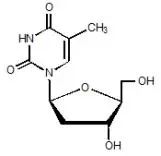
Drug Detail:Tyzeka (Telbivudine [ tel-biv-yoo-deen ])
Drug Class: Nucleoside reverse transcriptase inhibitors (NRTIs)
Highlights of Prescribing Information
Tyzeka® (telbivudine) tablets, for oral use
Tyzeka® (telbivudine) solution, for oral use
Initial U.S. Approval: 2006
WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS AND SEVERE ACUTE EXACERBATIONS OF HEPATITIS B
See full prescribing information for complete boxed warning.
-
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues (5.1).
- Severe acute exacerbations of hepatitis B have been reported in patients who discontinued anti-hepatitis B therapy, including Tyzeka. Hepatic function should be monitored closely in patients who discontinue therapy. Resumption of anti-hepatitis B therapy may be warranted (5.2).
Indications and Usage for Tyzeka
Tyzeka is an HBV nucleoside analogue reverse transcriptase inhibitor indicated for the treatment of chronic hepatitis B in adult patients with evidence of viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease (1.1).
Tyzeka Dosage and Administration
- Adults and Adolescents (16 years of age and older): 600 mg once daily, taken orally, with or without food (2.1).
- Renal Impairment: Dose adjustment required in patients with creatinine clearance less than 50 mL per min, (2.2) as follows:
| Creatinine Clearance (mL/min) | Tyzeka Dose | |
| Oral Solution
(100 mg/5 mL) | Tablet
(600 mg) |
|
| greater than or equal to 50 | 30 mL once daily | 1 tab every 24 hrs |
| 30 – 49 | 20 mL once daily | 1 tab every 48 hrs |
| less than 30 (not requiring dialysis) | 10 mL once daily | 1 tab every 72 hrs |
| End stage renal disease (ESRD) | 6 mL once daily | 1 tab every 96 hrs1 |
1When administered on hemodialysis days, administer Tyzeka after hemodialysis (2.2).
Dosage Forms and Strengths
- Tablet: 600 mg tablets.
- Oral Solution: 100 mg per 5 mL solution.
Contraindications
Combination of Tyzeka with pegylated interferon alfa-2a: Increased risk of peripheral neuropathy (4).
Warnings and Precautions
- Lactic acidosis and severe hepatomegaly with steatosis: If suspected, treatment should be suspended (5.1).
- Severe acute exacerbations of hepatitis B after discontinuation: Monitor hepatic function closely for at least several months (5.2, 6.1).
- Myopathy: Tyzeka should be interrupted if myopathy is suspected; and discontinued if confirmed. It is unknown whether risk of myopathy is increased with concomitant use of other medications associated with myopathy (5.3).
- Peripheral neuropathy: Risk increased when Tyzeka used in combination with alfa interferons, avoid concomitant use. Tyzeka should be interrupted if peripheral neuropathy is suspected; and discontinued if confirmed (4, 5.4, 7).
Adverse Reactions/Side Effects
In clinical trials, the most common adverse reactions (greater than or equal to 3%), of any severity, were: fatigue, increased creatine kinase (CK), headache, cough, diarrhea, abdominal pain, nausea, pharyngolaryngeal pain, arthralgia, pyrexia, rash, back pain, dizziness, myalgia, ALT increased, dyspepsia, insomnia, and abdominal distension (6.1). The most common adverse events resulting in Tyzeka discontinuation included increased CK, nausea, diarrhea, fatigue, myalgia, and myopathy (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Coadministration of Tyzeka with drugs that affect renal function may alter plasma concentrations of Tyzeka and/or coadministered drug (7).
- Coadministration of Tyzeka with pegylated interferon alfa-2a increases the risk and severity of peripheral neuropathy (7).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2013
Full Prescribing Information
1. Indications and Usage for Tyzeka
1.1 Chronic Hepatitis B
Tyzeka is indicated for the treatment of chronic hepatitis B in adult patients with evidence of viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease.
The following points should be considered when initiating therapy with Tyzeka:
- This indication is based on virologic, serologic, biochemical and histologic responses in nucleoside treatment naïve adult patients with HBeAg positive and HBeAg negative chronic hepatitis B with compensated liver disease [see Clinical Studies (14)].
- For HBeAg-positive patients, Tyzeka should only be initiated in patients with HBV DNA less than 9 log10 copies per mL and ALT greater than or equal to 2x Upper Limit of Normal (ULN) prior to treatment.
- For HBeAg-negative patients, Tyzeka should only be initiated in patients with HBV DNA less than 7 log10 copies per mL prior to treatment.
- On-treatment response should guide continued therapy [see Dosage and Administration (2.1) and Microbiology (12.4)].
- Tyzeka has not been evaluated in patients co-infected with HIV, HCV, or HDV.
- Tyzeka has not been evaluated in liver transplant recipients or in patients with decompensated liver disease.
- Tyzeka has not been studied in well-controlled trials for the treatment of patients with established nucleoside analog reverse transcriptase inhibitor-resistant hepatitis B virus infection, but is expected to be cross-resistant to lamivudine [see Microbiology (12.4)].
- The safety and efficacy of Tyzeka have not been evaluated in Black/African American or Hispanic patients [see Use in Specific Populations (8.9)].
2. Tyzeka Dosage and Administration
2.1 Adults and Adolescents (16 years of age and older)
Due to higher rates of resistance that may develop with longer term treatment among patients with incomplete viral suppression, treatment should only be initiated, if pre-treatment HBV DNA and ALT measurements are known, in the following patient populations:
For HBeAg-positive patients, HBV DNA should be less than 9 log10 copies per mL and ALT should be greater than or equal to 2x ULN prior to treatment with Tyzeka.
For HBeAg-negative patients, HBV DNA should be less than 7 log10 copies per mL prior to treatment with Tyzeka.
HBV DNA levels should be monitored at 24 weeks of treatment to assure complete viral suppression (HBV DNA less than 300 copies per mL). Alternate therapy should be initiated for patients who have detectable HBV DNA after 24 weeks of treatment. Optimal therapy should be guided by further resistance testing.
The recommended dose of Tyzeka for the treatment of chronic hepatitis B is 600 mg once daily, taken orally, with or without food.
Tyzeka oral solution (30 mL) may be considered for patients who have difficulty with swallowing tablets.
2.2 Renal Impairment
Tyzeka may be used for the treatment of chronic hepatitis B in patients with impaired renal function. No adjustment to the recommended dose of Tyzeka is necessary in patients whose creatinine clearance is greater than or equal to 50 mL per min. Adjustment of the total daily dose of Tyzeka oral solution or of the interval for administration of Tyzeka tablets is required in patients with creatinine clearance less than 50 mL per min including those with ESRD on hemodialysis (Table 1).
| Creatinine Clearance
(mL/min) | Tyzeka Oral Solution Dose
(5 mL = 100 mg) | Tyzeka Tablet Dose
(1 tablet = 600 mg) |
| greater than or equal to 50 | 30 mL once daily | 1 tablet every 24 hrs |
| 30-49 | 20 mL once daily | 1 tablet every 48 hrs |
| less than 30 (not requiring dialysis) | 10 mL once daily | 1 tablet every 72 hrs |
| ESRD | 6 mL once daily | 1 tablet every 96 hrs1 |
| 1When administered on hemodialysis days, Tyzeka should be administered after hemodialysis. | ||
2.3 Hepatic Impairment
No adjustment to the recommended dose of Tyzeka is necessary in patients with hepatic impairment.
2.4 Duration of Therapy
For patients with incomplete viral suppression (HBV DNA greater than or equal to 300 copies per mL) after 24 weeks of treatment, alternate therapy should be instituted. HBV DNA should be monitored every 6 months to assure continued response. If patients test positive for HBV DNA at any time after their initial response, alternate treatment should be instituted. Optimal therapy should be guided by resistance testing.
The optimal duration of therapy with Tyzeka for patients with chronic hepatitis B is unknown.
3. Dosage Forms and Strengths
4. Contraindications
Combination of Tyzeka with pegylated interferon alfa-2a is contraindicated because of increased risk of peripheral neuropathy [see Warnings and Precautions (5.4) and Drug Interactions (7)].
5. Warnings and Precautions
5.1 Lactic Acidosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination with antiretrovirals. Female gender, obesity, and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering HBV nucleoside analogue reverse transcriptase inhibitors to patients with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors. Treatment with Tyzeka should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.2 Exacerbations of Hepatitis B after Discontinuation of Treatment
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy, including Tyzeka. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, resumption of anti-hepatitis B therapy may be warranted [see Adverse Reactions (6.1)].
5.3 Myopathy
Cases of myopathy/myositis have been reported with Tyzeka use several weeks to months after starting therapy. Myopathy has also been reported with some other drugs in this class. Rhabdomyolysis has been reported during postmarketing use of Tyzeka [see Adverse Reactions (6.2)].
Uncomplicated myalgia has been reported in Tyzeka-treated patients [see Adverse Reactions (6.1)]. Myopathy, defined as persistent unexplained muscle aches and/or muscle weakness in conjunction with increases in creatine kinase (CK) values, should be considered in any patient with diffuse myalgias, muscle tenderness, or muscle weakness. Among patients with Tyzeka-associated myopathy, no pattern with regard to the degree or timing of CK elevations has been observed. In addition, the predisposing factors for the development of myopathy among Tyzeka recipients are unknown. Patients should be advised to report promptly unexplained muscle aches, pain, tenderness, or weakness. Tyzeka therapy should be interrupted if myopathy is suspected, and discontinued if myopathy is confirmed. It is unknown whether the risk of myopathy during treatment with drugs in this class is increased with concurrent administration of other drugs associated with myopathy, including but not limited to: corticosteroids, chloroquine, hydroxychloroquine, certain HMGCoA reductase inhibitors, fibric acid derivatives, penicillamine, zidovudine, cyclosporine, erythromycin, niacin, and certain azole antifungals. Physicians initiating concomitant treatment with any drug associated with myopathy should monitor patients closely for any signs or symptoms of unexplained muscle pain, tenderness, or weakness.
5.4 Peripheral Neuropathy
Peripheral neuropathy has been reported with Tyzeka alone or in combination with pegylated interferon alfa-2a and other interferons. In one clinical trial, an increased risk and severity of peripheral neuropathy was observed with the combination use of Tyzeka 600mg daily and pegylated interferon alfa-2a 180 micrograms once weekly compared to Tyzeka or pegylated interferon alfa-2a alone [see Contraindications (4) and Drug Interactions (7)]. Such risk cannot be excluded for other dose regimens of pegylated interferon alfa-2a, or other alfa interferons (pegylated or standard). The safety and efficacy of Tyzeka in combination with pegylated interferons or other interferons for the treatment of chronic hepatitis B have not been demonstrated. Patients should be advised to report any numbness, tingling, and/or burning sensations in the arms and/or legs, with or without gait disturbance. Tyzeka therapy should be interrupted if peripheral neuropathy is suspected, and discontinued if peripheral neuropathy is confirmed [see Adverse Reactions (6.1)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in other sections of the labeling:
- Lactic acidosis and severe hepatomegaly with steatosis [see Boxed Warning, Warnings and Precautions (5.1)]
- Severe acute exacerbations of hepatitis after discontinuation of treatment [see Boxed Warning, Warnings and Precautions (5.2)]
- Myopathy [see Warnings and Precautions (5.3)]
- Peripheral Neuropathy [see Warnings and Precautions (5.4)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to the rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Assessment of adverse reactions is primarily based on two trials (007 GLOBE and NV-02B-015) in which 1,699 subjects with chronic hepatitis B received double-blind treatment with Tyzeka 600 mg per day (n=847 subjects) or lamivudine (n=852 subjects) for 104 weeks. The median duration of therapy was 104 weeks for both treatment groups.
In the 104 week clinical trials, most adverse experiences reported with Tyzeka were classified as mild or moderate in severity and were not attributed to Tyzeka. Selected adverse events of any severity which were reported in greater than or equal to 3% of Tyzeka and lamivudine recipients are shown in Table 2. With the exception of increased creatine kinase (CK), which was reported more frequently among Tyzeka recipients, the adverse event profile was similar for the two drugs.
| Adverse Event (Preferred Term) | Tyzeka
N=847 n (%)b | Lamivudine
N=852 n (%)b |
| Fatigue | 106 (13) | 95 (11) |
| CK increased | 90 (11) | 52 (6) |
| Headache | 83 (10) | 95 (11) |
| Cough | 52 (6) | 45 (5) |
| Diarrhea | 50 (6) | 46 (5) |
| Abdominal pain, upper | 49 (6) | 52 (6) |
| Nausea | 45 (5) | 40 (5) |
| Pharyngolaryngeal pain | 38 (5) | 31 (4) |
| Arthralgia | 37 (4) | 38 (5) |
| Pyrexia | 34 (4) | 27 (3) |
| Rash | 33 (4) | 21 (3) |
| Back pain | 33 (4) | 32 (4) |
| Dizziness | 32 (4) | 43 (5) |
| Abdominal pain | 29 (3) | 31 (4) |
| Myalgia | 27 (3) | 17 (2) |
| ALT increased | 27 (3) | 31 (4) |
| Dyspepsia | 24 (3) | 39 (5) |
| Insomnia | 24 (3) | 22 (3) |
| Abdominal distension | 22 (3) | 19 (2) |
| Pruritus | 18 (2) | 23 (3) |
| Hepatitis B exacerbation | 17 (2) | 36 (4) |
| a Adverse events reported in greater than or equal to 3% subjects in either treatment group b n (%) = the number and proportion of subjects in whom adverse event was reported |
||
Moderate to severe (Grade 2-4) adverse events were reported in 239/847 (28%) of Tyzeka recipients and 229/852 (27%) of lamivudine recipients. The profile of adverse events of moderate to severe intensity was similar in both treatment groups and no individual adverse event was reported in greater than 2% of subjects in either treatment group.
Discontinuations due to adverse events were reported in 4% of Tyzeka recipients and 4% of lamivudine recipients. The most common adverse events resulting in Tyzeka discontinuation included increased CK, nausea, diarrhea, fatigue, myalgia, and myopathy.
Peripheral neuropathy was reported as an adverse event in less than 1% (2/847) of subjects receiving Tyzeka monotherapy [see Warnings and Precautions (5.4)]. Of Tyzeka-treated subjects less than 1% (5/847) were diagnosed with myopathy/myositis (presenting with muscular weakness) [see Warnings and Precautions (5.3)].
Laboratory Abnormalities
Frequencies of selected treatment-emergent laboratory abnormalities in the 007 GLOBE and NV-02B-015 trials are listed in Table 3.
| Test | Tyzeka
600 mg (n=847) | Lamivudine
100 mg (n=852) |
| Creatine Kinase (CK) greater than 7.0 x ULN | 13% | 4% |
| ALT greater than 10.0 x ULN and 2.0 x baselineb | 5% | 8% |
| ALT greater than 3 x baseline | 7% | 13% |
| AST (SGOT) greater than 3.0 x baseline | 6% | 10% |
| Lipase greater than 2.5 x ULN | 2% | 4% |
| Amylase greater than 3.0 x ULN | less than 1% | less than 1% |
| Total Bilirubin greater than 5.0 x ULN | less than 1% | less than 1% |
| Neutropenia (ANC less than or equal to 749/mm3) | 2% | 2% |
| Thrombocytopenia (Platelets less than or equal to 49,999/mm3) | less than 1% | less than 1% |
| a On-treatment value worsened from baseline to Grade 3 or Grade 4 during therapy | ||
| b American Association for the Study of Liver Diseases (AASLD) definition of acute hepatitis flare | ||
Creatine Kinase (CK) Elevations
Creatine kinase (CK) elevations were more frequent among subjects on Tyzeka treatment. By 104 weeks of treatment, Grade 1-4 CK elevations occurred in 79% of Tyzeka-treated subjects and 47% of lamivudine-treated subjects. Grade 3 or 4 CK elevations occurred in 13% of Tyzeka-treated subjects and 4% of lamivudine-treated subjects. Most CK elevations were asymptomatic, but the mean recovery time was longer for subjects on Tyzeka than subjects on lamivudine.
Among Tyzeka-treated subjects with Grade 1-4 CK elevations, 10% developed a musculoskeletal adverse event compared to 5% of lamivudine-treated subjects. A total of 2% (13/847) Tyzeka-treated subjects interrupted or discontinued trial drug due to CK elevation or musculoskeletal adverse events1.
______________________
1 Includes the Preferred Terms: back pain, chest wall pain, non-cardiac chest pain, chest discomfort, flank pain, muscle cramp, muscular weakness, musculoskeletal pain, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal stiffness, myalgia, myofascial pain syndrome, myopathy, myositis, neck pain, and pain in extremity.
______________________
ALT Flares During Treatment
The incidence of ALT flares, defined as ALT greater than 10 x ULN and greater than 2 x baseline, was similar in the two treatment arms (3%) in the first six months. After week 24, ALT flares were reported less frequently in the Tyzeka arm (2%) compared to the lamivudine arm (5%). Periodic monitoring of hepatic function is recommended during chronic hepatitis B treatment.
Exacerbations of Hepatitis after Discontinuation of Treatment
In the subset of subjects who discontinued treatment prematurely for reasons other than efficacy, or who elected not to continue Tyzeka in another clinical trial, 9/154 (6%) Tyzeka-treated and 10/180 (6%) lamivudine-treated subjects experienced an exacerbation of hepatitis (ALT elevation greater than 2 x baseline and greater than 10 x ULN) in the 4-month post-treatment period.
Results at 208 Weeks
After 104 weeks of blinded therapy in trials 007 GLOBE and NV-02B-015, 667 subjects received Tyzeka in an open-label extension trial, CLDT600A2303. Of those initially randomized to Tyzeka therapy, 78% of subjects (530/680) from trial 007 GLOBE and 82% (137/167) of subjects from trial NV-02B-015 enrolled into the extension trial and continued Tyzeka treatment for up to 208 weeks. The long-term Tyzeka safety population in trial CLDT600A2303 consisted of 655 subjects, including 518 subjects from trial 007 GLOBE and 137 subjects from trial NV-02B-015.
The overall safety profile from the pooled analysis up to 104 and 208 weeks was similar. Grade 3/4 CK elevations occurred in 16% of subjects (104/655) treated with Tyzeka in trial CLDT600A2303. Most grade 3/4 CK elevations were asymptomatic (74% of subjects without any muscle related adverse reaction) and transient (98% of episodes lasted one or two visits (visit interval 2 - 12 weeks) and 87% of subjects had one or two episodes). Most grade 3/4 CK elevations (93%) resolved spontaneously or returned to baseline levels. Two cases of myopathy and two cases of myositis were reported in the 655 Tyzeka-treated subjects.
Among the cohort of 655 subjects continuing Tyzeka for up to 208 weeks in trial CLDT600A2303, including the subgroup of patients (n=223) with mild renal impairment (eGFR 60-90 mL per min) at baseline, mean estimated GFR assessed by MDRD did not decline.
6.2 Postmarketing Experience
The following adverse reactions have been reported during post approval use of Tyzeka. Because these reactions were reported voluntarily from a population of unknown size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Musculoskeletal and Connective Tissue Disorders
Rhabdomyolysis
Nervous System Disorders
Paraesthesia, hypoaesthesia
Metabolism and Nutrition Disorders
Lactic acidosis
7. Drug Interactions
Tyzeka is excreted mainly by passive diffusion so the potential for interactions between Tyzeka and other drugs eliminated by renal excretion is low. However, because Tyzeka is eliminated primarily by renal excretion, coadministration of Tyzeka with drugs that alter renal function may alter plasma concentrations of Tyzeka [see Clinical Pharmacology (12.3)].
A clinical trial investigating the combination of Tyzeka, 600 mg daily, with pegylated interferon alfa-2a, 180 micrograms once weekly by subcutaneous administration, indicates that this combination is associated with an increased risk of peripheral neuropathy occurrence and severity, in comparison to Tyzeka or pegylated interferon alfa-2a alone [see Contraindications (4) and Warnings and Precautions (5.4)].

| TYZEKA
telbivudine tablet, film coated |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
