Drug Detail:Nexletol (Bempedoic acid)
Drug Class: Miscellaneous antihyperlipidemic agents
Highlights of Prescribing Information
NEXLETOL (bempedoic acid) tablets, for oral use
Initial U.S. Approval: 2020
Indications and Usage for Nexletol
NEXLETOL is an adenosine triphosphate-citrate lyase (ACL) inhibitor indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or established atherosclerotic cardiovascular disease who require additional lowering of LDL-C. (1)
Limitations of Use: The effect of NEXLETOL on cardiovascular morbidity and mortality has not been determined. (1)
Nexletol Dosage and Administration
Administer 180 mg orally once daily with or without food. (2.1)
Dosage Forms and Strengths
Tablets: 180 mg (3)
Contraindications
None. (4)
Warnings and Precautions
- Hyperuricemia: Elevations in serum uric acid have occurred. Assess uric acid levels periodically as clinically indicated. Monitor for signs and symptoms of hyperuricemia, and initiate treatment with urate-lowering drugs as appropriate. (5.1)
- Tendon Rupture: Tendon rupture has occurred. Discontinue NEXLETOL at the first sign of tendon rupture. Avoid NEXLETOL in patients who have a history of tendon disorders or tendon rupture. (5.2)
Adverse Reactions/Side Effects
Most common (incidence ≥ 2% and greater than placebo) adverse reactions are upper respiratory tract infection, muscle spasms, hyperuricemia, back pain, abdominal pain or discomfort, bronchitis, pain in extremity, anemia, and elevated liver enzymes. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Esperion at 833-377-7633 (833 ESPRMED) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Simvastatin: Avoid concomitant use of NEXLETOL with simvastatin greater than 20 mg. (7)
- Pravastatin: Avoid concomitant use of NEXLETOL with pravastatin greater than 40 mg. (7)
Use In Specific Populations
- Pregnancy: Based on mechanism of action, may cause fetal harm. (8.1)
- Lactation: Breastfeeding is not recommended with NEXLETOL. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Nexletol
NEXLETOL is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or established atherosclerotic cardiovascular disease who require additional lowering of LDL-C.
3. Dosage Forms and Strengths
NEXLETOL is available as:
- Tablets: 180 mg, white to off-white, oval shaped, debossed with "180" on one side and "ESP" on the other side.
5. Warnings and Precautions
5.1 Hyperuricemia
NEXLETOL inhibits renal tubular OAT2 and may increase blood uric acid levels [see Clinical Pharmacology (12.3)]. In clinical trials, 26% of NEXLETOL-treated patients with normal baseline uric acid values (versus 9.5% placebo) experienced hyperuricemia one or more times, and 3.5% of patients experienced clinically significant hyperuricemia reported as an adverse reaction (versus 1.1% placebo). Increases in uric acid levels usually occurred within the first 4 weeks of treatment initiation and persisted throughout treatment. After 12 weeks of treatment, the mean placebo-adjusted increase in uric acid compared to baseline was 0.8 mg/dL for patients treated with NEXLETOL.
Elevated blood uric acid may lead to the development of gout. Gout was reported in 1.5% of patients treated with NEXLETOL and 0.4% of patients treated with placebo. The risk for gout events was higher in patients with a prior history of gout (11.2% NEXLETOL versus 1.7% placebo), although gout also occurred more frequently than placebo in patients treated with NEXLETOL who had no prior gout history (1.0% NEXLETOL versus 0.3% placebo).
Advise patients to contact their healthcare provider if symptoms of hyperuricemia occur. Assess serum uric acid when clinically indicated. Monitor patients for signs and symptoms of hyperuricemia, and initiate treatment with urate-lowering drugs as appropriate.
5.2 Tendon Rupture
NEXLETOL is associated with an increased risk of tendon rupture or injury. In clinical trials, tendon rupture occurred in 0.5% of patients treated with NEXLETOL versus 0% of placebo-treated patients and involved the rotator cuff (the shoulder), biceps tendon, or Achilles tendon. Tendon rupture occurred within weeks to months of starting NEXLETOL. Tendon rupture may occur more frequently in patients over 60 years of age, in those taking corticosteroid or fluoroquinolone drugs, in patients with renal failure, and in patients with previous tendon disorders.
Discontinue NEXLETOL immediately if the patient experiences rupture of a tendon. Consider discontinuing NEXLETOL if the patient experiences joint pain, swelling, or inflammation. Advise patients to rest at the first sign of tendinitis or tendon rupture and to contact their healthcare provider if tendinitis or tendon rupture symptoms occur. Consider alternative therapy in patients with a history of tendon disorders or tendon rupture.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hyperuricemia [see Warnings and Precautions (5.1)]
- Tendon Rupture [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data described below reflect exposure to NEXLETOL in two placebo-controlled trials that included 2009 patients treated with NEXLETOL for 52 weeks (median treatment duration of 52 weeks) [see Clinical Studies (14)]. The mean age for NEXLETOL-treated patients was 65.4 years, 29% were women, 3% were Hispanic, 95% White, 3% Black, 1% Asian, and 1% other races. All patients received NEXLETOL 180 mg orally once daily plus maximally tolerated statin therapy alone or in combination with other lipid-lowering therapies. At baseline, 97% of patients had clinical atherosclerotic cardiovascular disease (ASCVD) and about 4% had a diagnosis of heterozygous familial hypercholesterolemia (HeFH). Patients on simvastatin 40 mg/day or higher were excluded from the trials.
Adverse reactions led to discontinuation of treatment in 11% of NEXLETOL-treated patients and 8% of placebo-treated patients. The most common reasons for NEXLETOL treatment discontinuation were muscle spasms (0.5% versus 0.3% placebo), diarrhea (0.4% versus 0.1% placebo), and pain in extremity (0.3% versus 0.0% placebo). Adverse reactions reported in at least 2% of NEXLETOL-treated patients and more frequently than in placebo-treated patients are shown in Table 1.
| Adverse Reaction | NEXLETOL + Statin and ± Other Lipid Lowering Therapies (N = 2009) % | Placebo (N = 999) % |
|---|---|---|
|
||
| Upper respiratory tract infection | 4.5 | 4.0 |
| Muscle spasms | 3.6 | 2.3 |
| Hyperuricemia* | 3.5 | 1.1 |
| Back pain | 3.3 | 2.2 |
| Abdominal pain or discomfort† | 3.1 | 2.2 |
| Bronchitis | 3.0 | 2.5 |
| Pain in extremity | 3.0 | 1.7 |
| Anemia | 2.8 | 1.9 |
| Elevated liver enzymes‡ | 2.1 | 0.8 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of NEXLETOL. Because these reactions are reported voluntarily from a populationof uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity: angioedema, wheezing, rash, and urticaria.
7. Drug Interactions
| Simvastatin | |
| Clinical Impact: | Concomitant use of NEXLETOL with simvastatin causes an increase in simvastatin concentration and may increase the risk of simvastatin-related myopathy [see Clinical Pharmacology (12.3)]. |
| Intervention: | Avoid concomitant use of NEXLETOL with simvastatin greater than 20 mg. |
| Pravastatin | |
| Clinical Impact: | Concomitant use of NEXLETOL with pravastatin causes an increase in pravastatin concentration and may increase the risk of pravastatin-related myopathy [see Clinical Pharmacology (12.3)]. |
| Intervention: | Avoid concomitant use of NEXLETOL with pravastatin greater than 40 mg. |
8. Use In Specific Populations
8.4 Pediatric Use
The safety and effectiveness of NEXLETOL have not been established in pediatric patients.
8.5 Geriatric Use
Of the 3009 patients in clinical trials of NEXLETOL, 1753 (58%) were 65 years and older, while 478 (16%) were 75 years and older. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients. However, greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dosage adjustment is necessary in patients with mild or moderate renal impairment. There is limited experience with NEXLETOL in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2), and NEXLETOL has not been studied in patients with end-stage renal disease (ESRD) receiving dialysis [see Clinical Pharmacology (12.3)].
10. Overdosage
There is no clinical experience with NEXLETOL overdose. In the event of an overdosage, contact Poison Control (1-800-222-1222) for latest recommendations.
11. Nexletol Description
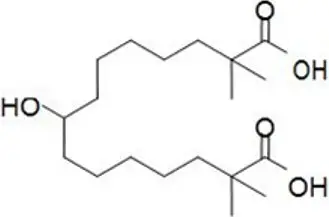
NEXLETOL tablets, for oral use, contain bempedoic acid, an adenosine triphosphate-citrate lyase (ACL) inhibitor. The chemical name for bempedoic acid is 8-hydroxy-2,2,14,14-tetramethyl-pentadecanedioic acid. The molecular formula is C19H36O5, and the molecular weight is 344.5 grams per mole. Bempedoic acid is a white to off-white crystalline powder that is highly soluble in ethanol, isopropanol and pH 8 phosphate buffer, and insoluble in water and aqueous solutions below pH 5.
Structural formula:
Each film-coated tablet of NEXLETOL contains 180 mg of bempedoic acid and the following inactive ingredients: colloidal silicon dioxide, hydroxyl propyl cellulose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The film coating comprises of partially hydrolyzed polyvinyl alcohol, polyethylene glycol, talc, and titanium dioxide.
12. Nexletol - Clinical Pharmacology
12.1 Mechanism of Action
Bempedoic acid is an adenosine triphosphate-citrate lyase (ACL) inhibitor that lowers low-density lipoprotein cholesterol (LDL-C) by inhibition of cholesterol synthesis in the liver. ACL is an enzyme upstream of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase in the cholesterol biosynthesis pathway. Bempedoic acid and its active metabolite, ESP15228, require coenzyme A (CoA) activation by very long-chain acyl-CoA synthetase 1 (ACSVL1) to ETC-1002-CoA and ESP15228-CoA, respectively. ACSVL1 is expressed primarily in the liver. Inhibition of ACL by ETC-1002-CoA results in decreased cholesterol synthesis in the liver and lowers LDL-C in blood via upregulation of low-density lipoprotein receptors.
12.2 Pharmacodynamics
Administration of bempedoic acid in combination with maximally tolerated statins, with or without other lipid modifying agents, decreases LDL-C, non-high density lipoprotein cholesterol (non-HDL-C), apolipoprotein B (apo B), and total cholesterol (TC) in patients with hyperlipidemia.
12.3 Pharmacokinetics
Bempedoic acid pharmacokinetic parameters are presented as the mean [standard deviation ± (SD)] unless otherwise specified. The steady-state maximum plasma concentration (Cmax) and area under the curve (AUC) following multiple-dose administration of bempedoic acid at 180 mg/day were 20.6 ± 6.1 µg/mL and 289.0 ± 96.4 µg∙h/mL, respectively. Bempedoic acid steady-state pharmacokinetics were generally linear over a range of > 60 mg to 220 mg (approximately 33% to 122% of the recommended dosage of 180 mg daily). There were no time-dependent changes in bempedoic acid pharmacokinetics following repeat administration at the recommended dosage, and bempedoic acid steady-state was achieved after 7 days. The mean accumulation ratio was approximately 2.3-fold.
The steady-state Cmax and AUC of the active metabolite (ESP15228) of bempedoic acid were 2.8 ± 0.9 µg/mL and 51.2 ± 17.2 µg∙h/mL, respectively. ESP15228 likely made a minor contribution to the overall clinical activity of bempedoic acid based on systemic exposure, relative potency, and pharmacokinetic properties.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Bempedoic acid was negative for mutagenicity in an in vitro Ames assay and negative for clastogenicity in the vitro human lymphocyte chromosome aberration assay. Bempedoic acid was negative in both in vivo mouse micronucleus and in vivo rat bone marrow micronucleus/liver comet assay. In a 2-year rat carcinogenicity study, Wistar rats were given oral doses of bempedoic acid at 3, 10 and 30 mg/kg/day. An increased incidence of liver hepatocellular adenomas and hepatocellular adenomas combined with carcinomas, thyroid gland follicular cell adenoma and follicular cell adenomas combined with carcinomas, and pancreatic islet cell adenomas combined with carcinomas were observed in male rats at the dose of 30 mg/kg/day (exposure equivalent to the maximum recommended human dose (MRHD), based on AUC). In a 2-year mice carcinogenicity study, CD-1 mice were given oral doses of bempedoic acid at 25, 75 and 150 mg/kg/day. Bempedoic acid-related increases in the incidence of liver hepatocellular adenomas, hepatocellular carcinomas and hepatocellular adenomas combined with carcinomas in male mice were observed at 75 and 150 mg/kg/day (exposures equivalent to the MRHD). Observations of liver and thyroid tumors are consistent with PPAR alpha agonism in rodents. The human relevance of pancreatic islet cell tumor findings is unknown.
In fertility and early embryofetal development study in rats, bempedoic acid was given orally to male and female rats at 10, 30 and 60 mg/kg/day. Males were dosed for 28 days prior to mating and females were dosed 14 days prior to mating through gestation day 7. No adverse effects on fertility were observed in females in the absence of maternal toxicity. No effects were observed on male fertility outcomes, but decreases in sperm counts were observed at 60 mg/kg/day (9 times the MRHD).
14. Clinical Studies
The efficacy of NEXLETOL was investigated in two multi-center, randomized, double-blind, placebo-controlled trials that enrolled 3009 adult patients with heterozygous familial hypercholesterolemia or established atherosclerotic cardiovascular disease who were on maximally tolerated statin therapy. Demographics and baseline disease characteristics were balanced between the treatment arms in all trials. In both trials, the maximum LDL-C lowering effects occurred at Week 4. These results were consistent across all subgroups studied in any of the trials, including age, gender, race, ethnicity, region, history of diabetes, baseline LDL-C, body mass index (BMI), HeFH status, and background therapies.
Study 1 (NCT02666664)
Study 1 was a multi-center, randomized, double-blind, placebo-controlled 52-week trial that evaluated safety and efficacy of bempedoic acid in patients with HeFH and/or ASCVD. Efficacy of NEXLETOL was evaluated at Week 12. The trial included 2230 patients randomized 2:1 to receive either NEXLETOL (n = 1488) or placebo (n = 742) as add-on to a maximally tolerated lipid lowering therapy. Maximally tolerated lipid lowering therapy was defined as a maximally tolerated statin dose alone or in combination with other lipid-lowering therapies. Patients were stratified by presence of HeFH and by baseline statin intensity. Patients on simvastatin 40 mg per day or higher and patients taking PCSK9 inhibitors were excluded from the trial.
Overall, the mean age at baseline was 66 years (range: 24 to 88 years), 61% were ≥ 65 years old, 27% women, 2% Hispanic, 96% White, 3% were Black, and 1% Asian. Ninety-five percent (95%) of patients had established atherosclerotic cardiovascular disease, and 5% of patients had HeFH. Twenty-nine percent (29%) of patients had diabetes at baseline. The mean baseline LDL-C was 103.2 mg/dL. At the time of randomization, all patients were receiving statin therapy and 50% were receiving high-intensity statin therapy.
The primary efficacy outcome measure of the study was the percent change from baseline to Week 12 in LDL-C. The difference between NEXLETOL and placebo in mean percent change in LDL-C from baseline to Week 12 was -18% (95% CI: -20%, -16%; p < 0.001). High-density lipoprotein (HDL) and triglycerides (TG) were examined as exploratory endpoints and were not included in the statistical hierarchy. The difference between NEXLETOL and placebo in mean percent change from baseline to Week 12 was -6% for HDL and median percent change from baseline to Week 12 was +3% for TG. For additional results see Table 2 and Figure 1.
| LDL-C*,† | Non-HDL-C† | apo B† | TC† | |
|---|---|---|---|---|
| apo B = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; TC = total cholesterol. | ||||
| Background statin: atorvastatin, simvastatin, pravastatin, | ||||
|
||||
| NEXLETOL ± Statin ± Other Lipid Lowering Therapies (180 mg/day; n = 1488‡) | -17 | -12 | -9 | -10 |
| Placebo (n = 742‡) | 2 | 2 | 3 | 1 |
| Mean Difference from Placebo (95% CI) | -18 (-20, -16) | -13 (-15, -12) | -12 (-14, -10) | -11 (-13, -10) |
Study 2 (NCT02991118)
Study 2 was a multi-center, randomized, double-blind, placebo-controlled, 52-week trial in patients with HeFH and/or ASCVD. Efficacy of NEXLETOL was evaluated at Week 12. The trial included 779 patients randomized 2:1 to receive either NEXLETOL (n = 522) or placebo (n = 257) as add-on to a maximally tolerated lipid lowering therapy. Maximally tolerated lipid lowering therapy was defined as a maximally tolerated statin dose alone or in combination with other lipid-lowering therapies. Patients were stratified by presence of HeFH and baseline statin intensity. Patients on simvastatin 40 mg/day or higher were excluded from the trial.
Overall, the mean age at baseline was 64 years (range: 28 to 91 years), 51% were ≥ 65 years old, 36% women, 8% Hispanic, 94% White, 5% were Black, and 1% Asian. Ninety-five percent (95%) of patients had established atherosclerotic cardiovascular disease, and 5% of patients had HeFH. Thirty percent (30%) of patients had diabetes at baseline. The mean baseline LDL-C was 120.4 mg/dL. At the time of randomization, 90% of patients were receiving statin therapy, 53% were receiving high-intensity statin therapy, and 0.3% were receiving PCSK9 inhibitors.
The primary efficacy outcome measure of the study was the percent change from baseline to Week 12 in LDL-C. The difference between NEXLETOL and placebo in mean percent change in LDL-C from baseline to Week 12 was -17 % (95% CI: -21%, -14%; p < 0.001). HDL and TG were exploratory endpoints and not included in the statistical hierarchy. The difference between NEXLETOL and placebo in mean percent change from baseline to Week 12 was -6% for HDL and the median percent change from baseline was -2% for TG. For additional results see Table 3 and Figure 1.
| LDL-C*,† | Non-HDL-C† | apo B† | TC† | |
|---|---|---|---|---|
| apo B = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; TC = total cholesterol. | ||||
| Background statin: atorvastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, pitavastatin, and lovastatin. | ||||
|
||||
| NEXLETOL ± Statin ± Other Lipid Lowering Therapies (180 mg/day; n = 522‡) | -15 | -11 | -9 | -10 |
| Placebo (n = 257‡) | 2 | 2 | 4 | 1 |
| Difference from Placebo (95% CI) | -17 (-21, -14) | -13 (-16, -10) | -13 (-16, -10) | -11 (-14, -9) |
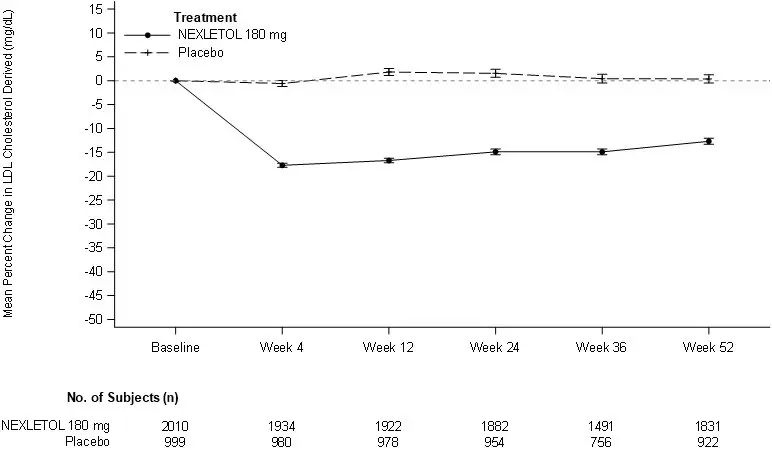
LDL-C derived is calculated from the Friedewald equation: LDL-C = TC - HDL-C - TG/5 in mg/dL.
The error bars represent standard error.

| NEXLETOL
bempedoic acid tablet, film coated |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Esperion Therapeutics, Inc. (029516312) |