Drug Detail:Rebyota ((fecal microbiota, live-jslm))
Drug Class: Miscellaneous uncategorized agents
Highlights of Prescribing Information
REBYOTA™ (fecal microbiota, live - jslm) suspension, for rectal use
Initial U.S. Approval: 2022
Indications and Usage for Rebyota
REBYOTA is indicated for the prevention of recurrence of Clostridioides difficile infection (CDI) in individuals 18 years of age and older, following antibiotic treatment for recurrent CDI. (1)
Limitation of Use:
REBYOTA is not indicated for treatment of CDI.
Rebyota Dosage and Administration
For rectal administration only.
Administer REBYOTA 24 to 72 hours after the last dose of antibiotics for CDI. (2)
Administer a single dose of 150 mL rectally of REBYOTA. (2)
Dosage Forms and Strengths
Suspension. A single dose is 150 mL. (3)
Contraindications
Severe allergic reactions (e.g. anaphylaxis) to any component of REBYOTA. (4)
Adverse Reactions/Side Effects
The most commonly reported (≥ 3%) adverse reactions occurring in adults following a single dose of REBYOTA were abdominal pain, (8.9%), diarrhea (7.2%), abdominal distention (3.9%), flatulence (3.3%), and nausea (3.3%) (Table 1).
To report SUSPECTED ADVERSE REACTIONS, contact Ferring Pharmaceuticals Inc. at 1-888-FERRING (1-888-337-7464) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2022
Full Prescribing Information
1. Indications and Usage for Rebyota
REBYOTA is indicated for the prevention of recurrence of Clostridioides difficile infection (CDI) in individuals 18 years of age and older following antibiotic treatment for recurrent CDI.
2. Rebyota Dosage and Administration
For rectal administration only.
2.2 Preparation
Prior to use, thaw REBYOTA completely by placing the carton in a refrigerator, 2°C to 8°C (36°F to 46°F), for approximately 24 hours. REBYOTA carton may be stored in the refrigerator at 2°C to 8°C (36°F to 46°F) and used within 5 days, including thawing time.
DO NOT thaw using a heat source such as a microwave or hot water.
Condensation is normal after thawing.
Remove the thawed REBYOTA carton from the refrigerator. Remove the bag containing thawed REBYOTA from the outer carton and the inner carton insert. DO NOT remove the bag containing thawed REBYOTA from the sealed outer bag. Locate an Administration Set (supplied), water-soluble lubricant (not included) and a disposable underpad (not included) (See Figure 1).
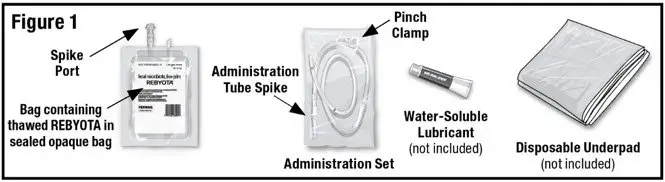
- 1.
- Open the administration set and close the pinch clamp by pushing the clamp until it is fully closed (see Figure 2).
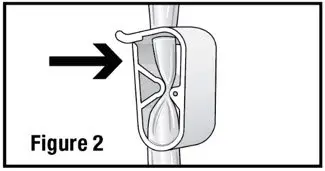
- 2.
- Remove the tab from the spike port of the bag containing thawed REBYOTA and remove the cap from the administration tube spike. Insert the administration tube spike through the spike port of the bag containing thawed REBYOTA (see Figure 3).
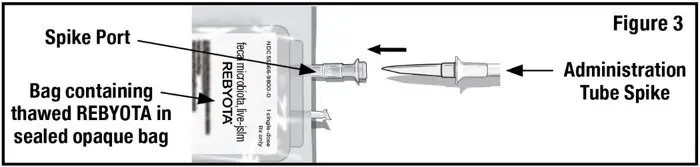
DO NOT remove air from the administration tube prior to insertion to avoid loss of REBYOTA.
2.3 Administration
Administer REBYOTA 24 to 72 hours after the last dose of antibiotics for CDI.
- 1.
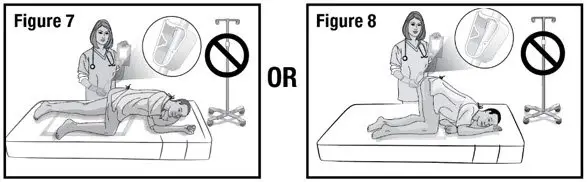
- Prepare the patient for administration by requesting they empty their bladder and bowel, if possible. Place the patient in the left-side position or the knee-chest position with a disposable underpad beneath the patient (see Figures 4 and 5).
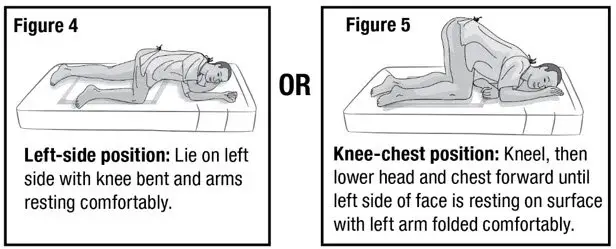
- 2.
- Apply water-soluble lubricant to the administration tube tip. Gently insert the administration tube tip into the rectum about 12 cm (5 inches) in a direction pointed slightly toward the navel (umbilicus) (see Figure 6).
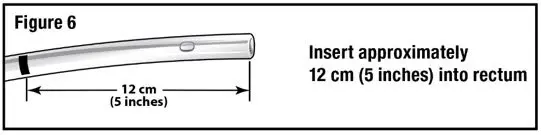
- 3.
- Hold the administration tube in place with one hand for the entire procedure to maintain the tube position in the rectum. With the other hand, open the pinch clamp on the administration tube, and then gradually raise the bag to allow delivery of REBYOTA via gravity flow (see Figure 7 and 8).
DO NOT allow the administration tube to sag or loop as this will prevent the entire dose from being delivered.
DO NOT squeeze the bag to deliver REBYOTA as this could be uncomfortable for the patient.
DO NOT hang the bag from an IV stand.
- 4.
- When the entire dose has been delivered, close the pinch clamp and then slowly withdraw the tube. Take care to prevent any residual REBYOTA remaining in the tube from leaking out.
NOTE: Some REBYOTA will remain in the tube after administration.
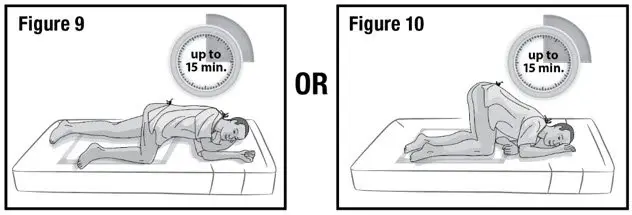
- 5.
- Keep the patient in the left-side position or the knee-chest position for up to 15 minutes to minimize any cramping that may occur (see Figure 9 and 10). There are no restrictions on the patient's use of the restroom.
Dispose of all components in medical waste.
4. Contraindications
Do not administer REBYOTA to individuals with a history of a severe allergic reaction (e.g. anaphylaxis) to any of the known product components [see Description 11].
5. Warnings and Precautions
5.1 Transmissible infectious agents
Because REBYOTA is manufactured from human fecal matter it may carry a risk of transmitting infectious agents. Any infection suspected by a physician possibly to have been transmitted by this product should be reported by the physician or other healthcare provider to Ferring Pharmaceuticals Inc.
6. Adverse Reactions/Side Effects
The most commonly reported (≥ 3%) adverse reactions occurring in adults following a single dose of REBYOTA were abdominal pain, (8.9%), diarrhea (7.2%), abdominal distention (3.9%), flatulence (3.3%), and nausea (3.3%).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of REBYOTA was evaluated in 2 randomized, double-blind clinical studies (Study 1: NCT03244644 and Study 2: NCT02299570) and 3 open-label clinical studies (NCT01925417, NCT02589847, NCT03931941) conducted in the United States and Canada. A total of 978 adults 18 years of age and older with a history of 1 or more recurrences of Clostridioides difficile (CDI) infection and whose symptoms were controlled 24 – 72 hours post-antibiotic treatment were enrolled and received 1 or more doses of REBYOTA; 595 of whom received a single dose of REBYOTA. In the 2 randomized, double-blind clinical studies, 131 adults were originally randomized to receive placebo and 48 crossed over to receive an open-label dose of REBYOTA after additional CDI recurrence. Overall, across the 5 studies, the median age of participants was 64 years and 67.2 % were female. The racial and ethnic distribution was as follows: 93.8% were white, and 2.4% were of Hispanic or Latino ethnicity. No meaningful differences in demographic characteristics occurred across the treatment groups. Study 1 and Study 2 excluded individuals with celiac disease, Inflammatory Bowel Disease, Irritable Bowel Syndrome, and chronic diarrhea. Individuals with these conditions were not excluded from one of the open-label studies (NCT03931941), and individuals with food allergies were not excluded from any of the 5 clinical studies.
Adverse Reactions
Across the 5 clinical studies, participants recorded solicited adverse events in a diary for the first 7 days after each dose of REBYOTA or placebo. Participants were monitored for all other adverse events by queries during scheduled visits, with duration of follow-up ranging from 6 to 24 months after the last dose. In Study 1, a multi-center, double-blind randomized (2:1), placebo-controlled trial conducted in the United States and Canada, 180 adults 18 years of age and older received a single dose of REBYOTA and 87 received placebo. Participants with a recurrence of CDI (rCDI) during the first 8 weeks after receipt of REBYOTA or placebo were censored from analysis at the time of rCDI. During the first two weeks following a dose of REBYOTA or placebo, 34 participants (18.9%) and 24 participants (27.6%) respectively, were censored. Overall, during the 8 week follow up period, 47 REBYOTA recipients (26.1%) and 30 placebo recipients (34.5%) were censored from analysis. In an analysis of solicited and unsolicited adverse events reported in Study 1, the most common adverse reactions (defined as adverse events assessed as definitely, possibly, or probably related to Investigational Product by the investigator) reported by ≥3% of REBYOTA recipients, and at a rate greater than that reported by placebo recipients, were abdominal pain, (8.9%), diarrhea (7.2%), abdominal distention (3.9%), flatulence (3.3%), and nausea (3.3%) (Table 1).
| Adverse Reaction | REBYOTA N=180 n (%) | Placebo N=87 n (%) |
|---|---|---|
|
||
| Abdominal Pain | 16 (8.9) | 6 (6.9) |
| Diarrhea | 13 (7.2) | 3 (3.4) |
| Abdominal distension | 7 (3.9) | 2 (2.3) |
| Flatulence | 6 (3.3) | 0 |
| Nausea | 6 (3.3) | 1 (1.1) |
Most adverse reactions occurred during the first 2 weeks after treatment. After this, the proportion of patients with adverse reactions declined in subsequent 2-week intervals. Beyond 2 weeks after treatment only a few single adverse reactions were reported. Most adverse drug reactions were mild to moderate in severity. No life-threatening adverse reaction was reported.
8. Use In Specific Populations
8.2 Lactation
REBYOTA is not absorbed systemically by the mother following rectal administration, and breastfeeding is not expected to result in exposure of the child to REBYOTA.
11. Rebyota Description
REBYOTA (fecal microbiota, live – jslm) is an opaque fecal microbiota suspension for rectal administration. REBYOTA is manufactured from human fecal matter sourced from qualified donors. The human fecal matter is tested for a panel of transmissible pathogens. Donors do not have dietary restrictions with respect to potential food allergens. The fecal microbiota suspension is the filtrate generated by processing the fecal matter in a pre-defined ratio with a solution of polyethylene glycol (PEG) 3350 and saline. Each 150mL dose of REBYOTA contains between 1×108 and 5×1010 colony forming units (CFU) per mL of fecal microbes including >1×105 CFU/mL of Bacteroides, and contains not greater than 5.97 grams of PEG3350 in saline.
14. Clinical Studies
The effectiveness of REBYOTA was evaluated using a Bayesian analysis of data from a randomized, double-blind, placebo-controlled, multicenter Phase 3 study (Study 1), which formally integrated treatment success rates from a placebo-controlled Phase 2 study (Study 2). Enrolled adults in both studies were 18 years of age or older and had a confirmed diagnosis of recurrent CDI (one or more episodes in Study 1; two or more episodes in Study 2) which was defined as diarrhea (passage of 3 or more loose bowel movements within a 24-hour period for 2 consecutive days) and a positive stool test for C. difficile toxin or toxigenic C. difficile, or had at least two episodes of severe CDI resulting in hospitalization within the last year. Enrolled adults were required to have completed at least 10 consecutive days of antibiotic therapy and have their CDI under control (<3 unformed/loose, i.e., Bristol Stool Scale type 6-7, stools/day for 2 consecutive days). A minimum of 24 hours to a maximum of 72 hours (Study 1) or 24 hours to a maximum of 48 hours (Study 2) antibiotic washout period was required prior to administration of the assigned study treatment. In Study 1, enrolled adults were randomized 2:1 to a single dose of REBYOTA or placebo respectively. In Study 2, randomization was 1:1:1 to receive two doses of REBYOTA, two doses of placebo, or one dose of REBYOTA and one dose of placebo, administered 7±2 days apart. Only data from the REBYOTA one-dose group and the placebo group are described below and were integrated in the Bayesian analysis.
In the integrated efficacy analysis set, the demographic profile and baseline recurrent CDI characteristics of treated adults were similar in the REBYOTA and placebo groups. In Study 1, a total of 262 adults were randomized and treated, of which 177 adults received REBYOTA and 85 received placebo. Adults had a mean age of 60.1 years with 45.4% of adults 65 years of age or older, were mainly white (92.0%) and female (69.1%). In this study, 32.8% of adults received REBYOTA or placebo for their first recurrence of CDI. In Study 1, 87.4% of adults had received vancomycin alone prior to treatment. In Study 2, 39 adults received one dose of REBYOTA and one dose of placebo and 43 adults received two doses of placebo. Adults in these two groups had a mean age of 59.8 years with 42.7% of adults 65 years of age or older, were mainly white (97.6%) and female (63.4%). In this study, 89.0% of adults had received vancomycin prior to treatment.
Treatment success was defined as the absence of CDI diarrhea within 8 weeks of blinded treatment. CDI diarrhea was defined as the passage of ≥ 3 unformed/loose stools in ≤ 24 hours for at least 2 consecutive days and a positive stool test for the presence of C difficile toxin at the time of the diarrhea.
In the Bayesian analysis, the estimated rate of treatment success was significantly higher in the REBYOTA group (70.6%) than in the Placebo group (57.5%) through 8 weeks after completing blinded treatment, resulting in a difference of 13.1 percentage points (95% Credible Interval: 2.3, 24.0) which corresponds to a 99.1% posterior probability that REBYOTA is superior to Placebo (Table 2).
| Parameter | REBYOTA Mean (95% CrI) | Placebo Mean (95% CrI) | Treatment Effect (REBYOTA – Placebo) Mean (95% CrI) |
|---|---|---|---|
| CrI=credible interval | |||
|
|||
| Model-Estimated Treatment Success (%) | 70.6 (64.1, 76.8) | 57.5 (48.1, 67.1) | 13.1 (2.3, 24.0) |
| Posterior Probability of Superiority | - | - | 0.991† |
Study 1 evaluated sustained clinical response which was defined as treatment success at 8 weeks and no CDI event through 6 months after the last dose during the blinded period. The difference in sustained clinical response rate (9.1%; 95% CI: -3.6%, 21.7%) was not statistically significant between the REBYOTA (65.5%) and the placebo groups (56.5%).
16. How is Rebyota supplied
16.1 How Supplied
REBYOTA and the administration set are shipped together in a box. Each box may contain up to 6 cartons of REBYOTA and up to 6 administration sets. Each carton of REBYOTA (NDC 55566-9800-2) contains a single dose.
17. Patient Counseling Information
Inform patients that REBYOTA is manufactured from human fecal matter and may contain food allergens.
Inform patients to notify their physician if persistent diarrhea, defined as 3 or more loose bowel movements within a 24-hour period for 2 consecutive days, returns following REBYOTA administration.
Patients should not take any oral antibiotic therapy for up to 8 weeks after administration of REBYOTA unless directed by their physician.

| REBYOTA
donor human stool suspension |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Ferring Pharmaceuticals Inc. (103722955) |
