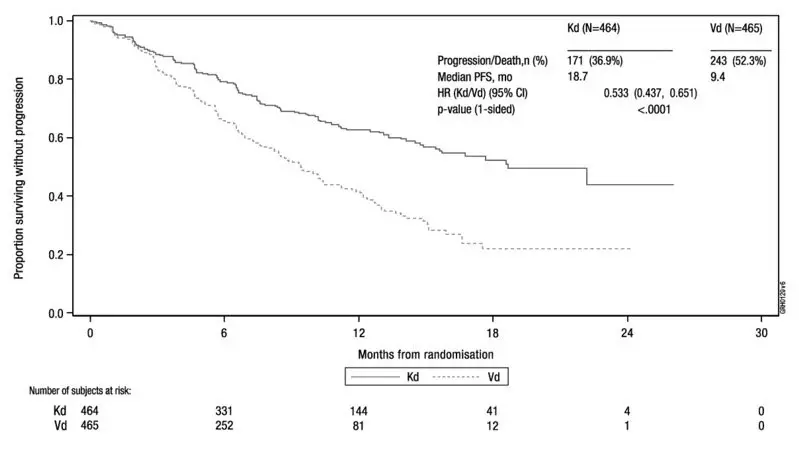 |
Other endpoints included OS and overall response rate (ORR).
A pre-planned OS analysis was performed after 189 deaths in the Kd arm and 209 deaths in the Vd arm. The median follow-up was approximately 37 months. A significantly longer OS was observed in patients in the Kd arm compared to patients in the Vd arm (HR = 0.79; 95% CI: 0.65, 0.96; P-value = 0.01). Results are provided in Table 25 and Figure 4.
| Kd (N = 464) | Vd (N = 465) |
|
|---|---|---|
| CI = confidence interval; CR = complete response; HR= hazard ratio; Kd = Kyprolis and dexamethasone; ORR = overall response rate; PFS = progression-free survival; PR = partial response; sCR = stringent CR; Vd = bortezomib and dexamethasone; VGPR = very good partial response; NE = non-estimable | ||
|
||
| PFS† | ||
| Number of events (%) | 171 (37) | 243 (52) |
| Median‡, Months (95% CI) | 18.7 (15.6, NE) | 9.4 (8.4, 10.4) |
| HR (Kd/Vd) (95% CI)§ | 0.53 (0.44, 0.65) | |
| P-value (1-sided)¶ | < 0.0001 | |
| Overall Survival | ||
| Number of deaths (%) | 189 (41) | 209 (45) |
| Median‡, Months (95% CI) | 47.6 (42.5, NE) | 40.0 (32.6, 42.3) |
| HR (Kd/Vd) (95% CI)§ | 0.79 (0.65, 0.96) | |
| P-value (1-sided)¶ | 0.01 | |
| Overall Response† | ||
| N with Response | 357 | 291 |
| ORR (%) (95% CI)# | 77 (73, 81) | 63 (58, 67) |
| P-value (1-sided)Þ | < 0.0001 | |
| Response Category, n (%) | ||
| sCR | 8 (2) | 9 (2) |
| CR | 50 (11) | 20 (4) |
| VGPR | 194 (42) | 104 (22) |
| PRß | 105 (23) | 158 (34) |
| CI = confidence interval; HR = hazard ratio; Kd = Kyprolis and dexamethasone; mo = month; OS = overall survival; Vd = bortezomib and dexamethasone |
|
Figure 4: Kaplan-Meier Plot of Overall Survival in ENDEAVOR |
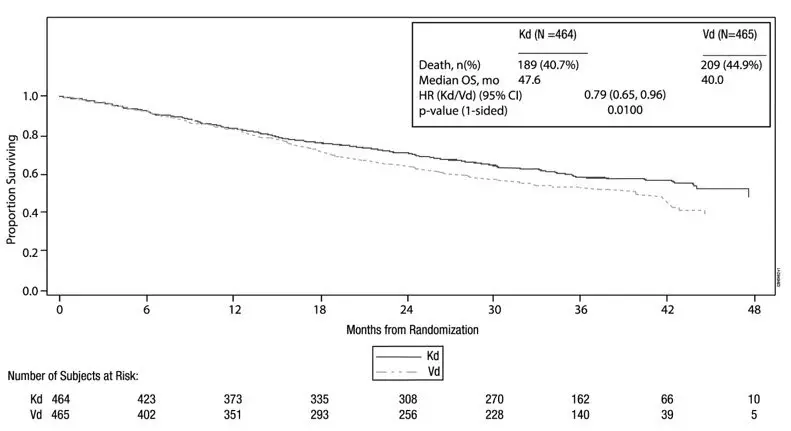 |
The median DOR in subjects achieving PR or better was 21.3 months (95% CI: 21.3, not estimable) in the Kd arm and 10.4 months (95% CI: 9.3, 13.8) in the Vd arm. The median time to response was 1 month (range < 1 to 8 months) in both arms.
A.R.R.O.W. (NCT02412878)
A.R.R.O.W. was a randomized, open-label, multicenter superiority trial of Kyprolis and dexamethasone (Kd) once weekly (20/70 mg/m2) versus Kd twice weekly (20/27 mg/m2) in patients with relapsed and refractory multiple myeloma who had received 2 to 3 prior lines of therapy. Patients were excluded if they had less than PR to at least one prior line; creatinine clearance < 30 mL/min; hepatic transaminases ≥ 3 × ULN; or left-ventricular ejection fraction < 40% or other significant cardiac conditions. A total of 478 patients were enrolled and randomized (240 in 20/70 mg/m2 arm; 238 in 20/27 mg/m2 arm). Randomization was stratified by current International Staging System stage (stage 1 versus stages 2 or 3), refractory to bortezomib treatment (yes versus no), and age (< 65 versus ≥ 65 years).
Arm 1 of this trial evaluated Kyprolis at a starting dose of 20 mg/m2, which was increased to 70 mg/m2 on Cycle 1, Day 8 onward. Arm 1 Kyprolis was administered once weekly as a 30-minute infusion on Days 1, 8 and 15, of each 28-day cycle. Arm 2 of this trial evaluated Kyprolis at a starting dose of 20 mg/m2, which was increased to 27 mg/m2 on Cycle 1, Day 8 onward. Arm 2 Kyprolis was administered twice weekly as a 10-minute infusion on Days 1, 2, 8, 9, 15, and 16 of each 28-day cycle. In both regimens, dexamethasone 40 mg was administered orally or intravenously on Days 1, 8, 15 for all cycles and on Day 22 for cycles 1 to 9 only. Concurrent use of thromboprophylaxis was optional, prophylaxis with an antiviral agent was recommended, and prophylaxis with a proton pump inhibitor was required. Treatment continued until disease progression or unacceptable toxicity.
The demographics and baseline characteristics are summarized in Table 26.
| Characteristics | Once weekly Kd 20/70 mg/m2 (N = 240) | Twice weekly Kd 20/27 mg/m2 (N = 238) |
|---|---|---|
| ECOG = Eastern Cooperative Oncology Group; FISH = Fluorescence in situ hybridization; ISS = International Staging System; Kd = Kyprolis and dexamethasone | ||
| Age, Years | ||
| Median (min, max) | 66 (39, 85) | 66 (35, 83) |
| < 65, n (%) | 104 (43) | 104 (44) |
| 65 – 74, n (%) | 90 (38) | 102 (43) |
| ≥ 75, n (%) | 46 (19) | 32 (13) |
| Sex, n (%) | ||
| Female | 108 (45) | 110 (46) |
| Male | 132 (55) | 128 (54) |
| Race, n (%) | ||
| White | 200 (83) | 202 (85) |
| Black | 3 (1) | 2 (1) |
| Asian | 30 (13) | 15 (6) |
| Other or Not Reported | 7 (3) | 19 (8) |
| ECOG Performance Status, n (%) | ||
| 0 | 118 (49) | 118 (50) |
| 1 | 121 (50) | 120 (50) |
| 2 | 1 (0.4) | 0 (0) |
| Creatinine Clearance (mL/min) | ||
| Median (min, max) | 70.80 (28, 212) | 73.20 (29, 181) |
| < 30, n (%) | 2 (1) | 1 (0.4) |
| 30 – < 50, n (%) | 48 (20) | 34 (14) |
| 50 – < 80, n (%) | 91 (38) | 111 (47) |
| ≥ 80, n (%) | 99 (41) | 91 (38) |
| FISH, n (%) | ||
| High-risk | 34 (14) | 47 (20) |
| Standard-risk | 47 (20) | 53 (22) |
| Unknown-risk | 159 (66) | 138 (58) |
| ISS Stage at Study Baseline, n (%) | ||
| ISS I | 94 (39) | 99 (42) |
| ISS II | 80 (33) | 81 (34) |
| ISS III | 63 (26) | 54 (23) |
| Number of Prior Regimens, n (%) | ||
| 2 | 116 (48) | 125 (53) |
| 3 | 124 (52) | 112 (47) |
| >3 | 0 (0) | 1 (0.4) |
| Prior Therapies, n (%) | ||
| Bortezomib | 236 (98) | 237 (100) |
| Transplantation | 146 (61) | 157 (66) |
| Thalidomide | 119 (50) | 119 (50) |
| Lenalidomide | 207 (86) | 194 (82) |
The efficacy of Kyprolis was evaluated by PFS using IMWG response criteria. Efficacy results are provided in Table 27 and Figure 5.
| CI = confidence interval; HR = hazard ratio; Kd = Kyprolis and dexamethasone; PFS = progression-free survival |
|
Figure 5: Kaplan-Meier Plot of Progression-Free Survival in A.R.R.O.W. |
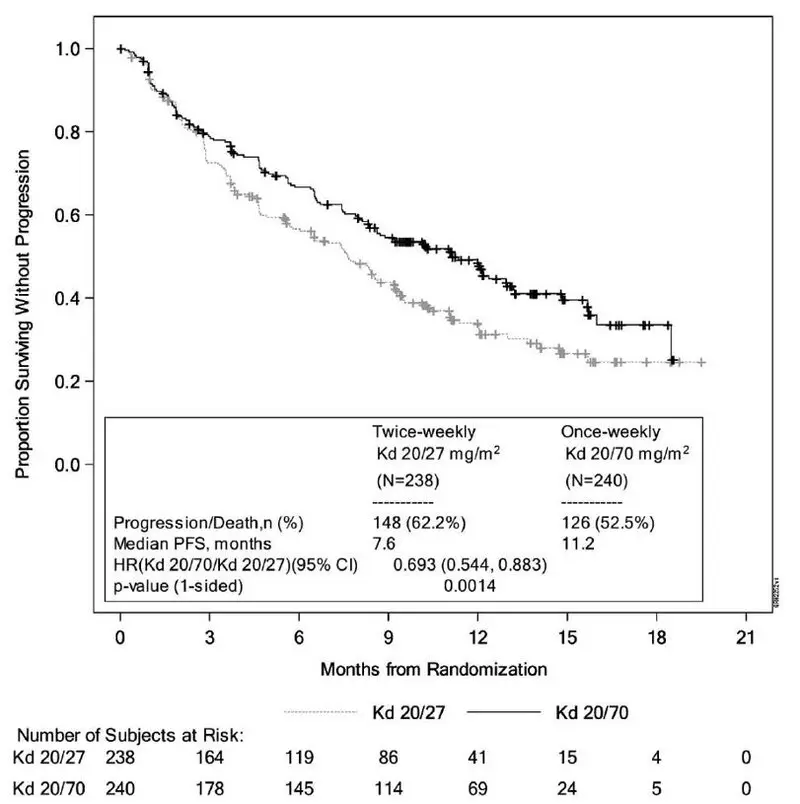 |
| Once weekly Kd 20/70 mg/m2 (N = 240) | Twice weekly Kd 20/27 mg/m2 (N = 238) |
|
|---|---|---|
| CI = confidence interval; CR = complete response; HR = hazard ratio; Kd = Kyprolis and dexamethasone; ORR = overall response rate; PFS = progression-free survival; PR = partial response; sCR = stringent complete response; VGPR = very good partial response | ||
|
||
| PFS | ||
| Number of events, n (%) | 126 (52.5) | 148 (62.2) |
| Median, Months (95% CI) | 11.2 (8.6, 13.0) | 7.6 (5.8, 9.2) |
| HR (95% CI) | 0.69 (0.54, 0.88) | |
| P-value (1-sided) | 0.0014 | |
| Overall Response* | ||
| N with Response | 151 | 97 |
| ORR (%) (95% CI) | 62.9 (56.5, 69.0) | 40.8 (34.5, 47.3) |
| P-value (1-sided) | < 0.0001 | |
| Response Category, n (%) | ||
| sCR | 4 (1.7) | 0 (0.0) |
| CR | 13 (5.4) | 4 (1.7) |
| VGPR | 65 (27.1) | 28 (11.8) |
| PR | 69 (28.8) | 65 (27.3) |
The median DOR in subjects achieving PR or better was 15 months (95% CI: 12.2, not estimable) in the Kd 20/70 mg/m2 arm and 13.8 months (95% CI: 9.5, not estimable) in the Kd 20/27 mg/m2 arm. The median time to response was 1.1 months in the Kd 20/70 mg/m2 arm and 1.9 months in the Kd 20/27 mg/m2 arm.
Kyprolis is not approved for twice weekly 20/27 mg/m2 administration in combination with dexamethasone alone.
14.3 In Combination with Daratumumab and Dexamethasone for Relapsed or Refractory Multiple Myeloma
The efficacy of Kyprolis in combination with daratumumab and dexamethasone or daratumumab and hyaluronidase-fihj and dexamethasone (DKd) was evaluated in three open-label clinical trials (CANDOR, EQUULEUS, and PLEIADES).
CANDOR (NCT03158688)
CANDOR was a randomized, open-label, multicenter trial which evaluated the combination of Kyprolis 20/56 mg/m2 twice weekly with intravenous daratumumab and dexamethasone (DKd) versus Kyprolis 20/56 mg/m2 twice weekly and dexamethasone (Kd) in patients with relapsed or refractory multiple myeloma who had received 1 to 3 prior lines of therapy. Patients who had the following were excluded from the trial: known moderate or severe persistent asthma within the past 2 years, known chronic obstructive pulmonary disease (COPD) with a FEV1 < 50% of predicted normal, and active congestive heart failure. Randomization was stratified by the ISS (stage 1 or 2 vs stage 3) at screening, prior proteasome inhibitor exposure (yes vs no), number of prior lines of therapy (1 vs ≥ 2), or prior cluster differentiation antigen 38 (CD38) antibody therapy (yes vs no).
Kyprolis was administered intravenously over 30 minutes at a dose of 20 mg/m2 in Cycle 1 on Days 1 and 2; at a dose of 56 mg/m2 in Cycle 1 on Days 8, 9, 15 and 16; and on Days 1, 2, 8, 9, 15 and 16 of each 28-day cycle thereafter. Dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 8, 9, 15 and 16 and then 40 mg orally or intravenously on Day 22 of each 28-day cycle. In the DKd arm, daratumumab was administered intravenously at a dose of 8 mg/kg in Cycle 1 on Days 1 and 2. Thereafter, daratumumab was administered intravenously at a dose of 16 mg/kg on Days 8, 15 and 22 of Cycle 1; Days 1, 8 and 15 and 22 of Cycle 2; Days 1 and 15 of Cycles 3 to 6; and Day 1 for the remaining cycles or until disease progression. For patients >75 years on a reduced dexamethasone dose of 20 mg, the entire 20 mg dose was given as a daratumumab pre-infusion medication on days when daratumumab was administered. Dosing of dexamethasone was otherwise split across days when Kyprolis was administered in both study arms. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 466 patients were randomized; 312 to the DKd arm and 154 to the Kd arm. The demographics and baseline characteristics are summarized in Table 28.
| Characteristics | DKd (N = 312) | Kd (N = 154) |
|---|---|---|
| DKd = Kyprolis, daratumumab, and dexamethasone; ECOG = Eastern Cooperative Oncology Group; FISH = Fluorescence in situ hybridization; ISS = International Staging System; Kd = Kyprolis and dexamethasone | ||
|
||
| Age at randomization (years) | ||
| Median (min, max) | 64 (29, 84) | 65 (35, 83) |
| Age group – n (%) | ||
| 18 – 64 years | 163 (52) | 77 (50) |
| 65 – 74 years | 121 (39) | 55 (36) |
| 75 years and older | 28 (9) | 22 (14) |
| Sex – n (%) | ||
| Male | 177 (57) | 91 (59) |
| Female | 135 (43) | 63 (41) |
| Race – n (%) | ||
| Asian | 46 (15) | 20 (13) |
| Black or African American | 7 (2.2) | 2 (1.3) |
| White | 243 (78) | 123 (80) |
| Other | 16 (5) | 9 (6) |
| Geographic region – n (%) | ||
| North America | 21 (7) | 12 (8) |
| Europe | 207 (66) | 103 (67) |
| Asia Pacific | 84 (27) | 39 (25) |
| ECOG performance status – n (%) | ||
| 0 or 1 | 295 (95) | 147 (95) |
| 2 | 15 (4.8) | 7 (4.5) |
| Missing | 2 (0.6) | 0 (0.0) |
| Risk group as determined by FISH – n (%) | ||
| High risk | 48 (15) | 26 (17) |
| Standard risk | 104 (33) | 52 (34) |
| Unknown | 160 (51) | 76 (49) |
| ISS stage per I × RS at screening – n (%) | ||
| I or II | 252 (81) | 127 (82) |
| III | 60 (19) | 27 (17) |
| Number of prior regimens – n (%)* | ||
| 1 | 144 (46) | 70 (45) |
| 2 | 99 (32) | 46 (30) |
| 3 | 69 (22) | 37 (24) |
| Prior Therapies | ||
| Lenalidomide | 123 (39) | 74 (48) |
| Refractory to lenalidomide | 99 (32) | 55 (36) |
| Bortezomib | 287 (92) | 134 (87) |
| Prior CD38 antibody therapy – n (%) | 1 (0.3) | 0 (0.0) |
| Prior stem cell transplant (ASCT) – n (%) | 195 (62) | 75 (49) |
Efficacy was assessed by an IRC evaluation of PFS using the IMWG response criteria. Efficacy results are provided in Table 29 and Figure 6. The median duration of response has not been reached for the DKd arm and was 16.6 months (13.9, NE) for the Kd arm. The median (min, max) time to response was 1.0 (1, 14) months for the DKd arm and 1.0 (1, 10) months for the Kd arm.
| CI = confidence interval; DKd = Kyprolis, daratumumab and dexamethasone; HR = hazard ratio; Kd= Kyprolis and dexamethasone |
|
Figure 6: Kaplan-Meier Plot of Progression-Free Survival in CANDOR |
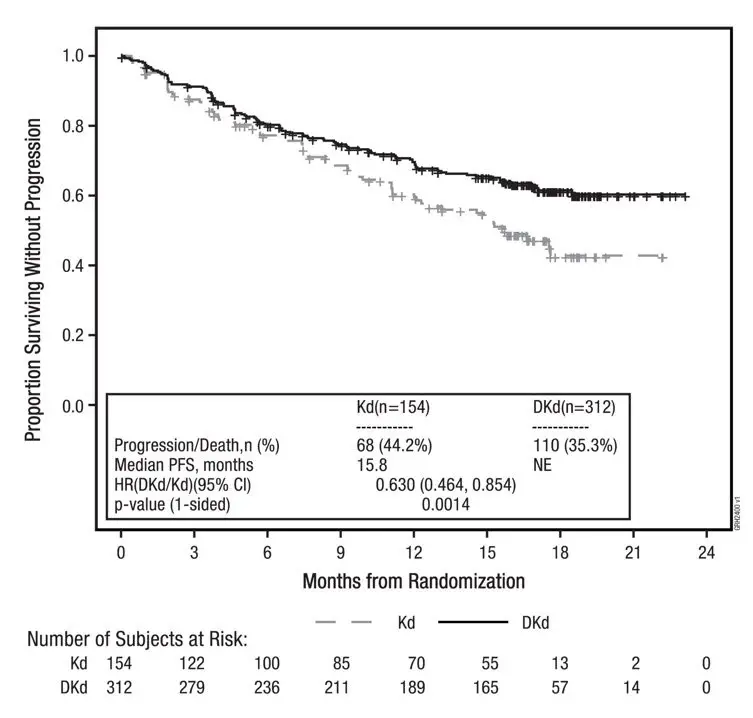 |
| DKd (N = 312) | Kd (N = 154) |
|
|---|---|---|
| CI = confidence interval; CR = complete response; HR = hazard ratio; DKd = Kyprolis, daratumumab, and dexamethasone; Kd = Kyprolis and dexamethasone; ORR = overall response rate; PFS = progression-free survival; PR = partial response; MRD [-] CR = minimal residual disease negative-complete response; NE = non-estimable; VGPR = very good partial response | ||
|
||
| PFS | ||
| Number of events (%) | 110 (35%) | 68 (44%) |
| Median, Months (95% CI) | NE (NE, NE) | 15.8 (12.1, NE) |
| HR (95% CI) | 0.63 (0.46, 0.85) | |
| P-value (1-sided)* | 0.0014 | |
| Overall Response | ||
| N with Response | 263 | 115 |
| ORR (%) (95% CI) | 84% (80%, 88%) | 75% (67%, 81%) |
| P-value (1-sided)† | 0.0040 | |
| CR | 89 (28%) | 16 (10%) |
| VGPR | 127 (41%) | 59 (38%) |
| PR | 47 (15%) | 40 (26%) |
| MRD [-] CR rate at 12 months n (%)‡ | 39 (12%) | 2 (1.3%) |
| (95% CI) | (9%, 17%) | (0.2%, 4.6%) |
| P-value (1-sided)† | < 0.0001 | |
| MRD [-] CR§ | 43 (14%) | 5 (3.2%) |
EQUULEUS (NCT01998971)
EQUULEUS was an open-label, multi-cohort trial which evaluated the combination of Kyprolis with intravenous daratumumab and dexamethasone in patients with relapsed or refractory multiple myeloma who had received 1 to 3 prior lines of therapy. Patients who had the following were excluded from the trial: known moderate or severe persistent asthma within the past 2 years, known chronic obstructive pulmonary disease (COPD) with a FEV1 < 50% of predicted normal, or active congestive heart failure (defined as New York Heart Association Class III-IV).
Kyprolis was administered intravenously over 30 minutes once weekly at a dose of 20 mg/m2 on Cycle 1 Day 1 and escalated to a dose of 70 mg/m2 on Cycle 1, Days 8 and 15; and on Days 1, 8, and 15 of each 28-day cycle. Ten patients were administered daratumumab at a dose of 16 mg/kg intravenously on Cycle 1, Day 1 and the remaining patients were administered daratumumab at a dose of 8 mg/kg intravenously on Cycle 1, Days 1 and 2. Thereafter, daratumumab was administered intravenously at a dose of 16 mg/kg on Days 8, 15 and 22 of Cycle 1; Days 1, 8, 15 and 22 of Cycle 2; Days 1 and 15 of Cycles 3 to 6; and then Day 1 for the remaining cycles of each 28-day cycle. In Cycles 1 and 2, dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 8, 9, 15, 16, 22 and 23; in cycles 3 to 6, dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 15 and 16 and at a dose of 40 mg on Day 8 and 22; and in cycles 7 and thereafter, dexamethasone 20 mg was administered orally or intravenously on Days 1 and 2 and at a dose of 40 mg on Days 8, 15, and 22. For patients > 75 years of age, dexamethasone 20 mg was administered orally or intravenously weekly after the first week. Treatment continued until disease progression or unacceptable toxicity.
The EQUULEUS trial enrolled 85 patients. The demographics and baseline characteristics are summarized in Table 30.
| Characteristics | Number of Patients (%) |
|---|---|
| ECOG = Eastern Cooperative Oncology Group; FISH = Fluorescence in situ hybridization; PI = proteasome inhibitor; IMiD = immunomodulatory agent. | |
| Age (years) | |
| Median (min, max) | 66 (38, 85) |
| Age group – n (%) | |
| < 65 years | 36 (42) |
| 65 - < 75 years | 41 (48) |
| ≥ 75 years | 8 (9) |
| Sex – n (%) | |
| Male | 46 (54) |
| Female | 39 (46) |
| Race – n (%) | |
| Asian | 3 (3.5) |
| Black or African American | 3 (3.5) |
| White | 68 (80) |
| ECOG Score, n (%) | |
| 0 | 32 (38) |
| 1 | 46 (54) |
| 2 | 7 (8) |
| FISH, n (%) | |
| N | 67 |
| Standard Risk | 54 (81) |
| High Risk | 13 (19) |
| Number of Prior regimens | |
| 1 | 20 (23) |
| 2 | 40 (47) |
| 3 | 23 (27) |
| > 3 | 2 (2.4) |
| Prior Therapies | |
| Bortezomib | 85 (100) |
| Lenalidomide | 81 (95) |
| Prior stem cell transplant (ASCT) | 62 (73) |
| Refractory to lenalidomide | 51 (60) |
| Refractory to both a PI and IMiD | 25 (29) |
Efficacy results were based on overall response rate using IMWG criteria. Efficacy results are provided in Table 31. The median time to response was 0.95 months (range: 0.9, 14.3). The median duration of response was 28 months (95% CI: 20.5, not estimable).
| Study Patients n (%) |
|
|---|---|
| CI = confidence interval; sCR = stringent complete response; CR = complete response; ORR = overall response rate; PR = partial response; VGPR = very good partial response | |
| Overall Response | |
| N with Response | 69 |
| ORR (%) (95% CI) | 81% (71, 89) |
| Response category, n (%) | |
| sCR | 18 (21%) |
| CR | 12 (14%) |
| VGPR | 28 (33%) |
| PR | 11 (13%) |
PLEIADES (NCT03412565)
The efficacy of Kyprolis with daratumumab and hyaluronidase-fihj plus dexamethasone (DKd) was evaluated in a single-arm cohort of PLEIADES, a multi-cohort, open-label trial. This cohort enrolled patients with relapsed or refractory multiple myeloma excluding patients with left ventricular ejection fraction (LVEF) less than 40%, myocardial infarction within 6 months, uncontrolled cardiac arrhythmia, or uncontrolled hypertension (systolic blood pressure >159 mmHg or diastolic >99 mmHg despite optimal treatment). Patients received Kyprolis administered by IV infusion at a dose of 20 mg/m2 on Cycle 1 Day 1 and if a dose of 20 mg/m2 was tolerated Kyprolis was administered at a dose of 70 mg/m2 as a 30-minute IV infusion on Cycle 1 Day 8 and Day 15, and then Day 1, 8 and 15 of each cycle; daratumumab and hyaluronidase-fihj1,800 mg/30,000 units administered subcutaneously once weekly from Weeks 1 to 8, once every 2 weeks from Weeks 9 to 24 and once every 4 weeks starting with Week 25 until disease progression or unacceptable toxicity; and dexamethasone 40 mg per week (or a reduced dose of 20 mg per week for patients ≥75 years or BMI <18.5). The major efficacy outcome measure was ORR.
A total of 66 patients received the DKd regimen. The median age was 61 years (range: 42, 84); 52% were male; 73% were White and 3% Black or African American; and 68% had ISS Stage I, 18% had ISS Stage II, and 14% had ISS Stage III disease. A total of 79% of patients had a prior ASCT; 91% of patients received a prior PI. All patients received 1 prior line of therapy with exposure to lenalidomide and 62% of patients were refractory to lenalidomide.
Efficacy results are summarized in Table 32. At a median follow-up of 9.2 months, the median duration of response had not been reached and an estimated 85.2% (95% CI: 72.5, 92.3) maintained response for at least 6 months and 82.5% (95% CI: 68.9, 90.6) maintained response for at least 9 months.
| DKd (N=66) |
|
|---|---|
| CI = confidence interval | |
|
|
| Overall response rate (sCR+CR+VGPR+PR), n (%)* | 56 (84.8%) |
| 95% CI (%) | (73.9%, 92.5%) |
| Stringent complete response (sCR) | 11 (16.7%) |
| Complete response (CR) | 14 (21.2%) |
| Very good partial response (VGPR) | 26 (39.4%) |
| Partial response (PR) | 5 (7.6%) |
14.4 In Combination with Isatuximab and Dexamethasone for Relapsed or Refractory Multiple Myeloma
IKEMA (NCT03275285)
The efficacy and safety of Kyprolis in combination with isatuximab and dexamethasone were evaluated in IKEMA, a multicenter, multinational, randomized, open-label, 2-arm, phase 3 study in patients with relapsed and/or refractory multiple myeloma. Patients had received one to three prior lines of therapy. Patients were eligible for inclusion if they had an ECOG status of 0-2, platelets ≥50,000 cells/mm3, absolute neutrophil count ≥1 × 109/L, creatinine clearance ≥15 mL/min/1.73 m2 (MDRD formula), AST ≤3 × ULN, and ALT ≤3 × ULN.
A total of 302 patients were randomized in a 3:2 ratio to receive either Kyprolis in combination with isatuximab and dexamethasone (Isa-Kd, 179 patients) or Kyprolis and dexamethasone (Kd, 123 patients). Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity. Isatuximab 10 mg/kg was administered as an intravenous infusion weekly in the first cycle and every two weeks thereafter. Kyprolis was administered as an intravenous infusion at the dose of 20 mg/m2 on days 1 and 2; 56 mg/m2 on days 8, 9, 15, and 16 of cycle 1; and at the dose of 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles of each 28-day cycle. Dexamethasone (intravenously on the days of isatuximab and/or Kyprolis infusions, and orally on the other days) 20 mg was given on days 1, 2, 8, 9, 15, 16, 22, and 23 for each 28-day cycle. On the days where both Kyprolis and isatuximab were administered, dexamethasone was administered first, followed by isatuximab infusion, then followed by Kyprolis infusion.
Overall, demographic and disease characteristics at baseline were similar between the two treatment groups. The median patient age was 64 years (range 33-90), 9% of patients were ≥ 75 years, 71% were White, 17% Asian, and 3% Black or African American. The proportion of patients with renal impairment (eGFR < 60 mL/min/1.73 m2) was 24% in the Isa-Kd group versus 15% in the Kd group. The International Staging System (ISS) stage at study entry was I in 53%, II in 31%, and III in 15% of patients. Overall, 24% of patients had high-risk chromosomal abnormalities at study entry; del(17p), t(4;14), t(14;16) were present in 11%, 14%, and 2% of patients, respectively. In addition, gain(1q21) was present in 42% of patients.
The median number of prior lines of therapy was 2 (range 1-4) with 44% of patients who received 1 prior line of therapy. Overall, 90% of patients received prior proteasome inhibitors, 78% received prior immunomodulators (including 43% who received prior lenalidomide), and 61% received prior stem cell transplantation. Overall, 33% of patients were refractory to prior proteasome inhibitors, 45% were refractory to prior immunomodulators (including 33% refractory to lenalidomide), and 21% were refractory to both a proteasome inhibitor and an immunomodulator.
The median duration of treatment was 80 weeks for the Isa-Kd group compared to 61 weeks for the Kd group.
Efficacy was based upon PFS. PFS results were assessed by an Independent Response Committee based on central laboratory data for M-protein and central radiologic imaging review using the IMWG criteria. The improvement in PFS represented a 45% reduction in the risk of disease progression or death in patients treated with Isa-Kd compared to patients treated with Kd.
Efficacy results are presented in Table 33.
| Endpoint | Isa-Kd N=179 | Kd N=123 |
|---|---|---|
| NR: not reached. | ||
|
||
| Progression-Free Survival‡ | ||
| Median (months) [95% CI] | NR [NR- NR] | 20.27 [15.77- NR] |
| Hazard ratio§ [95% CI] | 0.548 [0.366-0.822] | |
| p-value (stratified log-rank test) § | 0.0032 | |
| Overall Response Rate¶
Responders (sCR+CR+VGPR+PR) n (%) [95% CI]# | 155 (86.6) [80.7-91.2] | 102 (82.9) [75.1-89.1] |
| p-value (stratified Cochran-Mantel-Haenszel)§ | 0.3859 | |
| Complete Response (CR) n (%) | 71 (39.7) | 34 (27.6) |
| Very Good Partial Response (VGPR) n (%) | 59 (33) | 35 (28.5) |
| Partial Response (PR) n (%) | 25 (14) | 33 (26.8) |
|
Figure 7: Kaplan-Meier Plot of Progression-Free Survival in IKEMA |
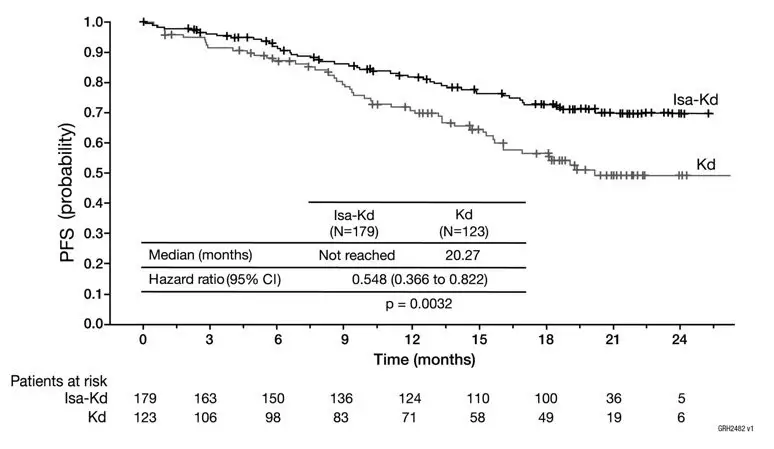 |
14.5 Monotherapy for Relapsed or Refractory Multiple Myeloma
Study PX-171-007 (NCT00531284)
Study PX-171-007 was a multicenter, open-label, dose escalation, single-arm trial that evaluated the safety of Kyprolis monotherapy as a 30-minute infusion in patients with relapsed or refractory multiple myeloma after 2 or more lines of therapy. Patients were excluded if they had a creatinine clearance < 20 mL/min; ALT ≥ 3 × upper limit of normal (ULN), bilirubin ≥ 1.5 × ULN; New York Heart Association Class III or IV congestive heart failure; or other significant cardiac conditions. A total of 24 subjects with multiple myeloma were enrolled at the maximum tolerated dose level of 20/56 mg/m2. Kyprolis was administered twice weekly for 3 consecutive weeks (Days 1, 2, 8, 9, 15, and 16) of a 28-day cycle. In Cycle 13 onward, the Day 8 and 9 Kyprolis doses could be omitted. Patients received Kyprolis at a starting dose of 20 mg/m2 on Days 1 and 2 of Cycle 1, which was increased to 56 mg/m2 for all subsequent doses. Dexamethasone 8 mg orally or intravenously was required prior to each Kyprolis dose in Cycle 1 and was optional in subsequent cycles. Treatment was continued until disease progression or unacceptable toxicity.
Efficacy was evaluated by ORR and DOR. ORR by investigator assessment was 50% (95% CI: 29, 71) per IMWG criteria (see Table 34). The median DOR in subjects who achieved a PR or better was 8.0 months (Range: 1.4, 32.5).
| Characteristics | Study Patients*
n (%) |
|---|---|
| CI = confidence interval; CR = complete response; PR = partial response; sCR = stringent complete response; VGPR = very good partial response | |
|
|
| Number of Patients (%) | 24 (100) |
| Overall Response† | 12 (50) |
| 95% CI‡ | (29, 71) |
| Response Category | |
| sCR | 1 (4) |
| CR | 0 (0) |
| VGPR | 4 (17) |
| PR | 7 (29) |
Study PX-171-003 A1 (NCT00511238)
Study PX-171-003 A1 was a single-arm, multicenter clinical trial of Kyprolis monotherapy by up to 10-minute infusion. Eligible patients were those with relapsed and refractory multiple myeloma who had received at least two prior therapies (including bortezomib and thalidomide and/or lenalidomide) and had ≤ 25% response to the most recent therapy or had disease progression during or within 60 days of the most recent therapy. Patients were excluded from the trial if they were refractory to all prior therapies or had a total bilirubin ≥ 2 × ULN; creatinine clearance < 30 mL/min; New York Heart Association Class III to IV congestive heart failure; symptomatic cardiac ischemia; myocardial infarction within the last 6 months; peripheral neuropathy Grade 3 or 4, or peripheral neuropathy Grade 2 with pain; active infections requiring treatment; or pleural effusion.
Kyprolis was administered intravenously up to 10 minutes on two consecutive days each week for three weeks, followed by a 12-day rest period (28-day treatment cycle), until disease progression, unacceptable toxicity, or for a maximum of 12 cycles. Patients received 20 mg/m2 at each dose in Cycle 1, and 27 mg/m2 in subsequent cycles. Dexamethasone 4 mg orally or intravenously was administered prior to Kyprolis doses in the first and second cycles.
A total of 266 patients were enrolled. Baseline patient and disease characteristics are summarized in Table 35.
| Characteristics | Number of Patients (%) |
|---|---|
| FISH = Fluorescence in situ hybridization; ISS = International Staging System | |
|
|
| Patient Characteristics | |
| Enrolled patients | 266 (100) |
| Median age, years (range) | 63 (37, 87) |
| Age group, < 65 / ≥ 65 (years) | 146 (55) / 120 (45) |
| Sex (male / female) | 155 (58) / 111 (42) |
| Race (White / Black / Asian / Other) | 190 (71) / 53 (20) / 6 (2) / 17 (6) |
| Disease Characteristics | |
| Number of Prior Regimens (median) | 5* |
| Prior Transplantation | 198 (74) |
| Refractory Status to Most Recent Therapy† | |
| Refractory: Progression during most recent therapy | 198 (74) |
| Refractory: Progression within 60 days after completion of most recent therapy | 38 (14) |
| Refractory: ≤ 25% response to treatment | 16 (6) |
| Relapsed: Progression after 60 days post treatment | 14 (5) |
| Years since diagnosis, median (range) | 5.4 (0.5, 22.3) |
| Plasma cell involvement (< 50% / ≥ 50% / unknown) | 143 (54) / 106 (40) / 17 (6) |
| ISS Stage at Study Baseline | |
| I | 76 (29) |
| II | 102 (38) |
| III | 81 (31) |
| Unknown | 7 (3) |
| Cytogenetics or FISH analyses | |
| Normal/Favorable | 159 (60) |
| Poor Prognosis | 75 (28) |
| Unknown | 32 (12) |
| Creatinine clearance < 30 mL/min | 6 (2) |
Efficacy was evaluated by ORR as determined by IRC assessment using IMWG criteria. Efficacy results are provided in Table 36. The median DOR was 7.8 months (95% CI: 5.6, 9.2).
| Characteristics | Study Patients*
n (%) |
|---|---|
| CI = confidence interval; CR = complete response; PR = partial response; VGPR = very good partial response | |
|
|
| Number of Patients (%) | 266 (100) |
| Overall Response† | 61 (23) |
| 95% CI‡ | (18, 28) |
| Response Category | |
| CR | 1 (< 1) |
| VGPR | 13 (5) |
| PR | 47 (18) |
Study PX-171-004 Part 2 (NCT00530816)
Study PX-171-004 Part 2 was a single-arm, multicenter clinical trial of Kyprolis monotherapy by up to 10-minute infusion. Eligible patients were those with relapsed or refractory multiple myeloma who were bortezomib-naïve, had received one to three prior lines of therapy and had ≤ 25% response or progression during therapy or within 60 days after completion of therapy. Patients were excluded from the trial if they were refractory to standard first-line therapy or had a total bilirubin ≥ 2 × ULN; creatinine clearance < 30 mL/min; New York Heart Association Class III to IV congestive heart failure; symptomatic cardiac ischemia; myocardial infarction within the last 6 months; active infections requiring treatment; or pleural effusion.
Kyprolis was administered intravenously up to 10 minutes on two consecutive days each week for three weeks, followed by a 12-day rest period (28-day treatment cycle), until disease progression, unacceptable toxicity, or for a maximum of 12 cycles. Patients received 20 mg/m2 at each dose in Cycle 1, and 27 mg/m2 in subsequent cycles. Dexamethasone 4 mg orally or intravenously was administered prior to Kyprolis doses in the first and second cycles.
A total of 70 patients were treated with this 20/27 mg/m2 regimen. Baseline patient and disease characteristics are summarized in Table 37.
| Characteristics | Number of Patients (%) |
|---|---|
| FISH = Fluorescence in situ hybridization; ISS = International Staging System | |
|
|
| Patient Characteristics | |
| Enrolled patients | 70 (100) |
| Median age, years (range) | 66 (45, 85) |
| Age group, < 65 / ≥ 65 (years) | 31 (44) / 39 (56) |
| Sex (male / female) | 44 (63) / 26 (37) |
| Race (White / Black / Asian / Hispanic / Other) | 52 (74) / 12 (17) / 3 (4) / 2 (3) / 1 (1) |
| Disease Characteristics | |
| Number of Prior Regimens (median) | 2* |
| Prior Transplantation | 47 (67) |
| Refractory Status to Most Recent Therapy† | |
| Refractory: Progression during most recent therapy | 28 (40) |
| Refractory: Progression within 60 days after completion of most recent therapy | 7 (10) |
| Refractory: ≤ 25% response to treatment | 10 (14) |
| Relapsed: Progression after 60 days post treatment | 23 (33) |
| No Signs of Progression | 2 (3) |
| Years since diagnosis, median (range) | 3.6 (0.7, 12.2) |
| Plasma cell involvement (< 50% / ≥ 50% / unknown) | 54 (77) / 14 (20) / 1 (1) |
| ISS Stage at Study Baseline, n (%) | |
| I | 28 (40) |
| II | 25 (36) |
| III | 16 (23) |
| Unknown | 1 (1) |
| Cytogenetics or FISH analyses | |
| Normal/Favorable | 57 (81) |
| Poor Prognosis | 10 (14) |
| Unknown | 3 (4) |
| Creatinine clearance < 30 mL/min | 1 (1) |
Efficacy was evaluated by ORR as determined by IRC assessment using IMWG criteria. Efficacy results are provided in Table 38. The median DOR was not reached.
| Characteristics | Study Patients*
n (%) |
|---|---|
| CI = confidence interval; CR = complete response; PR = partial response; VGPR = very good partial response | |
|
|
| Number of Patients (%) | 70 (100) |
| Overall Response† | 35 (50) |
| 95% CI‡ | (38, 62) |
| Response Category | |
| CR | 1 (1) |
| VGPR | 18 (26) |
| PR | 16 (23) |



| KYPROLIS
carfilzomib injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| KYPROLIS
carfilzomib injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| KYPROLIS
carfilzomib injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Onyx Pharmaceuticals, Inc. (789591724) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Amgen Technology (ireland) Unlimited Company | 896293920 | ANALYSIS(76075-101, 76075-102) , MANUFACTURE(76075-101, 76075-102) , PACK(76075-101, 76075-102) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Geneva Laboratories, Inc. | 091791129 | ANALYSIS(76075-101, 76075-102, 76075-103) | |
