Drug Detail:Qalsody (Tofersen)
Drug Class: Miscellaneous central nervous system agents
Highlights of Prescribing Information
QALSODY (tofersen) injection, for intrathecal use
Initial U.S. Approval: 2023
Indications and Usage for Qalsody
QALSODY is an antisense oligonucleotide indicated for the treatment of amyotrophic lateral sclerosis (ALS) in adults who have a mutation in the superoxide dismutase 1 (SOD1) gene. This indication is approved under accelerated approval based on reduction in plasma neurofilament light chain observed in patients treated with QALSODY. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s). (1)
Qalsody Dosage and Administration
QALSODY is administered intrathecally (2.1)
Dosing Information (2.1)
- Recommended dose: 100 milligrams (15 mL) per administration
- Initiate QALSODY treatment with 3 loading doses administered at 14day intervals. A maintenance dose should be administered once every 28 days thereafter.
Preparation and Administration Instructions (2.2)
- Allow to warm to room temperature prior to administration
- Administer within 4 hours of removal from vial
- Prior to administration, remove approximately 10 mL of cerebrospinal fluid
- Administer as an intrathecal bolus injection over 1 to 3 minutes
Dosage Forms and Strengths
Injection: 100 mg/15 mL (6.7 mg/mL) solution in a single-dose vial (3)
Contraindications
None (4)
Warnings and Precautions
- Myelitis and/ or Radiculitis: Serious events of myelitis and radiculitis have been reported. Monitor for symptoms; diagnostic workup and treatment should be initiated according to the standard of care. (5.1)
- Papilledema and Elevated Intracranial Pressure: Serious events of papilledema and elevated intracranial pressure have been reported. Monitor for symptoms; diagnostic workup and treatment should be initiated according to standard of care. (5.2)
- Aseptic Meningitis: Serious events of aseptic meningitis have been reported. Monitor for symptoms; diagnostic workup and treatment should be initiated according to standard of care. (5.3)
Adverse Reactions/Side Effects
The most common adverse reactions ( ≥ 10% of patients treated with QALSODY and greater than placebo) were pain, fatigue, arthralgia, cerebrospinal fluid white blood cell increased, and myalgia. (Section 6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Biogen at 1-877-725-7639 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2023
Full Prescribing Information
1. Indications and Usage for Qalsody
QALSODY is indicated for the treatment of amyotrophic lateral sclerosis (ALS) in adults who have a mutation in the superoxide dismutase 1 (SOD1) gene. This indication is approved under accelerated approval based on reduction in plasma neurofilament light chain (NfL) observed in patients treated with QALSODY [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s).
2. Qalsody Dosage and Administration
2.1. Dosing Information
Recommended Dosage
Administer QALSODY intrathecally using a lumbar puncture by, or under the direction of, healthcare professionals experienced in performing lumbar punctures.
The recommended dosage is 100 mg (15 mL) of QALSODY per administration.
Initiate QALSODY treatment with three (3) loading doses administered at 14-day intervals.
Administer a maintenance dose every 28 days thereafter.
2.2. Preparation and Administration Instructions
Use aseptic technique when preparing and administering QALSODY intrathecally. Prepare and administer QALSODY according to the following steps:
Preparation
Vial preparation instructions
- Allow refrigerated QALSODY vial to warm to room temperature (25°C/77°F) prior to administration without using external heat sources [see Storage and Handling (16.2)].
- Inspect the solution in the QALSODY vial prior to administration. Do not administer if particles are observed or the liquid in the vial is not clear and colorless to slightly yellow.
- Do not shake the QALSODY vial.
Procedural preparation instructions
- If indicated by the clinical condition of the patient, consider sedation.
- If indicated by the clinical condition of the patient, consider imaging to guide intrathecal administration of QALSODY.
- Prior to removing the vial's cap on the aluminum overseal, confirm readiness of the patient. An unopened QALSODY vial can be returned to the refrigerator [see Storage and Handling (16.2)].
- Evaluate patients prior to and after intrathecal injection for the presence of potential conditions related to lumbar puncture, to avoid serious procedural complications.
Administration
Prior to administration, remove approximately 10 mL of cerebrospinal spinal fluid (CSF) using a lumbar puncture needle.
Prior to administration, remove the plastic cap and attach a needle to the syringe, for the purpose of withdrawing QALSODY from the vial. Insert the needle into the vial through the center of the overseal and withdraw the required dose of 15 mL (equivalent to 100 mg) from the vial.
- Do not dilute QALSODY.
- External filters are not required.
Administer QALSODY using a lumbar puncture needle as an intrathecal bolus injection over 1 to 3 minutes.
- QALSODY contains no preservatives. Once drawn into the syringe, the solution should be administered immediately (within 4 hours of removal from the vial) at room temperature; otherwise, it must be discarded.
Any unused contents of the single-dose vial should be discarded.
3. Dosage Forms and Strengths
Injection: 100 mg/15 mL (6.7 mg/mL) as a clear and colorless to slightly yellow solution in a single-dose vial.
5. Warnings and Precautions
5.1. Myelitis and/or Radiculitis
Serious adverse reactions of myelitis and radiculitis have been reported in patients treated with QALSODY. Six patients treated with QALSODY experienced myelitis or radiculitis in the clinical studies. Two patients discontinued treatment with QALSODY and required symptomatic management with full resolution of symptoms. In the remaining 4 patients, symptoms resolved without discontinuation of QALSODY. If symptoms consistent with myelitis or radiculitis develop, diagnostic workup and treatment should be initiated according to the standard of care. Management may require interruption or discontinuation of QALSODY.
5.2. Papilledema and Elevated Intracranial Pressure
Serious adverse reactions of papilledema and elevated intracranial pressure have been reported in patients treated with QALSODY. Four patients developed elevated intracranial pressure and/or papilledema. All patients received treatment with standard of care with resolution of symptoms, and no events led to discontinuation of QALSODY. If symptoms consistent with papilledema or elevated intracranial pressure develop, diagnostic workup and treatment should be initiated according to the standard of care.
5.3. Aseptic Meningitis
Serious adverse reactions of aseptic meningitis (also called chemical meningitis or drug-induced aseptic meningitis) have been reported in patients treated with QALSODY. One patient experienced a serious adverse reaction of chemical meningitis, which led to discontinuation of QALSODY. One patient experienced a serious adverse reaction of aseptic meningitis, which did not lead to discontinuation of QALSODY. In addition, nonserious adverse drug reactions of CSF white blood cell increased, and CSF protein increased have also been reported with QALSODY [see Adverse Reactions (6.1)]. If symptoms consistent with aseptic meningitis develop, diagnostic workup and treatment should be initiated according to the standard of care.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- Myelitis and/or Radiculitis [see Warnings and Precautions (5.1)]
- Papilledema and Elevated Intracranial Pressure [see Warnings and Precautions (5.2)]
- Aseptic Meningitis [see Warnings and Precautions (5.3)]
6.1. Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of QALSODY cannot be directly compared to rates in clinical trials of other drugs and may not reflect the rates observed in practice.
The safety of QALSODY 100 mg was evaluated in 147 patients with SOD1-ALS. The median patient exposure was 119.4 weeks (range from 4 to 212 weeks). QALSODY was evaluated in the placebo-controlled Study 1 and in the open label extension Study 2. In Study 1 Part C, approximately 43% were female; 57% were male; 64% were White and 8% were Asian. The mean age at entry in Study 1 Part C was 49.8 years (range from 23 to 78 years).
The most common adverse reactions (≥ 10% of patients treated with QALSODY and greater than placebo) were pain, fatigue, arthralgia, CSF white blood cell increased, and myalgia. Table 1 shows the common adverse reactions that occurred in at least 5% of patients treated with QALSODY and at a 5% or higher frequency than placebo.
|
‡‡ Pain includes preferred terms of pain, back pain, and pain in extremity. |
||
|
* CSF white blood cell increased includes preferred terms of CSF white blood cell increased and pleocytosis. |
||
| Adverse Reaction | Study 1 Part C | |
| QALSODY
100 mg (n = 72) % | Placebo
(n = 36) % |
|
| Pain‡‡ | 42 | 22 |
| Fatigue | 17 | 6 |
| Arthralgia | 14 | 6 |
| CSF white blood cell increased* | 14 | 0 |
| Myalgia | 14 | 6 |
| CSF protein increased | 8 | 3 |
| Musculoskeletal stiffness | 6 | 0 |
| Neuralgia | 6 | 0 |
Less Common Adverse Reactions
Serious adverse reactions of myelitis and radiculitis; papilledema and elevated intracranial pressure; and aseptic meningitis have occurred in patients treated with QALSODY [see Warnings and Precautions (5.1, 5.2, 5.3)].
In the long-term extension study, nonserious adverse reactions of pyrexia have occurred with repeat administration of QALSODY.
8. Use In Specific Populations
8.1. Pregnancy
Risk Summary
There are no adequate data on developmental risks associated with the use of QALSODY in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Subcutaneous administration of tofersen (0, 3, 10, 30 mg/kg) every other day to pregnant mice during the period of organogenesis resulted in no adverse effects on embryofetal development. Plasma exposure at the highest dose tested (30 mg/kg) was approximately 4 times that in humans at the recommended human dose (RHD) of 100 mg.
Subcutaneous administration of tofersen (0, 3, 10, 30 mg/kg) every other day to pregnant rabbits during the period of organogenesis resulted in no adverse effects on embryofetal development. Plasma exposure at the highest dose tested (30 mg/kg) was approximately 20 times that in humans at the RHD.
Subcutaneous administration of tofersen (0, 3, 10, or 30 mg/kg) every other day to male and female mice prior to and during mating and continuing in females throughout organogenesis resulted in no adverse effects on pre- or postnatal development. Plasma exposures at the highest dose tested (30 mg/kg) were approximately 4 times that in humans at the RHD.
8.2. Lactation
Risk Summary
There are no data on the presence of tofersen or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. Tofersen was detected in the milk of lactating mice following subcutaneous administration. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for QALSODY and any potential adverse effects on the breastfed infant from QALSODY or from the underlying maternal condition.
8.5. Geriatric Use
A total of 13.5% (22/162) patients were 65 years of age and older and 1.2% (2/162) patients were 75 years of age and older at initiation of treatment in clinical studies for ALS in patients who have a mutation in the superoxide dismutase 1 (SOD1) gene [see Clinical Studies (14)]. No overall differences in safety or effectiveness were observed between these patients and younger patients, but a greater sensitivity of some older individuals cannot be ruled out. There is no evidence for special dosage considerations based on age when QALSODY is administered.
11. Qalsody Description
Tofersen, an antisense oligonucleotide, is a 20-base residue (20-mer) 5-10-5 MOE gapmer mixed backbone oligonucleotide. Of the nineteen internucleotide linkages, fifteen are 3′-O to 5′-O phosphorothioate diesters, and four are 3′-O to 5′-O phosphate diesters. Ten of the twenty sugar residues are 2-deoxy-D-ribose and the remainder are 2′-O-(2-methoxyethyl)-D-ribose (MOE). The residues are arranged so that there are five MOE nucleosides at the 5′ and 3′-ends of the molecule flanking a gap of ten 2′-deoxynucleosides. The cytosine and uridine bases are methylated at the 5-position. The structural formula is:
Figure 1: Structural Formula for Tofersen
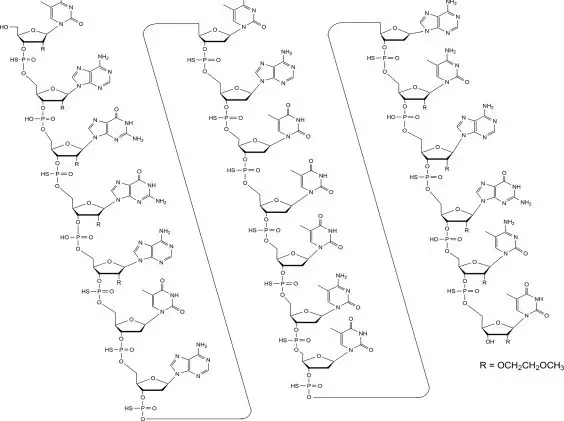
The molecular formula is C230 H317 N72 O123 P19 S15 and the molecular weight is 7127.86 atomic mass units (amu).
QALSODY is supplied as a sterile, preservative-free, clear, and colorless to slightly yellow solution in a Type I glass vial to be administered by intrathecal administration. Each vial of drug product contains a single dose of 100 mg tofersen at a concentration of 6.7 mg/mL in a formulation containing 0.21 mg/mL calcium chloride dihydrate, 0.11 mg/mL dibasic sodium phosphate, 0.16 mg/mL magnesium chloride hexahydrate, 0.03 mg/mL monobasic sodium phosphate, 0.22 mg/mL potassium chloride, 8.77 mg/mL sodium chloride, and water for injection. The pH of QALSODY is approximately 7.2 (range 6.7 to 7.7).
12. Qalsody - Clinical Pharmacology
12.1. Mechanism of Action
Tofersen is an antisense oligonucleotide that causes degradation of SOD1 mRNA through binding to SOD1 mRNA, which results in a reduction of SOD1 protein synthesis.
12.2. Pharmacodynamics
Effect of Tofersen on Total CSF SOD1 Protein
Total CSF SOD1, an indirect measure of target engagement, was evaluated in Study 1 Part C in SOD1-ALS patients [see Clinical Studies (14)].
At Week 28 in Study 1 Part C, a reduction in total CSF SOD1 protein of 35% (geometric mean ratio to baseline) in the tofersen-treated group versus a 2% decrease from baseline in the corresponding placebo subjects in the ITT population was observed (difference in geometric mean ratios for tofersen to placebo: 34%; nominal p<0.0001).
Effect of Tofersen on Neurofilament Proteins
Plasma NfL, a blood-based biomarker of axonal injury and neurodegeneration, was evaluated in Study 1 Part C in SOD1-ALS patients [see Clinical Studies (14)].
At Week 28 in Study 1 Part C, mean plasma NfL was reduced 55% (geometric mean ratio to baseline) in the QALSODY-treated subjects, compared to a 12% increase with placebo in ITT population (difference in geometric mean ratios for QALSODY to placebo: 60%; nominal p<0.0001). Plasma NfL declined until approximately Day 113, after which the reductions were sustained. The reductions in phosphorylated neurofilament heavy chain (pNfH) were similar compared to reductions in NfL, as were reductions in CSF compared to plasma.
12.3. Pharmacokinetics
Absorption
Intrathecal administration of QALSODY into the CSF allows tofersen to be distributed from the CSF to central nervous system tissues. The maximum CSF trough concentration occurred at the third dose, which was the last dose of the loading period. There was little to no accumulation for CSF tofersen with monthly dosing after the loading phase. Tofersen is transferred from CSF into the systemic circulation, with median time to maximum concentration (Tmax) plasma values ranging from 2 to 6 hours. There was no accumulation in plasma tofersen exposure following monthly maintenance dosing.
Distribution
Autopsy tissue from patients treated with tofersen (n=3) showed that tofersen administered intrathecally was distributed within the central nervous system tissues.
Elimination
Drug Interaction Studies
No clinical drug interaction studies have been performed. In vitro, QALSODY is not a substrate or inhibitor/inducer of major CYP enzymes or a substrate or inhibitor of major transporters.
12.6. Immunogenicity
As with all therapeutic oligonucleotides, there is a potential for immunogenicity.
Immunogenicity assay results are highly dependent on the sensitivity and specificity of the assay and may be influenced by several factors such as: assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to tofersen with the incidence of antibodies to other products may be misleading.
The immunogenic response to QALSODY was evaluated in 166 patients with post-baseline plasma samples for anti-drug antibodies (ADAs). Overall, 97 QALSODY-treated patients (58.4%) developed treatment-emergent ADAs, of which 14 were transient and 83 were persistent. The presence of anti-drug antibodies (ADA) appeared to decrease plasma tofersen clearance by 32%. Effects of ADA on CSF tofersen clearance is unknown. No discernible effects of ADAs on total SOD1 protein reduction or plasma NfL reduction have been observed. No discernible effects of ADAs on safety (incidence of AEs including hypersensitivity, anaphylactic reaction, and angioedema) have been observed. Medical review of individual cases of serious neurological events also showed no association with ADA status.
13. Nonclinical Toxicology
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Tofersen was negative in in vitro (bacterial reverse mutation and mammalian cell chromosomal aberration) and in vivo (mouse micronucleus) assays.
Impairment of Fertility
In a study to assess effects on fertility and reproductive function, tofersen (0, 3, 10, 30 mg/kg) was administered every other day to male and female mice prior to and during mating and continuing in females to gestation day (GD) 7. Adverse effects on male reproductive organs (seminiferous tubular degeneration, seminiferous tubule dilatation, spermatid retention, apoptosis of epithelial cells, increased cellular debris in the testes, and hypospermia in the epididymis) were observed at the highest dose tested; however, there were no adverse effects on functional endpoints. Plasma exposure at the no-effect dose (10 mg/kg) for adverse effects on male reproductive organs was approximately 2 times that in humans at the recommended human dose of 100 mg.
14. Clinical Studies
The efficacy of QALSODY was assessed in a 28-week randomized, double-blind, placebocontrolled clinical study in patients 23 to 78 years of age with weakness attributable to ALS and a SOD1 mutation confirmed by a central laboratory (Study 1 Part C, NCT02623699). One hundred eight (108) patients were randomized 2:1 to receive treatment with either QALSODY 100 mg (n = 72) or placebo (n = 36) for 24 weeks (3 loading doses followed by 5 maintenance doses). Concomitant riluzole and/or edaravone use was permitted for patients.
The prespecified primary analysis population (n = 60, modified intent to treat [mITT]) had a slow vital capacity (SVC) ≥ 65% of predicted value and met prognostic enrichment criteria for rapid disease progression, defined based on their pre-randomization ALS Functional Rating Scale–Revised (ALSFRS-R) decline slope and SOD1 mutation type.
The non-mITT population (n = 48) had a slow vital capacity (SVC) ≥ 50% of predicted value and did not meet the enrichment criteria for rapid disease progression.
Baseline disease characteristics in the overall intent-to-treat (ITT) population (combined mITT and non-mITT population) were generally similar in patients treated with QALSODY and patients who received placebo, with slightly shorter time from symptom onset and higher plasma NfL at baseline in the QALSODY group. At baseline, 62% of patients were taking riluzole, and 8% of patients were taking edaravone. Mean baseline ALSFRS-R score was 36.9 (5.9) in the QALSODY treatment group and 37.3 (5.8) in the placebo group. Median time from symptom onset was 11.4 months in the QALSODY treatment group and 14.6 months in the placebo group.
The primary efficacy analysis was the change from baseline to Week 28 in the ALSFRS-R total score in the mITT population, analyzed using the joint rank test to account for mortality in conjunction with multiple imputation (MI) to account for missing data for withdrawals other than death. Patients treated with QALSODY experienced less decline from baseline in the ALSFRS-R compared to placebo, but the results were not statistically significant (QALSODY-placebo adjusted mean difference [95% CI]: 1.2 [-3.2, 5.5]). Other clinical secondary outcomes also did not reach statistical significance.
Secondary endpoints of change from baseline at Week 28 in plasma NfL and CSF SOD1 protein were nominally statistically significant (see Table 2). NfL reduction was consistently observed for all subgroups based on sex, disease duration since symptom onset, site of onset, and riluzole/edaravone use.
|
Note 1: N is the number of patients with baseline value. |
||
|
Note 2: MI was used for missing data. Model included treatment, use of riluzole or edaravone, relevant baseline score and postbaseline values (natural log transformed data). Separate models for mITT and nonmITT were used and combined for ITT analyses. |
||
|
Note 3: Adjusted geometric mean ratios to baseline, treatment differences in adjusted geometric mean ratios to baseline and corresponding 95% CIs and nominal p-values were obtained from the ANCOVA model for change from baseline including treatment as a fixed effect and adjusting for the following covariates: baseline disease duration since symptom onset, relevant baseline score, and use of riluzole or edaravone. The analysis was based on natural log transformed data. |
||
| Biomarker Endpoints | QALSODY | Placebo |
| Plasma NfL | ||
| ITT population | N=72 | N=36 |
| Adjusted geometric mean ratio to baseline | 0.45 | 1.12 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) | 0.40 (0.33, 0.49) |
|
| Nominal p-value (ANCOVA+MI) | <0.0001 | |
| mITT population | N=39 | N=21 |
| Adjusted geometric mean ratio to baseline | 0.40 | 1.20 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) | 0.33 (0.25, 0.45) |
|
| Nominal p-value (ANCOVA+MI) | <0.0001 | |
| CSF SOD1 Protein | ||
| ITT population | N=72 | N=36 |
| Adjusted geometric mean ratio to baseline | 0.65 | 0.98 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) Nominal p-value (ANCOVA+MI) | 0.66 (0.57, 0.77) <0.0001 |
|
| mITT population | N=39 | N=21 |
| Adjusted geometric mean ratio to baseline | 0.71 | 1.16 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) | 0.62 (0.49, 0.78) |
|
| Nominal p-value (ANCOVA+MI) | <0.0001 | |
Figure 2: Plasma NfL Adjusted Geometric Mean Ratio to Baseline Values in Study 1 Part C by Study Week for the ITT Population
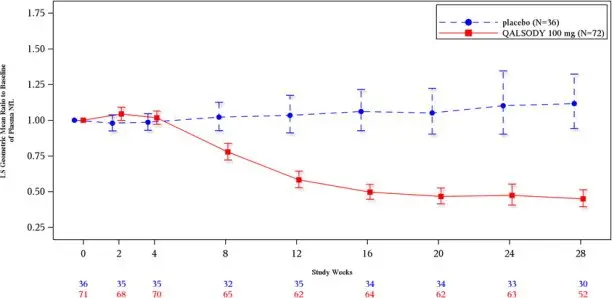
After completion of Study 1, patients had the option to enroll in an open-label extension study. At an interim analysis at 52 weeks, reductions in NfL were seen in patients previously receiving placebo who initiated QALSODY in the open-label extension study, similar to the reductions seen in patients treated with QALSODY in Study 1. Earlier initiation of QALSODY compared to placebo/delayed initiation of QALSODY was associated with trends for reduction in decline on ALSFRS-R, SVC percent-predicted, and hand-held dynamometry (HHD) megascore that were not statistically significant. Through all open-label follow-up at the time of the interim analysis, earlier initiation of QALSODY was also associated with a trend towards reduction of the risk of death or permanent ventilation, although it was not statistically significant. These exploratory analyses should be interpreted with caution given the limitations of data collected outside of a controlled study, which may be subject to confounding.
16. How is Qalsody supplied
16.1. How Supplied
QALSODY injection is a sterile, clear and colorless to slightly yellow solution supplied as 100 mg/15 mL (6.7 mg/mL) solution in a single-dose glass vial free of preservatives.
The NDC is 64406-109-01.
16.2. Storage and Handling
Store refrigerated between 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze.
If no refrigeration is available, QALSODY may be stored in its original carton, protected from light at or below 30°C (86°F) for up to 14 days.
If removed from the original carton, unopened vials of QALSODY can be removed from and returned to the refrigerator, if necessary, for not more than 6 hours per day at or below 30°C (86°F) for a maximum of 6 days (36 hours).
17. Patient Counseling Information
Myelitis and/or Radiculitis
Inform patients and caregivers that QALSODY could cause myelitis and radiculitis. Instruct patients and caregivers to contact their healthcare provider if symptoms consistent with these adverse reactions develop [see Warnings and Precautions (5.1)].
Papilledema and Elevated Intracranial Pressure
Inform patients and caregivers that QALSODY could cause papilledema and elevated intracranial pressure. Instruct patients and caregivers to contact their healthcare provider if symptoms consistent with these adverse reactions develop [see Warnings and Precautions (5.2)].
Aseptic Meningitis
Inform patients and caregivers that QALSODY could cause aseptic meningitis. Instruct patients and caregivers to contact their healthcare provider if symptoms consistent with meningitis develop [see Warnings and Precautions (5.3)].
57294-01
Manufactured by:
Biogen MA Inc.
Cambridge, MA 02142
QALSODY is a trademark of Biogen MA Inc.
© Biogen Inc.

| QALSODY
tofersen injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Biogen Inc. (121376230) |
