Drug Class: Antiviral combinations
Highlights of Prescribing Information
SYMTUZA ®(darunavir, cobicistat, emtricitabine, and tenofovir alafenamide) tablets, for oral use
Initial U.S. Approval: 2018
WARNING: POST TREATMENT ACUTE EXACERBATION OF HEPATITIS B
See full prescribing information for complete boxed warning.
Severe acute exacerbations of hepatitis B (HBV) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of SYMTUZA. Hepatic function should be monitored closely in these patients. If appropriate, anti-hepatitis B therapy may be warranted. ( 5.1)
Recent Major Changes
| Contraindications ( 4) | 04/2022 |
Indications and Usage for Symtuza
SYMTUZA is a four-drug combination of darunavir (DRV), a human immunodeficiency virus (HIV-1) protease inhibitor, cobicistat (COBI), a CYP3A inhibitor, and emtricitabine (FTC) and tenofovir alafenamide (TAF), both HIV-1 nucleoside analog reverse transcriptase inhibitors, and is indicated as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 40 kg:
- who have no prior antiretroviral treatment history or
- who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen for at least 6 months and have no known substitutions associated with resistance to darunavir or tenofovir. ( 1)
Symtuza Dosage and Administration
Testing: Prior to or when initiating SYMTUZA, test patients for HBV infection.
Prior to or when initiating SYMTUZA, and during treatment with SYMTUZA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. ( 2.1)
Recommended dosage: One tablet taken once daily with food in adults and pediatric patients, weighing at least 40 kg. ( 2.2)
Renal Impairment:SYMTUZA is not recommended in patients with estimated creatinine clearance below 30 mL/min. ( 2.3)
Hepatic Impairment: SYMTUZA is not recommended in patients with severe hepatic impairment. ( 2.4)
Dosage Forms and Strengths
Tablets: 800 mg of darunavir, 150 mg of cobicistat, 200 mg of emtricitabine, and 10 mg of tenofovir alafenamide (equivalent to 11.2 mg of tenofovir alafenamide fumarate). ( 3)
Contraindications
SYMTUZA is contraindicated to be co-administered with certain drugs for which altered plasma concentrations are associated with serious and/or life-threatening events or which may lead to loss of therapeutic effect of SYMTUZA and development of resistance. ( 4)
Warnings and Precautions
- Drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis) including some fatalities can occur with SYMTUZA. Monitor liver function before and during therapy, especially in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases. ( 5.2)
- Severe skin reactions, including Stevens-Johnson syndrome, toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms, and acute generalized exanthematous pustulosis may occur with SYMTUZA. Discontinue treatment if severe skin reaction develops. ( 5.3)
- Patients receiving SYMTUZA may develop new onset or exacerbations of immune reconstitution syndrome. ( 5.5)
- Monitor in patients with a known sulfonamide allergy. ( 5.7)
- Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. ( 5.8)
- Patients receiving SYMTUZA may develop new onset or exacerbation of diabetes mellitus/hyperglycemia and redistribution/accumulation of body fat. ( 5.9, 5.10)
- Patients with hemophilia may develop increase bleeding events. ( 5.11)
Adverse Reactions/Side Effects
The most common adverse reactions (all grades, incidence greater than or equal to 2%) were diarrhea, rash, nausea, fatigue, headache, abdominal discomfort, and flatulence. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Products, LP at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Co-administration of SYMTUZA with other drugs can alter the concentration of other drugs and other drugs may alter the concentrations of SYMTUZA components. Consult the full prescribing information prior to and during treatment for potential drug interactions. ( 4, 5.4, 7, 12.3)
Use In Specific Populations
- Pregnancy: SYMTUZA is not recommended during pregnancy due to substantially lower exposures of darunavir and cobicistat during pregnancy. ( 2.5, 8.1, 12.3)
- Lactation: Breastfeeding is not recommended. ( 8.2)
- Pediatrics: Not recommended for pediatric patients weighing less than 40 kg ( 8.4)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2023
Full Prescribing Information
WARNING: POST TREATMENT ACUTE EXACERBATION OF HEPATITIS B
Severe acute exacerbations of hepatitis B (HBV) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of SYMTUZA. Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue SYMTUZA. If appropriate, anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)] .
1. Indications and Usage for Symtuza
SYMTUZA is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 40 kg:
- who have no prior antiretroviral treatment history or
- who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen for at least 6 months and have no known substitutions associated with resistance to darunavir or tenofovir.
2. Symtuza Dosage and Administration
2.1 Testing Prior to Initiation of SYMTUZA
Prior to or when initiating SYMTUZA, test patients for hepatitis B (HBV) virus infection [see Warnings and Precautions (5.1)] .
Prior to or when initiating SYMTUZA, and during treatment with SYMTUZA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.6)] .
2.2 Recommended Dosage
SYMTUZA is a four-drug fixed-dose combination product containing 800 mg of darunavir (DRV), 150 mg of cobicistat (COBI), 200 mg of emtricitabine (FTC), and 10 mg of tenofovir alafenamide (TAF). The recommended dosage of SYMTUZA is one tablet taken orally once daily with food in adults and pediatric patients weighing at least 40 kg. For patients who are unable to swallow the whole tablet, SYMTUZA may be split into two pieces using a tablet-cutter, and the entire dose should be consumed immediately after splitting [see Clinical Pharmacology (12.3)] .
2.3 Not Recommended in Patients with Severe Renal Impairment
SYMTUZA is not recommended in patients with creatinine clearance below 30 mL per minute [see Use in Specific Populations (8.6)] .
2.4 Not Recommended in Patients with Severe Hepatic Impairment
SYMTUZA is not recommended for use in patients with severe hepatic impairment (Child-Pugh Class C) [see Use in Specific Populations (8.7)] .
2.5 Not Recommended During Pregnancy
SYMTUZA is not recommended during pregnancy because of substantially lower exposures of darunavir and cobicistat during the second and third trimesters [see Use in Specific Populations (8.1)and Clinical Pharmacology (12.3)].
SYMTUZA should not be initiated in pregnant individuals. An alternative regimen is recommended for those who become pregnant during therapy with SYMTUZA.
3. Dosage Forms and Strengths
Each SYMTUZA tablet contains darunavir ethanolate equivalent to 800 mg of darunavir, 150 mg of cobicistat, 200 mg of emtricitabine (FTC), and tenofovir alafenamide fumarate equivalent to 10 mg of tenofovir alafenamide (TAF). The yellow to yellowish-brown, capsule-shaped, film-coated tablet is debossed with "8121" on one side and "JG" on the other side.
4. Contraindications
Darunavir and cobicistat are both inhibitors of the cytochrome P450 3A (CYP3A) isoform. SYMTUZA should not be co-administered with medicinal products that are highly dependent on CYP3A for clearance and for which increased plasma concentrations are associated with serious and/or life-threatening events (narrow therapeutic index). Darunavir and cobicistat are both substrates of the cytochrome P450 3A (CYP3A) isoform. Co-administration of SYMTUZA with CYP3A inducers is expected to lower plasma concentrations of darunavir and cobicistat which may lead to loss of efficacy of darunavir and development of resistance. Examples of drugs that are contraindicated for co-administration with SYMTUZA due to the potential for serious and/or life-threatening events or loss of therapeutic effect [see Drug Interactions (7.5)] are listed below.
- Alpha 1-adrenoreceptor antagonist: alfuzosin
- Anticonvulsants: carbamazepine, phenobarbital, phenytoin
- Anti-gout: colchicine, in patients with renal and/or hepatic impairment
- Antimycobacterial: rifampin
- Antipsychotics: lurasidone, pimozide
- Cardiac Disorders: dronedarone, ivabradine, ranolazine
- Ergot derivatives, e.g., dihydroergotamine, ergotamine, methylergonovine
- Herbal product: St. John's wort ( Hypericum perforatum)
- Hepatitis C direct acting antiviral: elbasvir/grazoprevir
- Lipid modifying agents: lomitapide, lovastatin, simvastatin
- Opioid Antagonist: naloxegol
- PDE-5 inhibitor: sildenafil when used for treatment of pulmonary arterial hypertension
- Sedatives/hypnotics: orally administered midazolam, triazolam
5. Warnings and Precautions
5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
Patients with HIV-1 should be tested for the presence of chronic hepatitis B virus before initiating antiretroviral therapy [see Dosage and Administration (2.1)] . Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine and/or tenofovir disoproxil fumarate, and may occur with discontinuation of SYMTUZA. Patients coinfected with HIV-1 and HBV who discontinue SYMTUZA should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since post-treatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
5.2 Hepatotoxicity
Drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis) has been reported in clinical trials with darunavir, a component of SYMTUZA. Patients with pre-existing liver dysfunction, including chronic active hepatitis B or C, have an increased risk for liver function abnormalities including severe hepatic adverse reactions.
Post-marketing cases of liver injury, including some fatalities, have been reported with darunavir. These have generally occurred in patients with advanced HIV-1 disease taking multiple concomitant medications, having co-morbidities including hepatitis B or C co-infection, and/or developing immune reconstitution syndrome. A causal relationship with darunavir therapy has not been established.
Appropriate laboratory testing should be conducted prior to initiating therapy with SYMTUZA and patients should be monitored during treatment as clinically appropriate. Increased AST/ALT monitoring should be considered in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases, especially during the first several months of SYMTUZA treatment.
Evidence of new or worsening liver dysfunction (including clinically significant elevation of liver enzymes and/or symptoms such as fatigue, anorexia, nausea, jaundice, dark urine, liver tenderness, hepatomegaly) should prompt consideration of interruption or discontinuation of SYMTUZA.
5.3 Severe Skin Reactions
In patients receiving darunavir, a component of SYMTUZA, severe skin reactions may occur. These include conditions accompanied by fever and/or elevations of transaminases. Stevens-Johnson syndrome was reported with darunavir co-administered with cobicistat in clinical trials at a rate of 0.1%. During darunavir post-marketing experience, toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms (DRESS), and acute generalized exanthematous pustulosis have been reported. Discontinue SYMTUZA immediately if signs or symptoms of severe skin reactions develop. These can include but are not limited to severe rash or rash accompanied with fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, hepatitis, and/or eosinophilia.
Rash events of any cause and any grade occurred in 15% of subjects with no prior antiretroviral treatment history treated with SYMTUZA in the AMBER trial [see Adverse Reactions (6.1)] . Rash events were mild-to-moderate, often occurring within the first four weeks of treatment and resolving with continued dosing. The discontinuation rate due to rash in subjects using SYMTUZA was 2%.
5.4 Risk of Serious Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of SYMTUZA and other drugs may result in known or potentially significant drug interactions which may lead to [see Contraindications (4)and Drug Interactions (7.5)] :
- Clinically significant adverse reactions from greater exposures of concomitant drugs.
- Clinically significant adverse reactions from greater exposures of SYMTUZA.
- Loss of therapeutic effect of the concomitant drugs from lower exposures of active metabolite(s).
- Loss of therapeutic effect of SYMTUZA and possible development of resistance from lower exposures of SYMTUZA.
See Table 4for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during SYMTUZA therapy; review concomitant medications during SYMTUZA therapy; and monitor for the adverse reactions associated with concomitant medications [see Contraindications (4)and Drug Interactions (7)] .
When used with concomitant medications, SYMTUZA, which contains darunavir boosted with cobicistat, may result in different drug interactions than those observed or expected with darunavir co-administered with ritonavir. Complex or unknown mechanisms of drug interactions preclude extrapolation of drug interactions with darunavir co-administered with ritonavir to certain SYMTUZA interactions [see Drug Interactions (7)and Clinical Pharmacology (12.3)] .
5.5 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium aviuminfection, cytomegalovirus, Pneumocystis jiroveciipneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of antiretroviral treatment.
5.6 New Onset or Worsening Renal Impairment
Postmarketing cases of renal impairment, including acute renal failure, proximal renal tubulopathy (PRT), and Fanconi syndrome have been reported with TAF-containing products; while most of these cases were characterized by potential confounders that may have contributed to the reported renal events, it is also possible these factors may have predisposed patients to tenofovir-related adverse events [see Adverse Reactions (6.1, 6.2)] . SYMTUZA is not recommended in patients with estimated creatinine clearance below 30 mL per minute.
Patients taking tenofovir prodrugs who have impaired renal function and those taking nephrotoxic agents including non-steroidal anti-inflammatory drugs are at increased risk of developing renal-related adverse reactions.
Prior to or when initiating SYMTUZA and during treatment with SYMTUZA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. Discontinue SYMTUZA in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
Cobicistat, a component of SYMTUZA, produces elevations of serum creatinine due to inhibition of tubular secretion of creatinine without affecting glomerular filtration. This effect should be considered when interpreting changes in estimated creatinine clearance in patients initiating SYMTUZA, particularly in patients with medical conditions or receiving drugs needing monitoring with estimated creatinine clearance. The elevation is typically seen within 2 weeks of starting therapy and is reversible after discontinuation. Patients who experience a confirmed increase in serum creatinine of greater than 0.4 mg/dL should be closely monitored for renal safety.
5.7 Sulfa Allergy
Darunavir contains a sulfonamide moiety. Monitor patients with a known sulfonamide allergy after initiating SYMTUZA. In clinical studies with darunavir co-administered with ritonavir, the incidence and severity of rash were similar in subjects with or without a history of sulfonamide allergy.
5.8 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including emtricitabine, a component of SYMTUZA, and TDF, another prodrug of tenofovir, alone or in combination with other antiretrovirals. Treatment with SYMTUZA should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.9 Diabetes Mellitus/Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in HIV infected patients receiving HIV protease inhibitor (PI) therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued PI therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and causal relationships between HIV PI therapy and these events have not been established.
5.10 Fat Redistribution
Redistribution/accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
5.11 Hemophilia
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis in patients with hemophilia type A and B treated with HIV protease inhibitors (PIs). In some patients, additional factor VIII was given. In more than half of the reported cases, treatment with HIV PIs was continued or reintroduced if treatment had been discontinued. A causal relationship between PI therapy and these episodes has not been established.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in other sections of the labeling:
- Severe acute exacerbations of hepatitis B [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Severe skin reactions [see Warnings and Precautions (5.3)]
- Immune reconstitution syndrome [see Warnings and Precautions (5.5)]
- New onset or worsening renal impairment [see Warnings and Precautions (5.6)]
- Lactic acidosis/severe hepatomegaly with steatosis [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials in Adults
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of darunavir. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
7. Drug Interactions
7.1 Not Recommended With Other Antiretroviral Medications
SYMTUZA is a complete regimen for HIV-1 infection and co-administration with other antiretroviral medications for the treatment of HIV-1 infection is not recommended. For this reason, information regarding potential drug-drug interactions with other antiretroviral medications is not provided.
7.2 Potential for SYMTUZA to Affect Other Drugs
Darunavir co-administered with cobicistat is an inhibitor of CYP3A and CYP2D6. Cobicistat inhibits the following transporters: P-glycoprotein (P-gp), BCRP, MATE1, OATP1B1 and OATP1B3. Therefore, co-administration of SYMTUZA with drugs that are primarily metabolized by CYP3A and/or CYP2D6, or are substrates of P-gp, BCRP, MATE1, OATP1B1 or OATP1B3 may result in increased plasma concentrations of such drugs, which could increase or prolong their therapeutic effect and can be associated with adverse events. Co-administration of SYMTUZA with drugs that have active metabolite(s) formed by CYP3A may result in reduced plasma concentrations of these active metabolite(s), potentially leading to loss of their therapeutic effect (see Table 4).
7.3 Potential for Other Drugs to Affect SYMTUZA
Darunavir is metabolized by CYP3A. Cobicistat is metabolized by CYP3A and, to a minor extent, by CYP2D6. Co-administration of drugs that induce CYP3A activity are expected to increase the clearance of darunavir and cobicistat, resulting in lowered plasma concentrations which may lead to loss of therapeutic effect and development of resistance. Co-administration of SYMTUZA with other drugs that inhibit CYP3A may result in increased plasma concentrations of darunavir and cobicistat (see Table 4).
Tenofovir alafenamide (TAF) is a substrate of P-gp, BCRP, OATP1B1, and OATP1B3. Drugs that strongly affect P-gp activity may lead to changes in TAF absorption. Drugs that induce P-gp activity are expected to decrease the absorption of TAF, resulting in decreased plasma concentrations of TAF, which may lead to loss of therapeutic effect of SYMTUZA and development of resistance. Co-administration of SYMTUZA with other drugs that inhibit P-gp may increase the absorption and plasma concentrations of TAF (see Table 4).
7.4 Drugs Affecting Renal Function
Because emtricitabine and tenofovir are primarily excreted by the kidneys through glomerular filtration and active tubular secretion, co-administration of SYMTUZA with drugs that reduce renal function or compete for active tubular secretion may increase concentrations of emtricitabine, tenofovir, and other renally eliminated drugs and this may increase the risk of adverse reactions. Some examples of drugs that are eliminated by active tubular secretion include, but are not limited to, acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.6)] .
7.5 Significant Drug Interactions
Table 4 provides examples of established or potentially clinically significant drug interactions with SYMTUZA and recommended steps to prevent or manage these interactions. These recommendations are based on drug interaction trials conducted with the components of SYMTUZA, as individual agents or in combination, or are predicted interactions. No drug interaction trials have been performed with SYMTUZA or with all the components administered together. Drug interaction trials have been conducted with darunavir co-administered with ritonavir or cobicistat or with emtricitabine and tenofovir prodrugs. The table includes potentially significant interactions but is not all inclusive ,and therefore the label of each drug that is co- administered with SYMTUZA should be consulted for information related to the route of metabolism, interaction pathways, potential risks, and specific actions to be taken with regard to co- administration.
| Concomitant Drug Class: Drug Name
Examples | Effect on Concentration | Clinical Comment |
|---|---|---|
| This table is not all inclusive
↑ = increase, ↓ = decrease, ↔ = no effect |
||
| Alpha 1-adrenoreceptor antagonist:
alfuzosin | ↑ alfuzosin | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as hypotension. |
| Antibacterials:
clarithromycin, erythromycin, telithromycin | ↑ darunavir
↑ cobicistat ↑ antibacterial | Consider alternative antibiotics with concomitant use of SYMTUZA. |
| Anticancer agents:
dasatinib, nilotinib | ↑ anticancer agent | A decrease in the dosage or an adjustment of the dosing interval of dasatinib or nilotinib may be necessary when co-administered with SYMTUZA. Consult the dasatinib and nilotinib prescribing information for dosing instructions. |
| vinblastine, vincristine | For vincristine and vinblastine, consider temporarily withholding the cobicistat-containing antiretroviral regimen in patients who develop significant hematologic or gastrointestinal side effects when SYMTUZA is administered concurrently with vincristine or vinblastine. If the antiretroviral regimen must be withheld for a prolonged period, consider initiating a revised regimen that does not include a CYP3A or P-gp inhibitor. | |
| Anticoagulants:
Direct Oral Anticoagulants (DOACs) apixaban | ↑ apixaban | Due to potentially increased bleeding risk, dosing recommendations for co-administration of apixaban with SYMTUZA depend on the apixaban dose. Refer to apixaban dosing instructions for co-administration with P-gp and strong CYP3A inhibitors in apixaban prescribing information. |
| rivaroxaban | ↑ rivaroxaban | Co-administration of rivaroxaban with SYMTUZA is not recommended because it may lead to an increased bleeding risk. |
| dabigatran etexilate
edoxaban | ↑ dabigatran
↑ edoxaban | Refer to the dabigatran etexilate or edoxaban prescribing information for recommendations regarding co-administration. The specific recommendations are based on indication, renal function, and effect of the co-administered P-gp inhibitors on the concentration of dabigatran or edoxaban. Clinical monitoring is recommended when a DOAC not affected by CYP3A4 but transported by P-gp, including dabigatran etexilate and edoxaban, is co-administered with SYMTUZA. |
| Other Anticoagulants | ||
| warfarin | warfarin: effect unknown | Monitor international normalized ratio (INR) upon co-administration of SYMTUZA with warfarin. |
| Anticonvulsants:
carbamazepine, phenobarbital, phenytoin | ↓ cobicistat
↓ darunavir ↓ tenofovir alafenamide | Co-administration is contraindicated due to potential for loss of therapeutic effect and development of resistance. |
| Anticonvulsants with CYP3A induction effects that are NOT contraindicated:
e.g., eslicarbazepine, oxcarbazepine | ↓ cobicistat
↓ tenofovir alafenamide darunavir: effect unknown | Consider alternative anticonvulsant or antiretroviral therapy to avoid potential changes in exposures. If co-administration is necessary, monitor for lack or loss of virologic response. |
| Anticonvulsants that are metabolized by CYP3A:
e.g., clonazepam | ↑ clonazepam | Clinical monitoring of anticonvulsants is recommended. |
| Antidepressants:
Selective Serotonin Reuptake Inhibitors (SSRIs): e.g., paroxetine, sertraline | SSRIs: effects unknown | When co-administering with SSRIs, TCAs, or trazodone, careful dose titration of the antidepressant to the desired effect, including using the lowest feasible initial or maintenance dose, and monitoring for antidepressant response are recommended. |
| Tricyclic Antidepressants (TCAs):
e.g., amitriptyline, desipramine, imipramine, nortriptyline | ↑ TCAs | |
| Other antidepressants:
trazodone | ↑ trazodone | |
| Antifungals:
itraconazole, isavuconazole, ketoconazole, posaconazole | ↑ darunavir
↑ cobicistat | Monitor for increased darunavir or cobicistat and/or antifungal adverse reactions. |
| ↑ itraconazole
↑ isavuconazole ↑ ketoconazole ↔ posaconazole (not studied) | Specific dosing recommendations are not available for co-administration with these antifungals. Monitor for increased itraconazole or ketoconazole adverse reactions. | |
| voriconazole | voriconazole: effects unknown | Co-administration with voriconazole is not recommended unless benefit/risk assessment justifies the use of voriconazole. |
| Anti-gout:
colchicine | ↑ colchicine | Co-administration is contraindicated in patients with renal and/or hepatic impairment due to potential for serious and/or life-threatening reactions.
For patients without renal or hepatic impairment:
|
| Antimalarial:
artemether/lumefantrine | artemether: effect unknown
lumefantrine: effect unknown | Monitor for a potential decrease of antimalarial efficacy or potential QT prolongation. |
| Antimycobacterials:
rifampin | ↓ cobicistat
↓ darunavir ↓ tenofovir alafenamide | Co-administration is contraindicated due to potential for loss of therapeutic effect and development of resistance. |
| rifabutin | ↑ rifabutin
↓ TAF cobicistat: effects unknown darunavir: effects unknown | Co-administration of SYMTUZA with rifabutin is not recommended. If the combination is needed, the recommended dose of rifabutin is 150 mg every other day. Monitor for rifabutin-associated adverse reactions including neutropenia and uveitis. |
| rifapentine | ↓ darunavir
↓ TAF | Co-administration with rifapentine is not recommended. |
| Antipsychotics:
lurasidone | ↑ lurasidone | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions. |
| pimozide | ↑ pimozide | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| e.g., perphenazine, risperidone, thioridazine | ↑ antipsychotic | A decrease in the dose of antipsychotics that are metabolized by CYP3A or CYP2D6 may be needed when co-administered with SYMTUZA. |
| quetiapine | ↑ quetiapine | Initiation of SYMTUZA in patients taking quetiapine:Consider alternative antiretroviral therapy to avoid increases in quetiapine exposure. If co-administration is necessary, reduce the quetiapine dose to 1/6 of the current dose and monitor for quetiapine-associated adverse reactions. Refer to the quetiapine prescribing information for recommendations on adverse reaction monitoring.
Initiation of quetiapine in patients taking SYMTUZA:Refer to the quetiapine prescribing information for initial dosing and titration of quetiapine. |
| β-Blockers:
e.g., carvedilol, metoprolol, timolol | ↑ beta-blockers | Clinical monitoring is recommended for co-administration with beta-blockers that are metabolized by CYP2D6. |
| Calcium channel blockers:
e.g., amlodipine, diltiazem, felodipine, nifedipine, verapamil | ↑ calcium channel blockers | Clinical monitoring is recommended for co-administration with calcium channel blockers metabolized by CYP3A. |
| Cardiac Disorders: | ||
| ranolazine, ivabradine | ↑ ranolazine | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions. |
| dronedarone | ↑ dronedarone | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias |
| Other antiarrhythmics
e.g., amiodarone, disopyramide, flecainide, lidocaine (systemic), mexiletine, propafenone, quinidine | ↑ antiarrhythmics | Clinical monitoring is recommended upon co-administration with antiarrhythmics. |
| digoxin | ↑ digoxin | When co-administering with digoxin, titrate the digoxin dose and monitor digoxin concentrations. |
| Corticosteroids:
dexamethasone (systemic) | ↓ darunavir
↓ cobicistat | Co-administration with systemic dexamethasone or other systemic corticosteroids that induce CYP3A may result in loss of therapeutic effect and development of resistance to SYMTUZA. Consider alternative corticosteroids. |
| Corticosteroids primarily metabolized by CYP3A:
e.g., betamethasone budesonide ciclesonide fluticasone methylprednisolone mometasone triamcinolone | ↑ corticosteroids | Co-administration with corticosteroids (all routes of administration) of which exposures are significantly increased by strong CYP3A inhibitors can increase the risk for Cushing's syndrome and adrenal suppression.
Alternative corticosteroids including beclomethasone, prednisone, and prednisolone (for which PK and/or PD are less affected by strong CYP3A inhibitors relative to other steroids) should be considered, particularly for long term use. |
| Endothelin receptor antagonists:
bosentan | ↓ darunavir
↓ cobicistat ↑ bosentan | Initiation of bosentan in patients taking SYMTUZA: In patients who have been receiving SYMTUZA for at least 10 days, start bosentan at 62.5 mg once daily or every other day based upon individual tolerability.
Initiation of SYMTUZA in patients on bosentan:Discontinue use of bosentan at least 36 hours prior to initiation of SYMTUZA. After at least 10 days following the initiation of SYMTUZA, resume bosentan at 62.5 mg once daily or every other day based upon individual tolerability. Switching from darunavir co-administered with ritonavir to SYMTUZA in patients on bosentan:Maintain bosentan dose. |
| Ergot derivatives: | ||
| e.g., dihydroergotamine, ergotamine, methylergonovine | ↑ ergot derivatives | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues. |
| Hepatitis C virus (HCV):
Direct-Acting Antivirals: | ||
| elbasvir/grazoprevir | ↑ elbasvir/grazoprevir | Co-administration is contraindicated due to potential for the increased risk of alanine transaminase (ALT) elevations. |
| glecaprevir/pibrentasvir | ↑ glecaprevir
↑ pibrentasvir | Co-administration of SYTMUZA with glecaprevir/pibrentasvir is not recommended. |
| Herbal product: | ||
| St. John's wort ( Hypericum perforatum) | ↓ cobicistat
↓ darunavir ↓ tenofovir alafenamide | Co-administration is contraindicated due to potential for loss of therapeutic effect and development of resistance. |
| Hormonal contraceptives: | Additional or alternative (non-hormonal) forms of contraception should be considered when estrogen based contraceptives are co-administered with SYMTUZA. | |
| drosperinone/ethinylestradiol | ↑ drosperinone
↓ ethinylestradiol | For co-administration with drospirenone, clinical monitoring is recommended due to the potential for hyperkalemia. |
| other progestin/estrogen contraceptives | progestin: effects unknown
estrogen: effects unknown | No data are available to make recommendations on co-administration with oral or other hormonal contraceptives. |
| Immunosuppressants:
cyclosporine, sirolimus, tacrolimus | ↑ immunosuppressants | These immunosuppressant agents are metabolized by CYP3A. Therapeutic drug monitoring is recommended with concomitant use. |
| Immunosuppressant /neoplastic:
everolimus | Co-administration of everolimus and SYMTUZA is not recommended. | |
| irinotecan | Discontinue SYMTUZA at least 1 week prior to starting irinotecan therapy. Do not administer SYMTUZA with irinotecan unless there are no therapeutic alternatives. | |
| Inhaled beta agonist: | ||
| salmeterol | ↑ salmeterol | Co-administration with salmeterol is not recommended and may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations, and sinus tachycardia. |
| Lipid modifying agents:
HMG-CoA reductase inhibitors: | ||
| lovastatin, simvastatin | ↑ lovastatin
↑ simvastatin | Co-administration is contraindicated due to potential for serious reactions such as myopathy including rhabdomyolysis. |
| e.g., atorvastatin, fluvastatin, pitavastatin, pravastatin, rosuvastatin | ↑ atorvastatin
↑ fluvastatin ↑ pravastatin ↑ rosuvastatin pitavastatin: effect unknown | For atorvastatin, fluvastatin, pitavastatin, pravastatin, and rosuvastatin, start with the lowest recommended dose and titrate while monitoring for safety. |
Dosage recommendations with atorvastatin or rosuvastatin are as follows:
|
||
| Other lipid modifying agents:
lomitapide | ↑ lomitapide | Co-administration is contraindicated due to potential for markedly increased transaminases associated with increased plasma concentrations of lomitapide. |
| Narcotic analgesics metabolized by CYP3A:
e.g., fentanyl, oxycodone | ↑ fentanyl
↑ oxycodone | Careful monitoring of therapeutic effects and adverse reactions associated with CYP3A-metabolized narcotic analgesics (including potentially fatal respiratory depression) is recommended with co-administration. |
| tramadol | ↑ tramadol | A dose decrease may be needed for tramadol with concomitant use. |
| Narcotic analgesic for treatment of opioid dependence:
buprenorphine, buprenorphine/naloxone, methadone | buprenorphine or buprenorphine/ naloxone: effects unknown
methadone: effects unknown | Initiation of buprenorphine, buprenorphine/naloxone or methadone in patients taking SYMTUZA:Carefully titrate the dose of buprenorphine, buprenorphine/naloxone or methadone to the desired effect; use the lowest feasible initial or maintenance dose.
Initiation of SYMTUZA in patients taking buprenorphine, buprenorphine/naloxone, or methadone:A dose adjustment for buprenorphine, buprenorphine/naloxone, or methadone may be needed. Monitor clinical signs and symptoms. |
| Opioid Antagonist | ||
| naloxegol | ↑ naloxegol | Co-administration of SYMTUZA and naloxegol is contraindicated due to potential for precipitating opioid withdrawal symptoms. |
| Phosphodiesterase PDE-5 inhibitors: | ↑ PDE-5 inhibitors | |
| e.g., avanafil, sildenafil, tadalafil, vardenafil | Co-administration with avanafil is not recommended because a safe and effective avanafil dosage regimen has not been established.
Co-administration with PDE-5 inhibitors may result in an increase in PDE-5 inhibitor-associated adverse reactions including hypotension, syncope, visual disturbances, and priapism. Use of PDE-5 inhibitors for pulmonary arterial hypertension (PAH): Co-administration with sildenafil used for PAH is contraindicated due to potential for sildenafil associated adverse reactions (which include visual disturbances, hypotension, prolonged erection, and syncope). The following dose adjustments are recommended for use of tadalafil with SYMTUZA:
Sildenafil at a single dose not exceeding 25 mg in 48 hours, vardenafil at a single dose not exceeding 2.5 mg dose in 72 hours, or tadalafil at a single dose not exceeding 10 mg dose in 72 hours can be used with increased monitoring for PDE-5 inhibitor-associated adverse reactions. |
|
| Platelet aggregation inhibitor:
ticagrelor | ↑ticagrelor | Co-administration of SYMTUZA and ticagrelor is not recommended. |
| clopidogrel | ↓ clopidogrel active metabolite | Co-administration of SYMTUZA and clopidogrel is not recommended due to potential reduction of the antiplatelet activity of clopidogrel. |
| prasugrel | ↔ prasugrel active metabolite | No dose adjustment is needed when prasugrel is co-administered with SYMTUZA. |
| Sedatives/hypnotics:
orally administered midazolam, triazolam | ↑ midazolam
↑ triazolam | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as prolonged or increased sedation or respiratory depression. |
| metabolized by CYP3A:
e.g., buspirone, diazepam, estazolam, zolpidem | ↑ sedatives/hypnotics | With concomitant use, titration is recommended with sedatives/hypnotics metabolized by CYP3A and a lower dose of the sedatives/hypnotics should be considered with monitoring for increased and prolonged effects or adverse reactions. |
| parenterally administered midazolam | Co-administration of parenteral midazolam should be done in a setting that ensures close clinical monitoring and appropriate medical management in case of respiratory depression and/or prolonged sedation. Dose reduction for parenteral midazolam should be considered, especially if more than a single dose of midazolam is administered. | |
| Urinary antispasmodics | ||
| fesoterodine | ↑ fesoterodine | When fesoterodine is co-administered with SYMTUZA, do not exceed a fesoterodine dose of 4 mg once daily. |
| solifenacin | ↑ solifenacin | When solifenacin is co-administered with SYMTUZA, do not exceed a solifenacin dose of 5 mg once daily. |
8. Use In Specific Populations
8.1 Pregnancy
Data
8.4 Pediatric Use
The safety and effectiveness of SYMTUZA for the treatment of HIV-1 infection in pediatric patients weighing at least 40 kg was established through studies with components of SYMTUZA. Use of SYMTUZA in this group is supported by evidence from adequate and well-controlled studies of SYMTUZA in adults with additional pharmacokinetic, safety, and virologic data from studies of components of SYMTUZA (Trials GS-US-216-0128 and GS-US-292-0106) in pediatric subjects with HIV-1 infection aged 12 to less than 18 years [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.3)] .
The safety and effectiveness of SYMTUZA have not been established in pediatric patients weighing less than 40 kg.
Darunavir, a component of SYMTUZA is not recommended in pediatric patients below 3 years of age because of toxicity and mortality observed in juvenile rats dosed with darunavir.
8.5 Geriatric Use
Clinical trials of SYMTUZA included 35 subjects aged above 65 years of which 26 received SYMTUZA. No differences in safety or efficacy have been observed between elderly subjects and those aged 65 years or less. In general, caution should be exercised in the administration and monitoring of SYMTUZA in elderly patients, reflecting the greater frequency of decreased hepatic function and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)] .
8.6 Renal Impairment
SYMTUZA is not recommended in patients with severe renal impairment (creatinine clearance below 30 mL per minute). No dosage adjustment of SYMTUZA is required in patients with creatinine clearance greater than or equal to 30 mL per minute [see Clinical Pharmacology (12.3)] .
Cobicistat has been shown to decrease creatinine clearance without affecting actual renal glomerular function. Dosing recommendations are not available for drugs that require dosage adjustment for renal impairment when used in combination with SYMTUZA [see Warnings and Precautions (5.6)] .
8.7 Hepatic Impairment
No dosage adjustment of SYMTUZA is required in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. SYMTUZA has not been studied in patients with severe hepatic impairment (Child-Pugh Class C) and there are only limited data regarding the use of SYMTUZA components in this population. Therefore, SYMTUZA is not recommended for use in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)] .
10. Overdosage
Human experience of acute overdose with SYMTUZA is limited. There is no specific antidote for overdose with SYMTUZA. Treatment of overdose with SYMTUZA consists of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient.
Since darunavir and cobicistat are highly bound to plasma proteins, it is unlikely that they will be significantly removed by hemodialysis or peritoneal dialysis. Hemodialysis treatment removes approximately 30% of the emtricitabine dose over a 3-hour dialysis period starting within 1.5 hours of emtricitabine dosing (blood flow rate of 400 mL/min and a dialysate flow rate of 600 mL/min). Tenofovir is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%. It is not known whether emtricitabine or tenofovir can be removed by peritoneal dialysis.
11. Symtuza Description
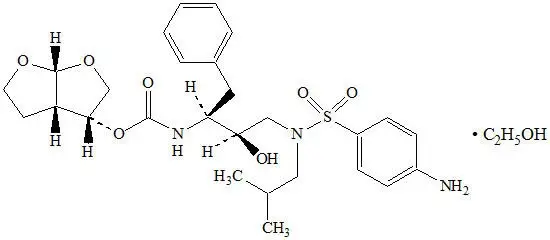
SYMTUZA ®(darunavir, cobicistat, emtricitabine, and tenofovir alafenamide) is a fixed-dose combination tablet.
- Darunavir is an inhibitor of the HIV-1 protease.
- Cobicistat is a mechanism-based inhibitor of cytochrome P450 (CYP) enzymes of the CYP3A family.
- Emtricitabine, a synthetic nucleoside analog of cytidine, is an HIV nucleoside analog reverse transcriptase inhibitor (HIV NRTI).
- Tenofovir alafenamide, an HIV NRTI, is converted in vivoto tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate.
SYMTUZA tablets are for oral administration. Each tablet contains darunavir ethanolate equivalent to 800 mg of darunavir, 150 mg of cobicistat, 200 mg of emtricitabine, and 11.2 mg of tenofovir alafenamide fumarate equivalent to 10 mg of tenofovir alafenamide. The tablets include the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. The tablets are film-coated with a coating material containing polyethylene glycol (macrogol), polyvinyl alcohol (partially hydrolyzed), talc, titanium dioxide, and yellow ferric oxide.
12. Symtuza - Clinical Pharmacology
12.1 Mechanism of Action
SYMTUZA is a fixed-dose combination of antiretroviral drugs darunavir (plus the CYP3A inhibitor cobicistat), emtricitabine, and tenofovir alafenamide [see Microbiology (12.4)] .
12.2 Pharmacodynamics
12.3 Pharmacokinetics
Specific Populations
Pregnancy and Postpartum
The exposure to total and unbound darunavir boosted with cobicistat after intake of darunavir/cobicistat as part of an antiretroviral regimen was substantially lower during the second and third trimesters of pregnancy compared with 6–12 weeks postpartum (see Table 9and Figure 1).
| Pharmacokinetics of total darunavir
(mean ± SD) | 2
ndTrimester of pregnancy
N=7 | 3
rdTrimester of pregnancy
N=6 | Postpartum (6–12 weeks)
N=6 |
|---|---|---|---|
| C max, ng/mL | 4340 ± 1616 | 4910 ± 970 | 7918 ± 2199 |
| AUC 24h, ng.h/mL | 47293 ± 19058 | 47991 ± 9879 | 99613 ± 34862 |
| C min, ng/mL | 168 ± 149 | 184 ± 99 | 1538 ± 1344 |
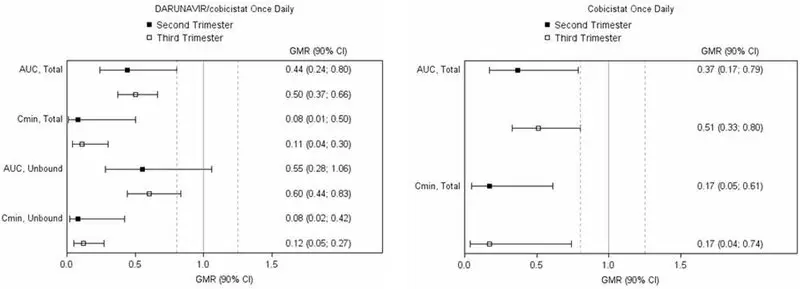
Legend: 90% CI: 90% confidence interval; GMR: geometric mean ratio (i.e., second or third trimester/postpartum). Solid vertical line: ratio of 1.0; dotted vertical lines: reference lines of 0.8 and 1.25.
12.4 Microbiology
Clinical Trials
Darunavir resistance-associated substitutions (V11I, V32I, L33F, I47V, I50V, I54L or M, T74P, L76V, I84V, and L89V) in HIV-1 protease were derived from clinical trial data of antiretroviral therapy experienced patients, which were all protease inhibitor-experienced patients. Baseline International AIDS Society-USA (IAS-USA)-defined PI resistance substitutions confer reduced virologic response to darunavir.
In the AMBER clinical trial of subjects with no prior antiretroviral treatment history, there were 7 subjects with protocol-defined virologic failure and with HIV-1 RNA ≥400 copies/mL at failure or later timepoints who had post-baseline resistance data in the SYMTUZA arm. None of the subjects had detectable emergent darunavir resistance-associated substitutions or other primary protease inhibitor resistance-associated substitutions and only one subject had emergent M184M/I/V, which confers resistance to emtricitabine and lamivudine. In the comparative PREZCOBIX + emtricitabine/tenofovir disoproxil fumarate arm, there were 2 protocol-defined virologic failures with post-baseline resistance data and neither had detectable resistance emergence.
In the EMERALD clinical trial of virologically-suppressed subjects who switched to SYMTUZA, 1 subject who rebounded and 2 subjects who discontinued early from the study had post-baseline resistance genotypes. None of the subjects had darunavir, primary protease inhibitor, emtricitabine, or tenofovir resistance-associated substitutions. In the control arm, there were 3 subjects who rebounded with post-baseline genotypes and no resistance-associated substitutions were observed.
13. Nonclinical Toxicology
13.2 Animal Toxicology and/or Pharmacology
Minimal to slight infiltration of mononuclear cells in the posterior uvea was observed in dogs with similar severity after 3 and 9 month administration of tenofovir alafenamide; reversibility was seen after a 3-month recovery period. No eye toxicity was observed in the dog at systemic exposures of 3.5 (TAF) and 0.62 (tenofovir) times the exposure seen in humans with the recommended daily dose of TAF in SYMTUZA.
14. Clinical Studies
14.1 Clinical Trial Results in Subjects with HIV-1 Infection with no Prior Antiretroviral Treatment History
The efficacy of SYMTUZA in HIV-1 subjects with no prior antiretroviral treatment history was evaluated in the Phase 3 trial TMC114FD2HTX3001 [NCT02431247, (AMBER)] in which subjects were randomized in a 1:1 ratio to receive either SYMTUZA (N=362) or a combination of PREZCOBIX and FTC/TDF (N=363) once daily. The median age was 34.0 years (range 18–71), 88.3% were male, 83% White, 11% Black, and 2% Asian. The mean baseline plasma HIV-1 RNA was 4.5 log 10copies/mL (range 1.3–6.7), and 18% had a baseline viral load ≥100,000 copies/mL. The median baseline CD4+ cell count was 453 cells/mm 3(range 38 to 1456 cells/mm 3).
Virologic outcomes at 48 weeks of treatment are presented in Table 11.
| SYMTUZA
N=362 | PREZCOBIX + FTC/TDF
N=363 |
|
|---|---|---|
| Virologic Response | ||
|
||
| HIV-1 RNA <50 copies/mL | 91% | 88% |
| Treatment difference * | 2.7 (95% CI: -1.6; 7.1) | |
| Virologic Failure † | 4% | 3% |
| No virologic data at Week 48 window ‡ | 4% | 8% |
| Reasons | ||
| Discontinued trial due to adverse event or death | 2% | 4% |
| Discontinued trial for other reasons § | 1% | 3% |
| Missing data during window but on trial | 1% | 1% |
The mean increase from baseline in CD4+ cell count at Week 48 was 189 and 174 cells/mm 3in the SYMTUZA and PREZCOBIX + FTC/TDF groups, respectively.
14.2 Clinical Trial Results in Virologically-Suppressed Subjects with HIV-1 Infection Who Switched to SYMTUZA
Phase 3 trial TMC114IFD3013 [NCT02269917, (EMERALD)] evaluated the efficacy of SYMTUZA in virologically-suppressed (HIV-1 RNA less than 50 copies/mL) subjects with HIV-1 infection. Subjects were virologically suppressed for at least 2 months and no more than once had a viral load elevation above 50 HIV-1 RNA copies/mL during the year prior to enrollment. Subjects were on a stable antiretroviral regimen (for at least 6 months), consisting of a bPI [either darunavir once daily or atazanavir (both boosted with ritonavir or cobicistat), or lopinavir with ritonavir] combined with emtricitabine and TDF. Subjects had no history of failure on darunavir treatment and no known or suspected darunavir resistance-associated substitutions. Emtricitabine or tenofovir resistance-associated substitutions were not specifically excluded by the protocol. They either switched to SYMTUZA (N=763) or continued their treatment regimen (N=378) (randomized 2:1). Subjects had a median age of 46 years (range 19–78), 82% were male, 75% White, 21% Black, and 2% Asian. The median baseline CD4+ cell count was 628 cells/mm 3(range 111–1921 cells/mm 3). Overall, 15% (N=169) of subjects had prior virologic failure. Five subjects had archived tenofovir resistance-associated substitutions and 53 subjects had archived emtricitabine resistance-associated substitutions, mainly at RT position M184. All of these subjects with emtricitabine resistance-associated substitutions had HIV-1 RNA<50 copies/mL at Week 48 (N=50) or at the last on-treatment viral load (N=3). Virologic outcomes are presented in Table 12. Prior virologic failure did not impact treatment outcomes.
| SYMTUZA
N=763 | bPI+FTC/TDF
N=378 |
|
|---|---|---|
|
||
| Virologic Failure* | 1% | 1% |
| Treatment difference † | 0.3 (95% CI: -0.7; 1.2) | |
| HIV-1 RNA <50 copies/mL | 95% | 94% |
| No virologic data at Week 48 window ‡ | 4% | 6% |
| Reasons | ||
| Discontinued trial due to adverse event or death | 1% | 1% |
| Discontinued trial for other reasons § | 3% | 4% |
| Missing data during window ‡but on trial | <1% | 1% |
The mean increase from baseline in CD4+ cell count at Week 48 was 20 cells/mm 3in subjects who switched to SYMTUZA and 8 cells/mm 3in subjects who stayed on their baseline PI + FTC/TDF.
14.3 Clinical Trial Results in Pediatric Subjects with HIV-1 Infection
The pharmacokinetic profile, safety, and antiviral activity of the components of SYMTUZA were evaluated in open-label clinical trials in pediatric subjects with HIV-1 infection aged 12 to less than 18 years: GS-US-216-0128 (N=7) and GS-US-292-0106 (N=50).
In the Phase 2/3 trial GS-US-216-0128, darunavir 800 mg and cobicistat 150 mg once daily with 2 NRTIs were evaluated in 7 virologically suppressed pediatric subjects aged 12 to less than 18 years and weighing at least 40 kg. Subjects had a median (range) age of 14 (12–16) years and a median (range) weight of 57 (45–78) kg. At baseline, plasma HIV-1 RNA was <50 copies/mL in all subjects, and the median (range) CD4+ cell count was 1,117 (658–2,416) cells/mm 3. At Week 48, the proportion of subjects who maintained HIV-1 RNA <50 copies/mL was 86%, and the median change in CD4+ cell count from baseline was -342 cells/mm 3(range -1,389 to 210 cells/mm 3). All 6 subjects with available data had CD4+ cell counts above 800 cells/mm 3at Week 48.
In the Phase 2/3 trial GS-US-292-0106, cobicistat 150 mg, emtricitabine 200 mg, and tenofovir alafenamide 10 mg, as part of a fixed-dose combination regimen together with elvitegravir 150 mg, were evaluated in 50 treatment-naïve pediatric subjects with HIV-1 aged 12 to less than 18 years and weighing at least 35 kg. Subjects had a median (range) age of 15 (12–17) years. At baseline, median (range) plasma HIV-1 RNA was 4.7 (3.3–6.5) log 10copies/mL, median (range) CD4+ cell count was 456 (95–1,110) cells/mm 3, and 22% had baseline plasma HIV-1 RNA >100,000 copies/mL). At Week 48, the proportion of subjects who had HIV-1 RNA <50 copies/mL was 92%, and the median increase in CD4+ cell count from baseline was 220 cells/mm 3.
The use of SYMTUZA in pediatric patients weighing less than 40 kg has not been established [see Use in Specific Populations (8.4)].
16. How is Symtuza supplied
SYMTUZA ®(darunavir, cobicistat, emtricitabine, and tenofovir alafenamide) tablets are supplied as yellow to yellowish-brown, capsule-shaped, film-coated tablets debossed with "8121" on one side and "JG" on the other side.
SYMTUZA is packaged in bottles of 30 tablets (NDC 59676-800-30), with a silica gel desiccant and child-resistant closure.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
PRINCIPAL DISPLAY PANEL - 30 Tablet Bottle Label
NDC 59676-800-30
Symtuza™
(darunavir, cobicistat,
emtricitabine, and tenofovir
alafenamide) tablets
800 mg / 150 mg /
200 mg / 10 mg
Each tablet contains darunavir
ethanolate equivalent to 800 mg
of darunavir, 150 mg of cobicistat,
200 mg of emtricitabine, and
tenofovir alafenamide fumarate
equivalent to 10 mg of tenofovir
alafenamide.
Rx only
30 Tablets

| SYMTUZA
darunavir, cobicistat, emtricitabine, and tenofovir alafenamide tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Janssen Products LP (804684207) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Cilag SpA | 542797928 | pack(59676-800) , manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Pharmaceuticals, Inc. | 868441320 | analysis(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Ortho LLC | 805887986 | pack(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Gilead Alberta ULC | 207452996 | api manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Esteve Quimica, SA | 633485529 | api manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Yuhan Chemical, Inc. | 687831958 | api manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Evonik Operations GmbH | 343170605 | api manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Union Quimico Farmaceutica SA (Uquifa) | 460940868 | api manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AMPAC Fine Chemicals | 073903937 | api manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Pharmaceutica NV | 400345889 | manufacture(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Pharmaceutical Sciences Unlimited Company | 985639841 | api manufacture(59676-800) , analysis(59676-800) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon, Inc. | 240769596 | manufacture(59676-800) , analysis(59676-800) | |
