Drug Detail:Lokelma (Sodium zirconium cyclosilicate [ soe-dee-um-zir-koe-nee-um-sye-kloe-sil-i-kate ])
Drug Class: Cation exchange resins
Highlights of Prescribing Information
LOKELMA® (sodium zirconium cyclosilicate) for oral suspension
Initial U.S. Approval: 2018
Recent Major Changes
Warnings and Precautions (5.4) 10/2021
Indications and Usage for Lokelma
LOKELMA is a potassium binder indicated for the treatment of hyperkalemia in adults. (1)
Limitation of Use
LOKELMA should not be used as an emergency treatment for life-threatening hyperkalemia because of its delayed onset of action. (1)
Lokelma Dosage and Administration
- •
- Recommended starting dose is 10 g administered three times a day for up to 48 hours. (2.1)
- •
- For maintenance treatment, recommended dose is 10 g once daily. Adjust dose at one-week intervals as needed (by 5 g daily) to obtain desired serum potassium target range. (2.1)
- Patients on Chronic Hemodialysis
- •
- Recommended starting dose is 5 g once daily on non-dialysis days. (2.2)
- See full Prescribing Information for additional dosing instructions, as well as reconstitution and administration instructions for the oral suspension.
Dosage Forms and Strengths
- •
- For oral suspension: 5 g per packet (3)
- •
- For oral suspension: 10 g per packet (3)
Contraindications
None. (4)
Warnings and Precautions
- •
- Gastrointestinal Adverse Events in Patients with Motility Disorders. (5.1)
- •
- Edema. (5.2)
- •
- Hypokalemia in patients on hemodialysis. (5.3)
- •
- LOKELMA has radio-opaque properties and, therefore, may give the appearance typical of an imaging agent during abdominal X-ray procedures. (5.4)
Adverse Reactions/Side Effects
Most common adverse reactions with LOKELMA: mild to moderate edema. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
In general, other oral medications should be administered at least 2 hours before or 2 hours after LOKELMA. (2.3, 7, 12.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2022
Full Prescribing Information
1. Indications and Usage for Lokelma
LOKELMA is indicated for the treatment of hyperkalemia in adults.
Limitation of Use
LOKELMA should not be used as an emergency treatment for life-threatening hyperkalemia because of its delayed onset of action [see Clinical Pharmacology (12.2) and Clinical Studies (14)].
2. Lokelma Dosage and Administration
2.1 Recommended Dosage
For initial treatment of hyperkalemia, the recommended dose of LOKELMA is 10 g administered three times a day for up to 48 hours. Administer LOKELMA orally as a suspension in water [see Dosage and Administration (2.3)].
For continued treatment, the recommended dose is 10 g once daily. Monitor serum potassium and adjust the dose of LOKELMA based on the serum potassium level and desired target range. During maintenance treatment, up-titrate based on the serum potassium level at intervals of 1-week or longer and in increments of 5 g. Decrease the dose of LOKELMA or discontinue if the serum potassium is below the desired target range. The recommended maintenance dose range is from 5 g every other day to 15 g daily.
2.2 Dosage Adjustment for Patients on Chronic Hemodialysis
For patients on chronic hemodialysis, administer LOKELMA only on non-dialysis days.
The recommended starting dose is 5 g once daily on non-dialysis days. Consider a starting dose of 10 g once daily on non-dialysis days in patients with serum potassium greater than 6.5 mEq/L. Monitor serum potassium and adjust the dose of LOKELMA based on the pre-dialysis serum potassium value after the long inter-dialytic interval and desired target range.
During initiation and after a dose adjustment, assess serum potassium after one week. The recommended maintenance dose range is from 5 g to 15 g once daily, on non-dialysis days.
Discontinue or decrease the dose of LOKELMA if:
- •
- serum potassium falls below the desired target range based on the pre-dialysis value after the long interdialytic interval, or;
- •
- the patient develops clinically significant hypokalemia
2.3 Reconstitution and Administration
In general, other oral medications should be administered at least 2 hours before or 2 hours after LOKELMA [see Drug Interactions (7)].
Instruct patients to empty the entire contents of the packet(s) into a drinking glass containing approximately 3 tablespoons of water or more if desired. Stir well and drink immediately. If powder remains in the drinking glass, add water, stir and drink immediately. Repeat until no powder remains to ensure the entire dose is taken.
3. Dosage Forms and Strengths
For oral suspension: 5 g or 10 g of white to grey powder in a foil-lined packet.
5. Warnings and Precautions
5.1 Gastrointestinal Adverse Events in Patients with Motility Disorders
Avoid use of LOKELMA in patients with severe constipation, bowel obstruction or impaction, including abnormal post-operative bowel motility disorders, because LOKELMA has not been studied in patients with these conditions and may be ineffective and may worsen gastrointestinal conditions.
5.2 Edema
Each 5 g dose of LOKELMA contains approximately 400 mg of sodium, but the extent of absorption by the patient is unknown. In clinical trials of LOKELMA in patients who were not on dialysis, edema was observed and was generally mild to moderate in severity and was more commonly seen in patients treated with 15 g once daily. Monitor for signs of edema, particularly in patients who should restrict their sodium intake or are prone to fluid overload (e.g., heart failure or renal disease). Advise patients to adjust dietary sodium, if appropriate. Increase the dose of diuretics as needed [see Adverse Reactions (6)].
In a clinical trial of LOKELMA in patients on chronic hemodialysis in which most patients were treated with doses of 5 to 10 g once daily on non-dialysis days, there was no difference in the mean change from baseline in interdialytic weight gain (a measure of fluid retention) between the LOKELMA and placebo groups.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail elsewhere in the label:
- •
- Edema [see Warnings and Precautions (5.2)].
6.1 Clinical Studies Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The total exposure to LOKELMA in the safety and efficacy clinical trials of patients not on dialysis with hyperkalemia was 1,760 patients with 652 patients exposed to LOKELMA for at least 6 months and 507 patients exposed for at least one year.
The population (n=1,009) in the placebo-controlled trials included patients aged 22 to 96 years, females (n=454), Caucasians (n=859) and Blacks (n=130). Patients had hyperkalemia in association with comorbid diseases such as chronic kidney disease, heart failure, and diabetes mellitus.
In placebo-controlled trials in which patients who were not on dialysis were treated with once daily doses of LOKELMA for up to 28 days, edema was reported in 4.4% of patients receiving 5 g, 5.9% of patients receiving 10 g and 16.1% of patients receiving 15 g LOKELMA compared to 2.4% of patients receiving placebo. In longer-term uncontrolled trials in which most patients were maintained on doses <15 g once daily, adverse reactions of edema (edema, generalized edema and peripheral edema) were reported in 8% to 11% of patients.
Laboratory Abnormalities
In clinical trials in patients who were not on dialysis, 4.1% of LOKELMA-treated patients developed hypokalemia with a serum potassium value less than 3.5 mEq/L, which resolved with dosage reduction or discontinuation of LOKELMA. In a clinical trial of LOKELMA in patients on chronic hemodialysis, 5% of patients developed pre-dialysis hypokalemia (serum potassium <3.5 mEq/L) in both the LOKELMA and placebo groups; 3% and 1% of patients developed a serum potassium < 3.0 mEq/L in the LOKELMA and placebo groups, respectively.
7. Drug Interactions
LOKELMA can transiently increase gastric pH. As a result, LOKELMA can change the absorption of co-administered drugs that exhibit pH-dependent solubility, potentially leading to altered efficacy or safety of these drugs when taken close to the time LOKELMA is administered. In general, other oral medications should be administered at least 2 hours before or 2 hours after LOKELMA [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)]. LOKELMA is not expected to impact systemic exposure of drugs that do not exhibit pH-dependent solubility and so spacing is not needed if it has been determined that the concomitant medication does not exhibit pH-dependent solubility.
11. Lokelma Description
LOKELMA is a powder for oral suspension. The active ingredient in LOKELMA is sodium zirconium cyclosilicate, a potassium binder. Sodium zirconium cyclosilicate is a non-absorbed zirconium silicate that preferentially exchanges potassium for hydrogen and sodium. LOKELMA is an odorless, insoluble white to grey powder for oral suspension. It has a mean particle size of 20 µm and includes no more than 3% of particles with a diameter below 3 µm. Each 5 g of sodium zirconium cyclosilicate contains 400 mg of sodium.
The chemical formula of sodium zirconium cyclosilicate is Na~1.5H~0.5ZrSi3O9•2–3H2O.
Figure 1: Crystal Structure of Sodium Zirconium Cyclosilicate
12. Lokelma - Clinical Pharmacology
12.1 Mechanism of Action
LOKELMA (sodium zirconium cyclosilicate) is a non-absorbed zirconium silicate that preferentially captures potassium in exchange for hydrogen and sodium. In vitro, LOKELMA has a high affinity for potassium ions, even in the presence of other cations such as calcium and magnesium. LOKELMA increases fecal potassium excretion through binding of potassium in the lumen of the gastrointestinal tract. Binding of potassium reduces the concentration of free potassium in the gastrointestinal lumen, thereby lowering serum potassium levels.
12.2 Pharmacodynamics
In a study in healthy adult subjects, LOKELMA administered as 5 g or 10 g once daily for four days caused a dose-dependent increase in fecal potassium excretion. Corresponding dose-dependent decreases in urinary potassium excretion and serum potassium were also observed.
In patients with hyperkalemia treated with LOKELMA 10 g three times a day for up to 48 hours, reductions in serum potassium were observed one hour after initiation of therapy; serum potassium concentrations continued to decline over the 48-hour treatment period [see Clinical Studies (14.2)]. In patients not continuing LOKELMA, potassium levels increased. Patients with higher starting serum potassium levels or receiving a higher dose have greater reductions in serum potassium.
LOKELMA causes a small dose-dependent increase in serum bicarbonate concentrations (1.1 mmol/L at 5 g once daily, 2.3 mmol/L at 10 g once daily and 2.6 mmol/L at 15 g once daily as compared with a mean increase of 0.6 mmol/L in patients treated with placebo). The clinical significance of this finding is unclear.
12.3 Pharmacokinetics
LOKELMA is an inorganic, insoluble compound that is not subject to enzymatic metabolism. In a clinical study in patients with hyperkalemia in which zirconium concentrations were measured in the urine and blood, zirconium concentrations were similar in treated and untreated patients (i.e., either undetectable or around the lower limit of quantification of the assay). An in vivo mass balance study in rats showed that LOKELMA was recovered in the feces with no evidence of systemic absorption.
Drug Interactions
Thirty-six (36) drugs were tested in-vitro to determine potential interactions with LOKELMA. Sixteen (16) drugs tested did not show an in vitro interaction with LOKELMA (allopurinol, apixaban, aspirin, captopril, cyclosporine, digoxin, ethinyl estradiol, lisinopril, magnesium, metformin, phenytoin, prednisone, propranolol, quinapril, spironolactone and ticagrelor).
Nine (9) of the 20 drugs that showed an in vitro interaction were subsequently tested in vivo with LOKELMA 10 gin healthy volunteers. Losartan, glipizide and levothyroxine did not show any changes in exposure when co-administered with LOKELMA. However, there was an increase in systemic exposure to weak acids such as furosemide and atorvastatin, and a decrease in systemic exposure to weak bases such as dabigatran when co-administered with LOKELMA, as shown in Figure 2. These changes are consistent with the hypothesis that LOKELMA, by elevating gastric pH, affects the systemic exposure of co-administered drugs whose solubility is pH-dependent [see Drug Interactions (7)].
In another drug-drug interaction study in healthy volunteers, co-administration of LOKELMA 15 g
decreased the systemic exposures of tacrolimus (Figure 2), likely due to LOKELMA’s action on elevating
gastric pH. In the same study, co-administration of LOKELMA and cyclosporine did not show a
clinically meaningful interaction.
Figure 2: Effects of LOKELMA 10 g or 15 g on the Pharmacokinetic Exposures of Other Orally Administered Medications
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The following tests for mutagenic potential of sodium zirconium cyclosilicate were negative: (1) the Ames (S. typhimurium and E. coli) test; (2) chromosomal aberration assay in Chinese Hamster Ovary (CHO) cells; and (3) in vivo rat micronucleus assay. Given that zirconium cyclosilicate is not genotoxic, not absorbed from the gastrointestinal tract, and did not cause local gastrointestinal alterations in a chronic toxicity study in dogs, carcinogenicity studies in animals to evaluate tumorigenic potential of sodium zirconium cyclosilicate were not deemed to be necessary.
Fertility in male and female rats has been assessed at doses up to a Human Equivalent Dose (HED) of 58 g per day (the maximum feasible dose) with no adverse effects.
14. Clinical Studies
14.1 Study 1
The effectiveness of LOKELMA in lowering serum potassium was demonstrated in a two-part, double-blind, randomized, placebo-controlled clinical trial (NCT01737697) in patients with hyperkalemia (5 to 6.5 mEq/L, mean potassium 5.3 mEq/L), Study 1.
In the first phase of the trial (the acute phase), 753 patients were randomized to receive one of four doses of LOKELMA (1.25, 2.5, 5 or 10 g) or placebo, administered three times daily for the initial 48 hours with meals.
The mean age of patients was 66 years, 59% of patients were men, and 86% were Caucasian. Approximately 60% of patients had chronic kidney disease, 10% had heart failure, 62% had diabetes mellitus and 67% were on renin angiotensin aldosterone system (RAAS) inhibitor therapy at baseline.
The primary endpoint in the acute phase was the difference in the exponential rate of change in serum potassium levels during the initial 48 hours of study drug treatment, comparing placebo-treated patients and LOKELMA-treated patients. The study met its primary endpoint demonstrating a greater reduction in serum potassium levels for the 2.5, 5 and 10 g (three times a day) dose groups compared to the placebo group (p<0.001). As displayed in Table 1 for the secondary endpoint of potassium change from baseline, LOKELMA showed dose-dependent reductions in serum potassium at 2.5, 5 and 10 g. In patients administered 10 g TID, the mean serum potassium reduction was -0.7 mEq/L at 48 hours. Patients with higher starting potassium levels had a greater response to LOKELMA. LOKELMA was effective in lowering potassium levels in patients with chronic kidney disease, heart failure, diabetes mellitus and those taking RAAS inhibitor therapy.
| Mean Serum Potassium Change mEq/L (95% Confidence Intervals) Sample Size | Placebo | 1.25 g TID | 2.5 g TID | 5 g TID | 10 g TID |
|---|---|---|---|---|---|
|
All Patients |
-0.2 |
-0.3 |
-0.5 |
-0.5 |
-0.7 |
|
Baseline Serum Potassium
|
-0.4 |
-0.3 |
-0.6 |
-0.9 |
-1.1 |
Patients who achieved a potassium level between 3.5 and 5 mEq/L after receiving LOKELMA during the acute phase were re-randomized to receive once daily placebo or 1.25, 2.5, 5 or 10 g of once daily LOKELMA for 12 days together with breakfast.
The primary endpoint in the maintenance phase was the difference in the exponential rate of change in serum potassium levels over the 12-day treatment interval, comparing patients receiving LOKELMA and patients receiving placebo. The study met the primary efficacy endpoint at the 5 and 10 g doses when compared with their respective placebo groups (p<0.01 and p<0.001).
14.2 Study 2
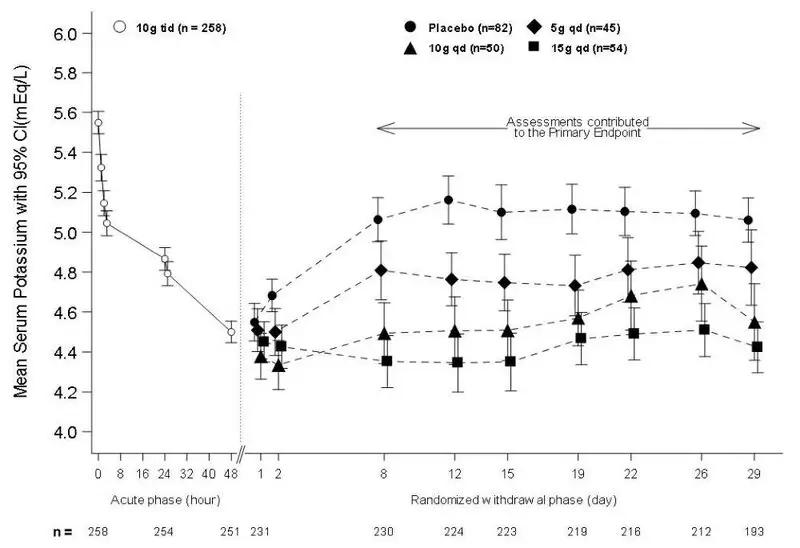
The efficacy of LOKELMA was also demonstrated in a two-part trial with an open-label acute phase and a month-long randomized, double-blind, placebo-controlled withdrawal phase (Study 2; NCT02088073).
In the open-label acute phase of Study 2, 258 patients with hyperkalemia (baseline mean 5.6 mEq/L, range 5.1 to 7.4 mEq/L) received 10 g of LOKELMA administered three times daily with meals for 48 hours. As shown in Figure 3, left, average serum potassium levels decreased from 5.6 to 4.5 mEq/L during treatment with LOKELMA in the acute phase.
Following the acute phase of the study, there was a double-blind randomized withdrawal phase where patients who achieved potassium levels between 3.5 and 5 mEq/L were randomized to one of three doses of LOKELMA administered once-daily for 28 days, or placebo just before breakfast. Of the patients enrolled in the acute phase, 92% achieved a potassium level within this range and were enrolled into the second phase of the trial.
The primary endpoint in the randomized withdrawal phase was the mean serum potassium value over the period from Day 8 to Day 29, comparing LOKELMA-treated and placebo-treated patients. All three doses (5, 10 and 15 g) of once daily LOKELMA maintained mean potassium at lower levels than placebo (mean serum potassium was 4.8, 4.5, and 4.4 mEq/L for the 5, 10 and 15 g dose groups, respectively, vs. 5.1 mEq/L in the placebo group, p≤0.001 for all doses, Figure 3, right). A greater proportion of patients had mean serum potassium levels in the normal range (3.5 to 5 mEq/L) while on LOKELMA than while on placebo (80%, 90% and 94% at the 5, 10 and 15 g doses, respectively, vs. 46% on placebo).
Figure 3: Study 2 - Mean Serum Potassium Levels in the Acute and Randomized Withdrawal Phases
Intent-to-Treat population includes subjects with at least one valid serum potassium measurement on or after Day 8
14.3 Eleven-Month Extension Study
Patients who completed the 28-day randomized withdrawal phase had the option to continue treatment with LOKELMA, taken just before breakfast, in an open-label extension phase for up to 11 months (n=123; NCT02107092). Figure 4 shows that the treatment effect on serum potassium was maintained during continued therapy.
Figure 4: 11-Month Open-Label Extension Phase of Study 2 - Mean Serum Potassium (mEq/L)
14.4 Study 3
LOKELMA was evaluated in an open-label 12-month study in 751 hyperkalemic patients (NCT02163499). The mean baseline potassium level in this study was 5.6 mEq/L. Following the acute phase treatment of LOKELMA 10 g three times a day, patients who achieved normokalemia (3.5-5.0 mEq/L) within 72 hours (n=746; 99%) entered the maintenance phase. For maintenance treatment, the initial dosage of LOKELMA was 5 g once daily and was adjusted to a minimum of 5 g every other day up to maximum of 15 g once daily, based on serum potassium level. The treatment effect on serum potassium was maintained during continued therapy.
14.5 Study 4
The effectiveness of LOKELMA in lowering serum potassium was studied in a double-blind, placebo-controlled trial of 196 chronic hemodialysis patients (mean age 58 years, range 20 to 86 years) with persistent pre-dialysis hyperkalemia (mean baseline potassium 5.8 mEq/L) who were randomized to receive LOKELMA 5 g or placebo once daily on non-dialysis days (NCT03303521). During the dose adjustment period (initial 4 weeks), the dose was adjusted weekly in 5 g increments up to 15 g once daily based on pre-dialysis serum potassium measurement after the long inter-dialytic interval to achieve a pre-dialysis serum potassium level between 4.0-5.0 mEq/L. The dose reached at the end of the dose-adjustment period was maintained throughout the subsequent 4-week evaluation period.
The primary endpoint in the trial was the proportion of responders, defined as patients who maintained a pre-dialysis serum potassium between 4.0 and 5.0 mEq/L on at least 3 out of 4 dialysis treatments after the long inter-dialytic interval and who did not receive rescue therapy during the evaluation period. A greater proportion of patients were responders in the LOKELMA arm as compared to placebo (41% vs 1%, respectively; p<0.001). The treatment effect on mean pre-dialysis serum potassium levels was maintained during continued treatment. Mean pre-dialysis serum potassium levels during the study are presented in Figure 5.
Figure 5: Mean Pre-Dialysis Serum Potassium Levels Over Time in Patients on Chronic Hemodialysis
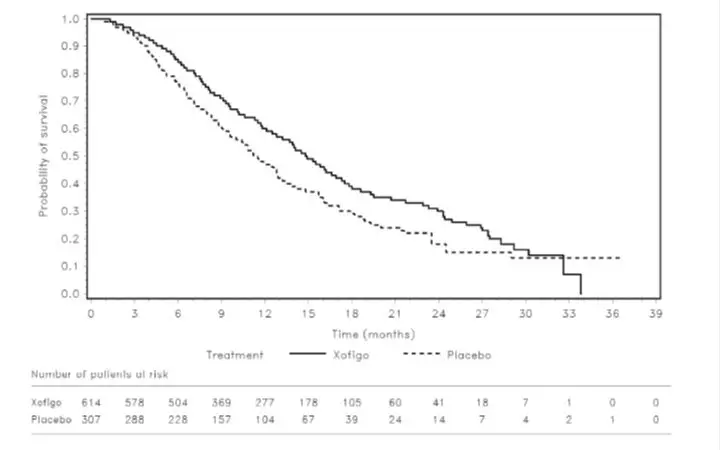
F/U - follow-up period
The displayed error bars correspond to 95% confidence intervals.
n = Number of patients with non-missing potassium measurements at a particular visit.
16. How is Lokelma supplied
LOKELMA (sodium zirconium cyclosilicate) for oral suspension is supplied as a white to grey powder in foil-lined packets as follows:
|
LOKELMA (grams) |
Single Packet |
Box of 11 Packets |
Box of 30 Packets |
|
5 |
NDC 0310-1105-01 |
NDC 0310-1105-39 |
NDC 0310-1105-30 |
|
10 |
NDC 0310-1110-01 |
NDC 0310-1110-39 |
NDC 0310-1110-30 |
Storage and Handling
Store LOKELMA at 15°C-30°C (59°F-86°F).
17. Patient Counseling Information
Dosing
Instruct the patient how to reconstitute LOKELMA for administration. Inform the patient that it is necessary to drink the full dose [see Dosage and Administration (2.3)].
Instruct dialysis patients who experience acute illness (e.g., decreased oral intake of food or fluids, diarrhea) to contact the health care provider. The dose of Lokelma may need to be adjusted [see Warnings and Precautions (5.3)].
Diagnostic Testing
Advise patients to notify their physician prior to an abdominal X-ray [see Warnings and Precautions (5.4)].
Drug Interactions
Advise patients who are taking other oral medications to separate dosing of LOKELMA by at least 2 hours (before or after) [see Drug Interactions (7)].
Diet
Advise patients to adjust dietary sodium, if appropriate [see Warnings and Precautions (5.2)].
U.S. Patent No: 6332985, 8808750, 8877255, 8802152, 9592253
© AstraZeneca 2021
Manufactured by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850


| LOKELMA
sodium zirconium cyclosilicate powder, for suspension |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| LOKELMA
sodium zirconium cyclosilicate powder, for suspension |
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
| Labeler - AstraZeneca Pharmaceuticals LP (054743190) |
| Registrant - AstraZeneca PLC (230790719) |
