Drug Detail:Rozlytrek (Entrectinib [ en-trek-ti-nib ])
Drug Class: Multikinase inhibitors
Highlights of Prescribing Information
ROZLYTREK (entrectinib) capsules, for oral use
Initial U.S. Approval: 2019
Recent Major Changes
| Indications and Usage (1.1, 1.2) | 7/2022 |
| Dosage and Administration (2.1) | 7/2022 |
Indications and Usage for Rozlytrek
ROZLYTREK is a kinase inhibitor indicated for the treatment of:
- Adult patients with ROS1-positive metastatic non-small cell lung cancer (NSCLC) as detected by an FDA-approved test. (1.1)
- Adult and pediatric patients 12 years of age and older with solid tumors that:
- have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion as detected by an FDA-approved test without a known acquired resistance mutation,
- are metastatic or where surgical resection is likely to result in severe morbidity, and
- have progressed following treatment or have no satisfactory alternative therapy.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. (1.2)
Rozlytrek Dosage and Administration
- Select patients for treatment based on the presence of ROS1 rearrangement(s) or NTRK gene fusion. (2.1)
- Recommended Dosage for ROS1-Positive Non-Small Cell Lung Cancer: 600 mg orally once daily. (2.2)
-
Recommended Dosage for NTRK Gene Fusion-Positive Solid Tumors:
- Adults: 600 mg orally once daily (2.3)
- Pediatric Patients 12 Years and Older: Recommended dosage is based on body surface area (BSA) as shown below (2.3)
- BSA greater than 1.50 m2: 600 mg once daily
- BSA 1.11 to 1.50 m2: 500 mg once daily
- BSA 0.91 to 1.10 m2: 400 mg once daily
Dosage Forms and Strengths
Capsules: 100 mg and 200 mg (3)
Contraindications
None. (4)
Warnings and Precautions
- Congestive Heart Failure (CHF): Assess left ventricular ejection fraction (LVEF) prior to initiation of ROZLYTREK in patients with symptoms or known risk factors for CHF. Monitor patients for clinical signs and symptoms of CHF. For patients with myocarditis, with or without a decreased ejection fraction, MRI or cardiac biopsy may be required to make the diagnosis. For new onset or worsening CHF, withhold ROZLYTREK, reassess LVEF and institute appropriate medical management. Reduce dose or permanently discontinue ROZLYTREK based on severity of CHF or worsening LVEF. (2.4, 5.1)
- Central Nervous System (CNS) Effects: CNS adverse reactions including cognitive impairment, mood disorders, dizziness, and sleep disturbances can occur with ROZLYTREK. Withhold and then resume at same or reduced dose upon improvement or permanently discontinue ROZLYTREK based on severity. (2.4, 5.2)
- Skeletal Fractures: ROZLYTREK increases the risk of fractures. Promptly evaluate patients with signs or symptoms of fractures. (5.3)
- Hepatotoxicity: Monitor liver tests, including ALT and AST, every 2 weeks during the first month of treatment, then monthly thereafter, and as clinically indicated. Withhold or permanently discontinue ROZLYTREK based on severity. If withheld, resume ROZLYTREK at same or reduced dose based on severity. (2.4, 5.4)
- Hyperuricemia: Assess serum uric acid levels prior to initiation and periodically during treatment with ROZLYTREK. Monitor patients for signs and symptoms of hyperuricemia. Initiate treatment with urate-lowering medications as clinically indicated and withhold ROZLYTREK for signs and symptoms of hyperuricemia. Resume at same or reduced dose upon improvement based on severity. (2.4, 5.5)
- QT Interval Prolongation: Monitor patients who have or who are at risk for QTc interval prolongation. Assess QT interval and electrolytes at baseline and periodically during treatment. Withhold and then resume at same or reduced dose, or permanently discontinue ROZLYTREK based on severity. (2.4, 5.6)
- Vision Disorders: Withhold for new visual changes or changes that interfere with activities of daily living until improvement or stabilization. Conduct an ophthalmological evaluation as appropriate. Resume at same or reduced dose upon improvement or stabilization. (2.4, 5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and use of effective contraception. (5.8, 8.1, 8.3)
Adverse Reactions/Side Effects
The most common adverse reactions (≥ 20%) were fatigue, constipation, dysgeusia, edema, dizziness, diarrhea, nausea, dysesthesia, dyspnea, myalgia, cognitive impairment, increased weight, cough, vomiting, pyrexia, arthralgia, and vision disorders. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
-
Moderate and Strong CYP3A Inhibitors:
- For adult and pediatric patients 12 years and older with a BSA greater than 1.50 m2, reduce the dose of ROZLYTREK if coadministration of moderate or strong CYP3A inhibitors cannot be avoided. (2.5, 7.1)
- For pediatric patients 12 years and older with a BSA less than or equal to 1.50 m2, avoid coadministration with ROZLYTREK. (7.1)
- Moderate and Strong CYP3A Inducers: Avoid coadministration with ROZLYTREK. (7.1)
Use In Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Rozlytrek
1.1 ROS1-Positive Non-Small Cell Lung Cancer
ROZLYTREK is indicated for the treatment of adult patients with ROS1-positive metastatic non-small cell lung cancer (NSCLC), as detected by an FDA-approved test.
1.2 NTRK Gene Fusion-Positive Solid Tumors
ROZLYTREK is indicated for the treatment of adult and pediatric patients 12 years of age and older with solid tumors that:
- have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion, as detected by an FDA-approved test without a known acquired resistance mutation,
- are metastatic or where surgical resection is likely to result in severe morbidity, and
- have either progressed following treatment or have no satisfactory alternative therapy.
This indication is approved under accelerated approval based on tumor response rate and durability of response [see Clinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
2. Rozlytrek Dosage and Administration
2.1 Patient Selection
Select patients for the treatment of metastatic NSCLC with ROZLYTREK based on the presence of ROS1 rearrangement(s) in tumor specimens [see Clinical Studies (14.1)].
Information on FDA-approved tests for the detection of ROS1 rearrangement(s) in NSCLC is available at http://www.fda.gov/CompanionDiagnostics.
Select patients for treatment of locally advanced or metastatic solid tumors with ROZLYTREK based on the presence of a NTRK gene fusion [see Clinical Studies (14.2)].
Information on FDA-approved tests for the detection of NTRK gene fusion(s) in solid tumors is available at http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage for ROS1-Positive Non-Small Cell Lung Cancer
The recommended dosage of ROZLYTREK is 600 mg orally once daily with or without food until disease progression or unacceptable toxicity.
2.3 Recommended Dosage for NTRK Gene Fusion-Positive Solid Tumors
Pediatric Patients 12 Years and Older (Adolescents)
The recommended dosage of ROZLYTREK is based on body surface area (BSA) as shown in Table 1 below. Take ROZLYTREK orally once daily with or without food until disease progression or unacceptable toxicity.
| Body Surface Area (BSA) | Recommended Dosage (Orally once daily) |
|---|---|
| Greater than 1.50 m2 | 600 mg |
| 1.11 to 1.50 m2 | 500 mg |
| 0.91 to 1.10 m2 | 400 mg |
2.4 Dosage Modifications for Adverse Reactions
The recommended dosage reductions for adverse reactions are provided in Table 2.
| Action | Adults and Pediatric Patients 12 Years and Older with BSA Greater than 1.50 m2
(Orally once daily) | Pediatric Patients 12 Years and Older with BSA of 1.11 to 1.50 m2
(Orally once daily) | Pediatric Patients 12 Years and Older with BSA of 0.91 to 1.10 m2
(Orally once daily) |
|---|---|---|---|
|
|||
| First dose reduction | 400 mg | 400 mg | 300 mg |
| Second dose reduction* | 200 mg | 200 mg | 200 mg |
Table 3 describes dosage modifications for specific adverse reactions.
| Adverse Reaction | Severity* | Dosage Modification |
|---|---|---|
|
||
| Congestive Heart Failure [see Warnings and Precautions (5.1)] | Grade 2 or 3 |
|
| Grade 4 |
|
|
| Central Nervous System Effects [see Warnings and Precautions (5.2)] | Intolerable Grade 2 |
|
| Grade 3 |
|
|
| Grade 4 |
|
|
| Hepatotoxicity [see Warnings and Precautions (5.4)] | Grade 3 |
|
| Grade 4 |
|
|
| ALT or AST greater than 3 times ULN with concurrent total bilirubin greater than 1.5 times ULN (in the absence of cholestasis or hemolysis). |
|
|
| Hyperuricemia [see Warnings and Precautions (5.5)] | Symptomatic or Grade 4 |
|
| QT Interval Prolongation [see Warnings and Precautions (5.6)] | QTc greater than 500 ms |
|
| Torsade de pointes; polymorphic ventricular tachycardia; signs/symptoms of serious arrhythmia |
|
|
| Vision Disorders [see Warnings and Precautions (5.7)] | Grade 2 or above |
|
| Anemia or Neutropenia [see Adverse Reactions (6.1)] | Grade 3 or 4 |
|
| Other Clinically Relevant Adverse Reactions | Grade 3 or 4 |
|
2.6 Administration
Swallow capsules whole. Do not open, crush, chew, or dissolve the contents of the capsule.
If a patient misses a dose, instruct patients to make up that dose unless the next dose is due within 12 hours.
If a patient vomits immediately after taking a dose, instruct patients to repeat that dose.
3. Dosage Forms and Strengths
Hard capsules:
- 100 mg: Size 2 yellow opaque body and cap, with "ENT 100" printed in blue ink on body.
- 200 mg: Size 0 orange opaque body and cap, with "ENT 200" printed in blue ink on body.
5. Warnings and Precautions
5.1 Congestive Heart Failure
Among the 355 patients who received ROZLYTREK across clinical trials, congestive heart failure (CHF) occurred in 3.4% of patients, including Grade 3 (2.3%) [see Adverse Reactions (6.1)]. In clinical trials, baseline cardiac function and routine cardiac monitoring other than electrocardiograms (ECGs) were not conducted and eligibility criteria excluded patients with symptomatic CHF, myocardial infarction, unstable angina, and coronary artery bypass graft within 3 months of study entry. Among the 12 patients with CHF, the median time to onset was 2 months (range: 11 days to 12 months). ROZLYTREK was interrupted in 6 of these patients (50%) and discontinued in 2 of these patients (17%). CHF resolved in 6 patients (50%) following interruption or discontinuation of ROZLYTREK and institution of appropriate medical management. In addition, myocarditis in the absence of CHF was documented in 0.3% of patients.
Assess left ventricular ejection fraction (LVEF) prior to initiation of ROZLYTREK in patients with symptoms or known risk factors for CHF. Monitor patients for clinical signs and symptoms of CHF, including shortness of breath and edema. For patients with myocarditis, with or without a decreased ejection fraction, MRI or cardiac biopsy may be required to make the diagnosis. For patients with new onset or worsening CHF, withhold ROZLYTREK, institute appropriate medical management, and reassess LVEF. Based on the severity of CHF or worsening LVEF, resume ROZLYTREK at a reduced dose upon recovery to baseline or permanently discontinue [see Dosage and Administration (2.4)].
5.2 Central Nervous System Effects
A broad spectrum of central nervous system (CNS) adverse reactions occurred in patients receiving ROZLYTREK, including cognitive impairment, mood disorders, dizziness, and sleep disturbances.
Among the 355 patients who received ROZLYTREK across clinical trials, 96 (27%) experienced cognitive impairment; symptoms occurred within 3 months of starting ROZLYTREK in 74 (77%). Cognitive impairment included cognitive disorders (8%), confusional state (7%), disturbance in attention (4.8%), memory impairment (3.7%), amnesia (2.5%), aphasia (2.3%), mental status changes (2%), hallucinations (1.1%), and delirium (0.8%). Grade 3 cognitive adverse reactions occurred in 4.5% of patients. Among the 96 patients with cognitive impairment, 13% required a dose reduction, 18% required dose interruption and 1% discontinued ROZLYTREK due to cognitive adverse reactions.
Among the 355 patients who received ROZLYTREK across clinical trials, 36 (10%) experienced mood disorders. The median time to onset of mood disorders was 1 month (range: 1 day to 9 months). Mood disorders occurring in ≥ 1% of patients included anxiety (4.8%), depression (2.8%) and agitation (2%). Grade 3 mood disorders occurred in 0.6% of patients. One completed suicide was reported 11 days after treatment had ended. Among the 36 patients who experienced mood disorders, 6% required a dose reduction, 6% required dose interruption and no patients discontinued ROZLYTREK due to mood disorders.
Dizziness occurred in 136 (38%) of the 355 patients. Among the 136 patients who experienced dizziness, Grade 3 dizziness occurred in 2.2% of patients. Ten percent of patients required a dose reduction, 7% required dose interruption and 0.7% discontinued ROZLYTREK due to dizziness.
Among the 355 patients who received ROZLYTREK across clinical trials, 51 (14%) experienced sleep disturbances. Sleep disturbances included insomnia (7%), somnolence (7%), hypersomnia (1.1%), and sleep disorder (0.3%). Grade 3 sleep disturbances occurred in 0.6% of patients. Among the 51 patients who experienced sleep disturbances, 6% required a dose reduction and no patients discontinued ROZLYTREK due to sleep disturbances.
The incidence of CNS adverse reactions was similar in patients with and without CNS metastases; however, the incidence of dizziness (38% vs 31%), headache (21% vs 13%), paresthesia (20% vs 6%), balance disorder (13% vs 4%), and confusional state (11% vs 2%) appeared to be increased in patients with CNS metastases who had received prior CNS irradiation (N = 90) compared to those who did not (N = 48).
Advise patients and caregivers of these risks with ROZLYTREK. Advise patients not to drive or operate hazardous machinery if they are experiencing CNS adverse reactions. Withhold and then resume at same or reduced dose upon improvement, or permanently discontinue ROZLYTREK based on severity [see Dosage and Administration (2.4)].
5.3 Skeletal Fractures
ROZLYTREK increases the risk of fractures. In an expanded safety population that included 338 adult patients and 30 pediatric patients who received ROZLYTREK across clinical trials, 5% of adult patients and 23% of pediatric patients experienced fractures [see Use in Specific Population (8.4)]. In adult patients, some fractures occurred in the setting of a fall or other trauma to the affected area, while in pediatric patients all fractures occurred in patients with minimal or no trauma. In general, there was inadequate assessment for tumor involvement at the site of fracture; however, radiologic abnormalities possibly indicative of tumor involvement were reported in some patients. In both adult and pediatric patients, most fractures were hip or other lower extremity fractures (e.g., femoral or tibial shaft). In a limited number of patients, bilateral femoral neck fractures occurred. The median time to fracture was 3.8 months (range 0.3 to 18.5 months) in adults and 4.0 months (range: 1.8 months to 7.4 months) in pediatric patients. ROZLYTREK was interrupted in 41% of adults and 43% of pediatric patients due to fractures. No patients discontinued ROZLYTREK due to fractures.
Promptly evaluate patients with signs or symptoms (e.g., pain, changes in mobility, deformity) of fractures. There are no data on the effects of ROZLYTREK on healing of known fractures and risk of future fractures.
5.4 Hepatotoxicity
Among the 355 patients who received ROZLYTREK, increased AST of any grade occurred in 42% of patients and increased ALT of any grade occurred in 36%. Grade 3 – 4 increased AST or ALT occurred in 2.5% and 2.8% of patients, respectively; the incidence may be underestimated as 4.5% of patients had no post-treatment liver function tests [see Adverse Reactions (6.1)]. The median time to onset of increased AST was 2 weeks (range: 1 day to 29.5 months). The median time to onset of increased ALT was 2 weeks (range: 1 day to 9.2 months). Increased AST or ALT leading to dose interruptions or reductions occurred in 0.8% and 0.8% of patients, respectively. ROZLYTREK was discontinued due to increased AST or ALT in 0.8% patients.
Monitor liver tests, including ALT and AST, every 2 weeks during the first month of treatment, then monthly thereafter, and as clinically indicated. Withhold or permanently discontinue ROZLYTREK based on the severity. If withheld, resume ROZLYTREK at the same or reduced dose [see Dosage and Administration (2.4)].
5.5 Hyperuricemia
Among 355 patients who received ROZLYTREK across clinical trials, 32 patients (9%) experienced hyperuricemia reported as adverse reactions with symptoms, as well as elevated uric acid levels. Grade 4 hyperuricemia occurred in 1.7% of patients, including one patient who died due to tumor lysis syndrome. Among the 32 patients with hyperuricemic adverse reactions, 34% required urate-lowering medication to reduce uric acid levels, 6% required dose reduction and 6% required dose interruption. Hyperuricemia resolved in 73% of patients following initiation of urate-lowering medication without interruption or dose reduction of ROZLYTREK. No patients discontinued ROZLYTREK due to hyperuricemia.
Assess serum uric acid levels prior to initiating ROZLYTREK and periodically during treatment. Monitor patients for signs and symptoms of hyperuricemia. Initiate treatment with urate-lowering medications as clinically indicated and withhold ROZLYTREK for signs and symptoms of hyperuricemia. Resume ROZLYTREK at same or reduced dose upon improvement of signs or symptoms based on severity [see Dosage and Administration (2.4)].
5.6 QT Interval Prolongation
Among the 355 patients who received ROZLYTREK across the clinical trials, 3.1% of patients with at least one post-baseline ECG assessment experienced QTcF interval prolongation of > 60 ms after starting ROZLYTREK and 0.6% had a QTcF interval > 500 ms [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)].
Monitor patients who already have or who are at significant risk of developing QTc interval prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, severe or uncontrolled heart failure and those taking other medicinal products associated with QT prolongation. Assess QT interval and electrolytes at baseline and periodically during treatment, adjusting frequency based upon risk factors such as congestive heart failure, electrolyte abnormalities, or concomitant medications known to prolong the QTc interval. Based on the severity of QTc interval prolongation, withhold ROZLYTREK and then resume at same or reduced dose, or permanently discontinue [see Dosage and Administration (2.4)].
5.7 Vision Disorders
Among the 355 patients who received ROZLYTREK across clinical trials, vision changes occurred in 21% of patients, including Grade 1 (17%), Grade 2 (2.8%) and Grade 3 (0.8%) [see Adverse Reactions (6.1)]. Vision disorders occurring in ≥ 1% included blurred vision (9%), photophobia (5%), diplopia (3.1%), visual impairment (2%), photopsia (1.1%), cataract (1.1%), and vitreous floaters (1.1%).
For patients with new visual changes or changes that interfere with activities of daily living, withhold ROZLYTREK until improvement or stabilization and conduct an ophthalmological evaluation as clinically appropriate. Upon improvement or stabilization, resume ROZLYTREK at same or reduced dose [see Dosage and Administration (2.4)].
5.8 Embryo-Fetal Toxicity
Based on literature reports in humans with congenital mutations leading to changes in TRK signaling, findings from animal studies, and its mechanism of action, ROZLYTREK can cause fetal harm when administered to a pregnant woman. Administration of entrectinib to pregnant rats resulted in malformations at exposures approximately 2.7 times the human exposure at the 600 mg dose based on area under the curve (AUC).
Advise pregnant women of the potential risk to a fetus. Advise female patients of reproductive potential to use effective contraception during treatment with ROZLYTREK and for 5 weeks following the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with ROZLYTREK and for 3 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Congestive Heart Failure [see Warnings and Precautions (5.1)]
- Central Nervous System Effects [see Warnings and Precautions (5.2)]
- Skeletal Fractures [see Warnings and Precautions (5.3)]
- Hepatotoxicity [see Warnings and Precautions (5.4)]
- Hyperuricemia [see Warnings and Precautions (5.5)]
- QT Interval Prolongation [see Warnings and Precautions (5.6)]
- Vision Disorders [see Warnings and Precautions (5.7)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Data in WARNINGS AND PRECAUTIONS and below reflect exposure to ROZLYTREK in 355 patients, including 172 (48%) patients exposed for 6 months or longer and 84 (24%) patients exposed for 1 year or longer. ROZLYTREK was studied in one dose-finding trial in adults [ALKA (n = 57)], one dose-finding and activity-estimating trial in adults [STARTRK-1 (n = 76)], one dose-finding and activity-estimating trial in pediatric and adult patients [STARTRK-NG (n = 16)], and one single arm, activity-estimating trial in adults [STARTRK-2 (n = 206)].
The population characteristics were: median age 55 years (range: 4 to 86 years); 5% (n = 17) were less than 18 years of age; 55% were female; and 66% were White, 23% were Asian, and 5% were Black; 3% were Hispanic/Latino. The most common tumors (≥ 5%) were lung (56%), sarcoma (8%), and colon (5%). ROS1 gene fusions were present in 42% and NTRK gene fusions were present in 20%. Most adults (75%) received ROZLYTREK 600 mg orally once daily. The doses ranged from 100 mg/m2 to 1600 mg/m2 once daily in adults and 250 mg/m2 to 750 mg/m2 once daily in pediatric patients. ROZLYTREK is not indicated for pediatric patients less than 12 years of age [see Use in Specific Populations (8.4)].
Serious adverse reactions occurred in 39% of patients. The most frequent serious adverse reactions (≥ 2%) were pneumonia (3.9%), dyspnea (3.7%), pleural effusion (3.4%), sepsis (2.5%), pulmonary embolism (2.3%), respiratory failure (2%), and pyrexia (2%). Grade 3 or 4 adverse reactions occurred in 60% of patients; the most common (≥ 2%) were lung infection (5%), increased weight (7%), dyspnea (6%), fatigue/asthenia (5%), cognitive disorders (4.5%), syncope (2.5%), pulmonary embolism (3.4%), hypoxia (3.4%), pleural effusion (3.1%), hypotension (2.8%), diarrhea (2%), and urinary tract infection (2.5%). Fatal events included dyspnea (0.6%), pneumonia (0.6%), sepsis (0.6%), completed suicide (0.3%), large intestine perforation (0.3%) and tumor lysis syndrome (0.3%). One patient developed Grade 4 myocarditis after one dose of ROZLYTREK which resolved after discontinuation of ROZLYTREK and administration of high-dose corticosteroids.
Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received ROZLYTREK. The most frequent adverse reactions (< 1% each) that resulted in permanent discontinuation were pneumonia, cardio-respiratory arrest, dyspnea, and fatigue.
Dose interruptions due to adverse reactions occurred in 46% of patients. The most frequent adverse reactions (≥ 2%) that resulted in interruption were increased blood creatinine (4%), fatigue (3.7%), anemia (3.1%), diarrhea (2.8%), pyrexia (2.8%), dizziness (2.5%), dyspnea (2.3%), nausea (2.3%), pneumonia (2.3%), cognitive disorder (2%) and neutropenia (2%).
Dose reductions due to adverse reactions occurred in 29% of patients who received ROZLYTREK. The most frequent adverse reactions resulting in dose reductions (≥ 1%) were dizziness (3.9%), increased blood creatinine (3.1%), fatigue (2.3%), anemia (1.7%), and increased weight (1.4%).
The most common adverse reactions (≥ 20%) were fatigue, constipation, dysgeusia, edema, dizziness, diarrhea, nausea, dysesthesia, dyspnea, myalgia, cognitive impairment, increased weight, cough, vomiting, pyrexia, arthralgia and vision disorders.
Table 4 summarizes the adverse reactions observed in these 355 patients.
| Adverse Reactions | ROZLYTREK N = 355 |
|
|---|---|---|
| All Grades (%) | Grade ≥ 3* (%) | |
|
||
| General | ||
| Fatigue† | 48 | 5 |
| Edema‡ | 40 | 1.1 |
| Pyrexia | 21 | 0.8 |
| Gastrointestinal | ||
| Constipation | 46 | 0.6 |
| Diarrhea | 35 | 2.0 |
| Nausea | 34 | 0.3 |
| Vomiting | 24 | 0.8 |
| Abdominal pain§ | 16 | 0.6 |
| Nervous System | ||
| Dysgeusia | 44 | 0.3 |
| Dizziness¶ | 38 | 0.8 |
| Dysesthesia# | 34 | 0.3 |
| Cognitive impairmentÞ | 27 | 4.5 |
| Peripheral sensory neuropathyß | 18 | 1.1 |
| Headache | 18 | 0.3 |
| Ataxiaà | 17 | 0.8 |
| Sleepè | 14 | 0.6 |
| Mood disordersð | 10 | 0.6 |
| Respiratory, Thoracic and Mediastinal | ||
| Dyspnea | 30 | 6* |
| Cough | 24 | 0.3 |
| Musculoskeletal and Connective Tissue | ||
| Myalgiaø | 28 | 1.1 |
| Arthralgia | 21 | 0.6 |
| Muscular weakness | 12 | 0.8 |
| Back pain | 12 | 1 |
| Pain in extremity | 11 | 0.3 |
| Metabolism and Nutritional | ||
| Increased weight | 25 | 7 |
| Decreased appetite | 13 | 0.3 |
| Dehydration | 10 | 1.1 |
| Eye | ||
| Vision disordersý | 21 | 0.8 |
| Infections | ||
| Urinary tract infection | 13 | 2.3 |
| Lung infection£ | 10 | 6* |
| Vascular | ||
| Hypotension¥ | 18 | 2.8 |
| Skin and Subcutaneous Tissue | ||
| RashΠ| 11 | 0.8 |
Clinically relevant adverse reactions occurring in ≤ 10% of patients include dysphagia (10%), fall (8%), pleural effusion (8%), fractures (6%), hypoxia (4.2%), pulmonary embolism (3.9%), syncope (3.9%), congestive heart failure (3.4%), and QT prolongation (3.1%).
Table 5 summarizes the laboratory abnormalities.
| Laboratory Abnormality | ROZLYTREK NCI CTCAE Grade |
|
|---|---|---|
| All Grades (%)* | Grade 3 or 4 (%)* | |
| AST: Aspartate Aminotransferase; ALT: Alanine Aminotransferase | ||
|
||
| Hematology | ||
| Anemia | 67 | 9 |
| Lymphopenia | 40 | 12 |
| Neutropenia | 28 | 7 |
| Chemistry | ||
| Increased creatinine† | 73 | 2.1 |
| Hyperuricemia | 52 | 10 |
| Increased AST | 44 | 2.7 |
| Increased ALT | 38 | 2.9 |
| Hypernatremia | 35 | 0.9 |
| Hypocalcemia | 34 | 1.8 |
| Hypophosphatemia | 30 | 7 |
| Increased lipase | 28 | 10 |
| Hypoalbuminemia | 28 | 2.9 |
| Increased amylase | 26 | 5.4 |
| Hyperkalemia | 25 | 1.5 |
| Increased alkaline phosphatase | 25 | 0.9 |
| Hyperglycemia‡ | NE‡ | 3.8 |
8. Use In Specific Populations
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
The safety and effectiveness of ROZLYTREK in pediatric patients aged 12 years and older with solid tumors that have an NTRK gene fusion have been established. The effectiveness of ROZLYTREK in adolescent patients was established based on extrapolation of data from three open-label, single-arm clinical trials in adult patients with solid tumors harboring an NTRK gene fusion (ALKA, STARTRK-1, and STARTRK-2) and pharmacokinetic data in adolescents enrolled in STARTRK-NG. ROZLYTREK doses based on body surface area in pediatric patients 12 years and older resulted in similar systemic exposure compared to that in adults who received a ROZLYTREK dose of 600 mg [see Dosage and Administration (2.3), Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.2)].
There is limited clinical experience with ROZLYTREK in pediatric patients. The safety of ROZLYTREK in pediatric patients 12 years of age and older was established based on extrapolation of data in adults and data from 30 pediatric patients enrolled in STARTRK-NG. Of these 30 patients, 7% were < 2 years (n = 2), 77% were 2 to < 12 years (n = 23), 17% were 12 to < 18 years (n = 5); 57% had metastatic disease (n = 17) and 44% had locally advanced disease (n = 13); and all patients had received prior treatment for their cancer, including surgery, radiotherapy, or systemic therapy. The most common cancers were neuroblastoma (47%), primary CNS tumors (30%), and sarcoma (10%). The median duration of exposure for all pediatric patients was 4.2 months (range: 0.2 to 22.7 months).
Due to the small number of pediatric and adult patients, the single arm design of clinical studies of ROZLYTREK, and confounding factors such as differences in susceptibility to infections between pediatric and adult patients, it is not possible to determine whether the observed differences in the incidence of adverse reactions to ROZLYTREK are related to patient age or other factors. In an expanded safety database that included 338 adult patients and 30 pediatric patients who received ROZLYTREK across clinical trials, the Grade 3 or 4 adverse reactions and laboratory abnormalities that occurred more frequently (≥ 5%) in pediatric patients (n = 30) compared with adults (n = 338) were neutropenia (27% vs 2%), bone fractures (23% vs 5%), increased weight (20% vs 7%), thrombocytopenia (10% vs 0.3%), lymphopenia (7% vs 1%), increased gamma-glutamyl transferase (7% vs 0%), and device-related infection (7% vs 0.3%). Three pediatric patients discontinued ROZLYTREK due to an adverse reaction (Grade 4 pulmonary edema, Grade 3 dyspnea, and Grade 4 pancreatitis).
The safety and effectiveness of ROZLYTREK in pediatric patients less than 12 years of age with solid tumors who have an NTRK gene fusion have not been established.
The safety and effectiveness of ROZLYTREK in pediatric patients with ROS1-positive NSCLC have not been established.
8.5 Geriatric Use
Of the 355 patients who received ROZLYTREK across clinical trials, 25% were 65 years or older, and 5% were 75 years of age or older. Clinical studies of ROZLYTREK did not include sufficient numbers of geriatric patients to determine whether they respond differently from younger patients.
8.6 Renal Impairment
No dose adjustment is recommended for patients with mild or moderate renal impairment (CLcr 30 to < 90 mL/min calculated by Cockcroft-Gault equation). ROZLYTREK has not been studied in patients with severe renal impairment (CLcr < 30 mL/min) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The effect of moderate hepatic impairment (total bilirubin > 1.5 – 3.0 times ULN with any aspartate aminotransferase) or severe hepatic impairment (total bilirubin >3.0 times ULN with any aspartate aminotransferase) on the safety of ROZLYTREK at the recommended dosage is unknown. Consider the risk-benefit profile of ROZLYTREK prior to determining whether to administer ROZLYTREK to patients with moderate to severe hepatic impairment. Monitor for ROZLYTREK adverse reactions in patients with hepatic impairment more frequently since these patients may be at increased risk for adverse reactions [see Clinical Pharmacology (12.3)].
11. Rozlytrek Description
Entrectinib is a kinase inhibitor. The molecular formula for entrectinib is C31H34F2N6O2 and the molecular weight is 560.64 Daltons. The chemical name is N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methylpiperazin-1-yl)-2-(tetrahydro-2H-pyran-4-ylamino) benzamide. The chemical structure of entrectinib is as follows:
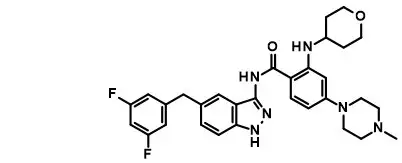
Entrectinib is white to pale pink powder.
ROZLYTREK (entrectinib) capsules for oral use are supplied as printed hard-shell capsules containing 100 mg (yellow opaque HPMC capsule) or 200 mg of entrectinib (orange opaque HPMC capsule). Inactive ingredients are tartaric acid, lactose anhydrous, hypromellose, crospovidone, microcrystalline cellulose, colloidal silicon dioxide, and magnesium stearate.
The yellow opaque capsule shell contains hypromellose, titanium dioxide, and yellow iron oxide. The orange opaque capsule shell contains hypromellose, titanium dioxide, and FD&C yellow #6. The printing ink contains shellac, propylene glycol, strong ammonia solution, and FD&C blue #2 aluminum lake.
12. Rozlytrek - Clinical Pharmacology
12.1 Mechanism of Action
Entrectinib is an inhibitor of the tropomyosin receptor tyrosine kinases (TRK) TRKA, TRKB, and TRKC (encoded by the neurotrophic tyrosine receptor kinase [NTRK] genes NTRK1, NTRK2, and NTRK3, respectively), proto-oncogene tyrosine-protein kinase ROS1 (ROS1), and anaplastic lymphoma kinase (ALK) with IC50 values of 0.1 to 2 nM. Entrectinib also inhibits JAK2 and TNK2 with IC50 values > 5 nM. The major active metabolite of entrectinib, M5, showed similar in vitro activity against TRK, ROS1, and ALK.
Fusion proteins that include TRK, ROS1, or ALK kinase domains can drive tumorigenic potential through hyperactivation of downstream signaling pathways leading to unconstrained cell proliferation. Entrectinib demonstrated in vitro and in vivo inhibition of cancer cell lines derived from multiple tumor types harboring NTRK, ROS1, and ALK fusion genes.
Entrectinib demonstrated steady-state brain-to-plasma concentration ratios of 0.4 – 2.2 in multiple animal species (mice, rats, and dogs) and demonstrated in vivo anti-tumor activity in mice with intracranial implantation of TRKA- and ALK-driven tumor cell lines.
12.2 Pharmacodynamics
Entrectinib exposure-response relationships and the time course of pharmacodynamic responses are unknown.
12.3 Pharmacokinetics
The pharmacokinetics for entrectinib and its pharmacologically active major circulating metabolite M5 were characterized in adult patients with ROS1-positive NSCLC, NTRK gene fusion-positive solid tumors, and healthy subjects. The pharmacokinetics of entrectinib and M5 are linear and are not dose-dependent or time-dependent. Steady state is achieved within one week for entrectinib and two weeks for M5 following daily administration of ROZLYTREK. The pharmacokinetic parameters for entrectinib and M5 are described in Table 6.
| Parameter | Entrectinib Mean* (% CV) | M5 Mean* (% CV) |
|---|---|---|
|
||
| AUCD1 (nM*h) | 31800 (48%) | 10200 (82%) |
| AUCss (nM*h) | 48000 (77%) | 24000 (97%) |
| CmaxD1 (nM) | 2250 (58%) | 622 (79%) |
| Cmaxss (nM) | 3130 (80%) | 1250 (90%) |
| Racc(AUC) | 1.55 (49%) | 2.84 (93%) |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were not conducted with entrectinib. Entrectinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay; however, an in vitro assay in cultured human peripheral blood lymphocytes did demonstrate a potential for abnormal chromosome segregation (aneugenicity). Entrectinib was not clastogenic or aneugenic in the in vivo micronucleus assay in rats and did not induce DNA damage in a comet assay in rats.
Dedicated fertility studies were not conducted with entrectinib. With the exception of dose-dependent decreases in prostate weight in male dogs, there were no effects on male and female reproductive organs observed in general toxicology studies conducted in rats and dogs at doses resulting in exposures of up to approximately 3.2 fold the human exposure (AUC) at the 600 mg dose.
14. Clinical Studies
14.1 ROS1-Positive Non-Small Cell Lung Cancer
The efficacy of ROZLYTREK was evaluated in a pooled subgroup of patients with ROS1-positive metastatic NSCLC who received ROZLYTREK at various doses and schedules (90% received ROZLYTREK 600 mg orally once daily) and were enrolled in one of three multicenter, single-arm, open-label clinical trials: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267). To be included in this pooled subgroup, patients were required to have histologically confirmed, recurrent or metastatic, ROS1-positive NSCLC, ECOG performance status ≤ 2, measurable disease per RECIST v 1.1, ≥ 18 months of follow-up from first post-treatment tumor assessment, and no prior therapy with a ROS1 inhibitor. Identification of ROS1 gene fusion in tumor specimens was prospectively determined in local laboratories using either a fluorescence in situ hybridization (FISH), next-generation sequencing (NGS), or polymerase chain reaction (PCR) laboratory-developed tests. All patients were assessed for CNS lesions at baseline. The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR) according to RECIST v1.1 as assessed by blinded independent central review (BICR). Intracranial response according to RECIST v1.1 was assessed by BICR. Tumor assessments with imaging were performed every 8 weeks.
Efficacy was assessed in 92 patients with ROS1-positive NSCLC. The median age was 53 years (range: 27 to 86); female (65%); White (48%), Asian (45%), and Black (5%); and Hispanic or Latino (2.4%); never smoked (59%); and ECOG performance status 0 or 1 (88%). Ninety-nine percent of patients had metastatic disease, including 42% with CNS metastases; 96% had adenocarcinoma; 65% received prior platinum-based chemotherapy for metastatic or recurrent disease and no patient had progressed in less than 6 months following platinum-based adjuvant or neoadjuvant therapy. ROS1 positivity was determined by NGS in 79%, FISH in 16%, and PCR in 4%. Twenty-five percent had central laboratory confirmation of ROS1 positivity using an analytically validated NGS test.
Efficacy results are summarized in Table 7.
| Confidence Interval (CI) calculated using the Clopper-Pearson method. | |
|
|
| Efficacy Parameters | ROZLYTREK N = 92 |
| Overall Response Rate (95% CI) | 74% (64, 83) |
| Complete Response | 15% |
| Partial Response | 59% |
| Duration of Response (DOR)* | N = 68 |
| Range (months) | 2.4, 55.2† |
| % DOR ≥ 9 months | 75% |
| % DOR ≥ 12 months | 57% |
| % DOR ≥ 18 months | 38% |
Among the 92 patients, 10 had measurable CNS metastases at baseline as assessed by BICR and had not received radiation therapy to the brain within 2 months prior to study entry. Responses in intracranial lesions were observed in 7 of these 10 patients.
14.2 NTRK Gene Fusion-Positive Solid Tumors
The efficacy of ROZLYTREK was evaluated in a pooled subgroup of adult patients with unresectable or metastatic solid tumors with a NTRK gene fusion enrolled in one of three multicenter, single-arm, open-label clinical trials: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267). To be included in this pooled subgroup, patients were required to have progressed following systemic therapy for their disease, if available, or would have required surgery causing significant morbidity for locally advanced disease; measurable disease per RECIST v1.1; at least 2 years of follow-up from first post-treatment tumor assessment; and no prior therapy with a TRK inhibitor. Patients received ROZLYTREK at various doses and schedules (94% received ROZLYTREK 600 mg orally once daily) until unacceptable toxicity or disease progression. Identification of positive NTRK gene fusion status was prospectively determined in local laboratories or a central laboratory using various nucleic acid-based tests. The major efficacy outcome measures were ORR and DOR, as determined by a BICR according to RECIST v1.1. Intracranial response according to RECIST v1.1 as evaluated by BICR. Tumor assessments with imaging were performed every 8 weeks.
Efficacy was assessed in the first 54 adult patients with solid tumors with an NTRK gene fusion enrolled into these trials. The median age was 58 years (range: 21 to 83); female (59%); White (80%), Asian (13%) and Hispanic or Latino (7%); and ECOG performance status 0 (43%) or 1 (46%). Ninety-six percent of patients had metastatic disease, including 22% with CNS metastases, and 4% had locally advanced, unresectable disease. All patients had received prior treatment for their cancer including surgery (n = 43), radiotherapy (n = 36), or systemic therapy (n = 48). Forty patients (74%) received prior systemic therapy for metastatic disease with a median of 1 prior systemic regimen and 17% (n = 9) received 3 or more prior systemic regimens. The most common cancers were sarcoma (24%), lung cancer (19%), salivary gland tumors (13%), breast cancer (11%), thyroid cancer (9%), and colorectal cancer (7%). A total of 52 (96%) patients had an NTRK gene fusion detected by NGS and 2 (4%) had an NTRK gene fusion detected by other nucleic acid-based tests. Eighty-three percent of patients had central laboratory confirmation of NTRK gene fusion using an analytically validated NGS test.
Efficacy results are summarized in Tables 8, 9, and 10.
|
|
| Efficacy Parameter | ROZLYTREK N = 54 |
| Overall Response Rate (95% CI) | 59% (45, 72) |
| Complete Response | 13% |
| Partial Response | 46% |
| Duration of Response* | N = 32 |
| Range (months) | 2.8, 47.8† |
| % with duration ≥ 6 months | 72% |
| % with duration ≥ 9 months | 66% |
| % with duration ≥ 12 months | 56% |
| Tumor Type | Patients N = 54 | ORR | DOR | |
|---|---|---|---|---|
| % | 95% CI | Range (months) |
||
| MASC: mammary analogue secretory carcinoma; NA = not applicable; PR = partial response. | ||||
|
||||
| Sarcoma | 13 | 46% | 19%, 75% | 2.8, 33.6* |
| Non-small cell lung cancer | 10 | 60% | 26%, 88% | 3.7, 47.8* |
| Salivary (MASC) | 7 | 86% | 42%, 100% | 2.8, 38.5* |
| Breast cancer | 6 | 83% | 36%, 100% | 4.2, 42.3* |
| Thyroid cancer | 5 | 60% | NA | 7.9, 31.5* |
| Colorectal cancer | 4 | 25% | NA | 15.1 |
| Neuroendocrine cancers | 3 | CR | NA | 32.9* |
| Pancreatic cancer | 3 | PR, PR | NA | 7.1, 12.9 |
| Gynecological cancers | 2 | PR | NA | 38.2 |
| Cholangiocarcinoma | 1 | PR | NA | 9.3 |
| NTRK Partner | Patients N = 54 | ORR | DOR | |
|---|---|---|---|---|
| % | 95% CI | Range (months) | ||
| PR = partial response; PD = progressive disease; SD = stable disease; NA = not applicable; NE = not evaluable. | ||||
|
||||
| ETV6 – NTRK3 | 25 | 72% | 51%, 88% | 2.8, 47.8* |
| TPM3 – NTRK1 | 4 | 50% | 7%, 93% | 2.8, 15.1 |
| TPR – NTRK1 | 4 | 100% | 40%, 100% | 5.6, 33.6* |
| LMNA – NTRK1 | 2 | PR, PD | NA | 4.2 |
| SQSTM1 – NTRK1 | 2 | PR, PD | NA | 18.8* |
| PEAR1 – NTRK1 | 2 | SD, NE | NA | NA |
| EML4 – NTRK3 | 2 | PR, NE | NA | 13.2 |
| CD74 – NTRK1 | 1 | PR | NA | 10.4 |
| PLEKHA6 – NTRK1 | 1 | PR | NA | 9.3 |
| CDC42BPA – NTRK1 | 1 | PR | NA | 29.4 |
| EPS15L1 – NTRK1 | 1 | PR | NA | 3.7 |
| RBPMS – NTRK3 | 1 | PR | NA | 4.6 |
| ERC1 – NTRK1 | 1 | SD | NA | NA |
| PDIA3 – NTRK1 | 1 | SD | NA | NA |
| TRIM33 – NTRK1 | 1 | SD | NA | NA |
| AKAP13 – NTRK3 | 1 | SD | NA | NA |
| KIF7 – NTRK3 | 1 | SD | NA | NA |
| FAM19A2 – NTRK3 | 1 | PD | NA | NA |
| CGN – NTRK1 | 1 | NE | NA | NA |
| SQSTM1 – NTRK2 | 1 | NE | NA | NA |
Among the subset of patients who received prior systemic therapy for metastatic disease, the ORR was 53%, similar to that seen in the overall population. Among the 54 adult patients, 4 had measurable CNS metastases at baseline as assessed by BICR and had not received radiation therapy to the brain within 2 months of study entry. Responses in intracranial lesions were observed in 3 of these 4 patients.
16. How is Rozlytrek supplied
- 100 mg hard capsules: Size 2 yellow opaque, with "ENT 100" printed in blue ink; available in:
HDPE bottles of 30 capsules: NDC 50242-091-30 - 200 mg hard capsules: Size 0 orange opaque, with "ENT 200" printed in blue ink; available in:
HDPE bottles of 90 capsules: NDC 50242-094-90
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
| PATIENT INFORMATION ROZLYTREK® (roz lye' trek) (entrectinib) capsules |
|||
|---|---|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Issued: 7/2022 | ||
| What is the most important information I should know about ROZLYTREK? | |||
ROZLYTREK may cause serious side effects, including:
|
|||
|
|
||
|
|||
|
|
||
| See "What are the possible side effects of ROZLYTREK?" for more information about side effects. | |||
| What is ROZLYTREK? | |||
ROZLYTREK is a prescription medicine used to treat:
|
|||
| It is not known if ROZLYTREK is safe and effective for use in children less than 12 years of age. | |||
Before taking ROZLYTREK, tell your healthcare provider about all your medical conditions, including if you:
|
|||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, or herbal supplements. | |||
| Certain other medicines may affect how ROZLYTREK works causing side effects. Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine. | |||
How should I take ROZLYTREK?
|
|||
What should I avoid while taking ROZLYTREK?
|
|||
| What are the possible side effects of ROZLYTREK? | |||
ROZLYTREK may cause serious side effects, including:
|
|||
| The most common side effects of ROZLYTREK include: | |||
|
|
|
|
| These are not all the possible side effects of ROZLYTREK. For more information, ask your healthcare provider or pharmacist. | |||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||
How should I store ROZLYTREK?
|
|||
| Keep ROZLYTREK and all medicines out of the reach of children. | |||
| General information about the safe and effective use of ROZLYTREK. | |||
| Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ROZLYTREK for a condition for which it was not prescribed. Do not give ROZLYTREK to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ROZLYTREK that is written for health professionals. | |||
| What are the ingredients in ROZLYTREK? | |||
| Active ingredient: entrectinib | |||
| Inactive ingredients: tartaric acid, lactose anhydrous, hypromellose, crospovidone, microcrystalline cellulose, colloidal silicon dioxide, and magnesium stearate. Yellow opaque capsule shell contains: hypromellose, titanium dioxide, and yellow iron oxide. Orange opaque capsule shell contains: hypromellose, titanium dioxide, and FD&C Yellow No. 6. Printing ink contains: shellac, propylene glycol, strong ammonia solution, and FD&C Blue No. 2 aluminum lake. | |||
| Distributed by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 | |||
| ROZLYTREK® is a registered trademark of Genentech, Inc. ©2022 Genentech, Inc. | |||
| For more information, go to www.ROZLYTREK.com or call 1-877-436-3683. | |||


| ROZLYTREK
entrectinib capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| ROZLYTREK
entrectinib capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Genentech, Inc. (080129000) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| F. Hoffmann-La Roche Ltd | 485244961 | ANALYSIS(50242-091, 50242-094) , LABEL(50242-091, 50242-094) , PACK(50242-091, 50242-094) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| F. Hoffmann-La Roche AG | 482242971 | API MANUFACTURE(50242-091, 50242-094) , ANALYSIS(50242-091, 50242-094) | |
