Drug Detail:Xalkori (Crizotinib [ kriz-oh-ti-nib ])
Drug Class: Multikinase inhibitors
Highlights of Prescribing Information
XALKORI® (crizotinib) capsules, for oral use
Initial U.S. Approval: 2011
Recent Major Changes
| Indications and Usage (1) | 7/2017 |
| Dosage and Administration, Patient Selection (2.1) | 7/2017 |
| Dosage and Administration, Recommended Dosing (2.2) | 2/2018 |
Indications and Usage for Xalkori
XALKORI is a kinase inhibitor indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) whose tumors are anaplastic lymphoma kinase (ALK) or ROS1-positive as detected by an FDA-approved test. (1)
Xalkori Dosage and Administration
- Recommended Dose: 250 mg orally twice daily. (2.2)
- Moderate Hepatic Impairment: 200 mg orally twice daily. (2.2)
- Severe Hepatic Impairment: 250 mg orally once daily. (2.2)
- Severe Renal Impairment: 250 mg orally once daily. (2.2)
Dosage Forms and Strengths
Capsules: 250 mg and 200 mg. (3)
Contraindications
None. (4)
Warnings and Precautions
- Hepatotoxicity: Fatal hepatotoxicity occurred in 0.1% of patients. Monitor with periodic liver testing. Temporarily suspend, dose reduce, or permanently discontinue XALKORI. (5.1)
- Interstitial Lung Disease (ILD)/Pneumonitis: Occurred in 2.9% of patients. Permanently discontinue in patients with ILD/pneumonitis. (5.2)
- QT Interval Prolongation: Occurred in 2.1% of patients. Monitor electrocardiograms and electrolytes in patients who have a history of or predisposition for QTc prolongation, or who are taking medications that prolong QT. Temporarily suspend, dose reduce, or permanently discontinue XALKORI. (5.3)
- Bradycardia: XALKORI can cause bradycardia. Monitor heart rate and blood pressure regularly. Temporarily suspend, dose reduce, or permanently discontinue XALKORI. (5.4)
- Severe Visual Loss: Reported in 0.2% of patients. Discontinue XALKORI in patients with severe visual loss. Perform an ophthalmological evaluation. (5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and use of effective contraception. (5.6, 8.1, 8.3)
Adverse Reactions/Side Effects
The most common adverse reactions (≥25%) are vision disorders, nausea, diarrhea, vomiting, edema, constipation, elevated transaminases, fatigue, decreased appetite, upper respiratory infection, dizziness, and neuropathy. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Strong CYP3A Inhibitors: Avoid coadministration with XALKORI. (7.1)
- Strong CYP3A Inducers: Avoid coadministration with XALKORI. (7.2)
- CYP3A Substrates: Avoid coadministration of CYP3A substrates with narrow therapeutic indices with XALKORI. (7.3)
Use In Specific Populations
Lactation: Do not breastfeed while taking XALKORI. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2018
Full Prescribing Information
1. Indications and Usage for Xalkori
XALKORI is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) whose tumors are anaplastic lymphoma kinase (ALK) or ROS1-positive as detected by an FDA-approved test [see Clinical Studies (14.1 and 14.2)].
2. Xalkori Dosage and Administration
2.1 Patient Selection
Select patients for the treatment of metastatic NSCLC with XALKORI based on the presence of ALK or ROS1 positivity in tumor specimens [see Indications and Usage (1) and Clinical Studies (14.1, 14.2)].
Information on FDA-approved tests for the detection of ALK and ROS1 rearrangements in NSCLC is available at http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.htm.
2.2 Recommended Dosing
The recommended dose of XALKORI is 250 mg orally twice daily until disease progression or no longer tolerated by the patient.
The recommended dose of XALKORI in patients with pre-existing moderate hepatic impairment [any aspartate aminotransferase (AST) and total bilirubin >1.5 times the upper limit of normal (ULN) and ≤3 times ULN] is 200 mg orally twice daily. The recommended dose of XALKORI in patients with pre-existing severe hepatic impairment [any AST and total bilirubin >3 times ULN] is 250 mg orally once daily [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
The recommended dose of XALKORI in patients with severe renal impairment [creatinine clearance (CLcr) <30 mL/min] not requiring dialysis is 250 mg orally once daily [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
XALKORI may be taken with or without food. Swallow capsules whole. If a dose of XALKORI is missed, make up that dose unless the next dose is due within 6 hours. If vomiting occurs after taking a dose of XALKORI, take the next dose at the regular time.
2.3 Dose Modification
Reduce dose as below, if 1 or more dose reductions are necessary due to adverse reactions of Grade 3 or 4 severity, as defined by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.0:
- First dose reduction: XALKORI 200 mg taken orally twice daily
- Second dose reduction: XALKORI 250 mg taken orally once daily
- Permanently discontinue if unable to tolerate XALKORI 250 mg taken orally once daily
Dose reduction guidelines are provided in Tables 1 and 2.
| Grade | XALKORI Dosing |
|---|---|
|
|
| Grade 3 | Withhold until recovery to Grade 2 or less, then resume at the same dose schedule |
| Grade 4 | Withhold until recovery to Grade 2 or less, then resume at next lower dose |
| Criteria | XALKORI Dosing |
|---|---|
|
|
| Alanine aminotransferase (ALT) or aspartate aminotransferase (AST) elevation greater than 5 times upper limit of normal (ULN) with total bilirubin less than or equal to 1.5 times ULN | Withhold until recovery to baseline or less than or equal to 3 times ULN, then resume at reduced dose. |
| ALT or AST elevation greater than 3 times ULN with concurrent total bilirubin elevation greater than 1.5 times ULN (in the absence of cholestasis or hemolysis) | Permanently discontinue. |
| Any grade drug-related interstitial lung disease/pneumonitis | Permanently discontinue. |
| QT corrected for heart rate (QTc) greater than 500 ms on at least 2 separate electrocardiograms (ECGs) | Withhold until recovery to baseline or to a QTc less than 481 ms, then resume at reduced dose. |
| QTc greater than 500 ms or greater than or equal to 60 ms change from baseline with Torsade de pointes or polymorphic ventricular tachycardia or signs/symptoms of serious arrhythmia | Permanently discontinue. |
| Bradycardia* (symptomatic, may be severe and medically significant, medical intervention indicated) | Withhold until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. Evaluate concomitant medications known to cause bradycardia, as well as antihypertensive medications. If contributing concomitant medication is identified and discontinued, or its dose is adjusted, resume at previous dose upon recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. If no contributing concomitant medication is identified, or if contributing concomitant medications are not discontinued or dose modified, resume at reduced dose upon recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. |
| Bradycardia*,† (life-threatening consequences, urgent intervention indicated) | Permanently discontinue if no contributing concomitant medication is identified. If contributing concomitant medication is identified and discontinued, or its dose is adjusted, resume at 250 mg once daily upon recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above, with frequent monitoring. |
| Visual Loss (Grade 4 Ocular Disorder) | Discontinue during evaluation of severe vision loss. |
Monitor complete blood counts including differential white blood cell counts monthly and as clinically indicated, with more frequent repeat testing if Grade 3 or 4 abnormalities are observed, or if fever or infection occurs.
3. Dosage Forms and Strengths
- 250 mg capsules: hard gelatin capsule, size 0, pink opaque cap and body, with "Pfizer" on the cap and "CRZ 250" on the body.
- 200 mg capsules: hard gelatin capsule, size 1, white opaque body and pink opaque cap, with "Pfizer" on the cap and "CRZ 200" on the body.
5. Warnings and Precautions
5.1 Hepatotoxicity
Drug-induced hepatotoxicity with fatal outcome occurred in 2 (0.1%) of the 1719 patients treated with XALKORI across clinical trials. Concurrent elevations in alanine aminotransferase (ALT) or AST greater than or equal to 3 times the ULN and total bilirubin greater than or equal to 2 times the ULN, with normal alkaline phosphatase, occurred in 10 patients (<1%) treated with XALKORI. Elevations in ALT or AST greater than 5 times the ULN occurred in 187 (11.2%) and 95 (5.7%) patients, respectively. Seventeen patients (1.0%) required permanent discontinuation due to elevated transaminases. Transaminase elevations generally occurred within the first 2 months of treatment.
Monitor liver function tests, including ALT, AST, and total bilirubin, every 2 weeks during the first 2 months of treatment, then once a month, and as clinically indicated, with more frequent repeat testing for increased liver transaminases, alkaline phosphatase, or total bilirubin in patients who develop transaminase elevations. Temporarily suspend, dose reduce, or permanently discontinue XALKORI as described in Table 2 [see Dosage and Administration (2.3) and Adverse Reactions (6)].
5.2 Interstitial Lung Disease (Pneumonitis)
Severe, life-threatening, or fatal interstitial lung disease (ILD)/pneumonitis can occur in patients treated with XALKORI. Across clinical trials (n=1719), 50 XALKORI-treated patients (2.9%) had ILD of any grade, 18 patients (1.0%) had Grade 3 or 4 ILD, and 8 patients (0.5%) had fatal ILD. Interstitial lung disease generally occurred within 3 months after the initiation of XALKORI.
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis. Exclude other potential causes of ILD/pneumonitis, and permanently discontinue XALKORI in patients diagnosed with drug-related ILD/pneumonitis [see Dosage and Administration (2.3) and Adverse Reactions (6)].
5.3 QT Interval Prolongation
QTc prolongation can occur in patients treated with XALKORI. Across clinical trials, 34 of 1616 patients (2.1%) had QTcF (corrected QT for heart rate by the Fridericia method) greater than or equal to 500 ms and 79 of 1582 patients (5.0%) had an increase from baseline QTcF greater than or equal to 60 ms by automated machine-read evaluation of ECGs.
Avoid use of XALKORI in patients with congenital long QT syndrome. Monitor ECGs and electrolytes in patients with congestive heart failure, bradyarrhythmias, electrolyte abnormalities, or who are taking medications that are known to prolong the QT interval. Permanently discontinue XALKORI in patients who develop QTc greater than 500 ms or greater than or equal to 60 ms change from baseline with Torsade de pointes or polymorphic ventricular tachycardia or signs/symptoms of serious arrhythmia. Withhold XALKORI in patients who develop QTc greater than 500 ms on at least 2 separate ECGs until recovery to a QTc less than or equal to 480 ms, then resume XALKORI at a reduced dose as described in Table 2 [see Dosage and Administration (2.3) and Clinical Pharmacology (12.2)].
5.4 Bradycardia
Symptomatic bradycardia can occur in patients receiving XALKORI. Across clinical trials, bradycardia occurred in 219 (12.7%) of 1719 patients treated with XALKORI. Grade 3 syncope occurred in 2.4% of XALKORI-treated patients and in 0.6% of the chemotherapy-treated patients.
Avoid using XALKORI in combination with other agents known to cause bradycardia (e.g., beta-blockers, non-dihydropyridine calcium channel blockers, clonidine, and digoxin) to the extent possible. Monitor heart rate and blood pressure regularly. In cases of symptomatic bradycardia that is not life-threatening, hold XALKORI until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above, re-evaluate the use of concomitant medications, and adjust the dose of XALKORI. Permanently discontinue for life-threatening bradycardia due to XALKORI; however, if associated with concomitant medications known to cause bradycardia or hypotension, hold XALKORI until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above, and if concomitant medications can be adjusted or discontinued, restart XALKORI at 250 mg once daily with frequent monitoring [see Dosage and Administration (2.3) and Adverse Reactions (6)].
5.5 Severe Visual Loss
Across all clinical trials, the incidence of Grade 4 visual field defect with vision loss was 0.2% (4/1719). Optic atrophy and optic nerve disorder have been reported as potential causes of vision loss.
Discontinue XALKORI in patients with new onset of severe visual loss (best corrected vision less than 20/200 in one or both eyes). Perform an ophthalmological evaluation consisting of best corrected visual acuity, retinal photographs, visual fields, optical coherence tomography (OCT) and other evaluations as appropriate for new onset of severe visual loss. There is insufficient information to characterize the risks of resumption of XALKORI in patients with a severe visual loss; a decision to resume XALKORI should consider the potential benefits to the patient.
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action, XALKORI can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of crizotinib in pregnant rats during organogenesis at exposures similar to those observed with the maximum recommended human dose resulted in embryotoxicity and fetotoxicity. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with XALKORI and for at least 45 days following the final dose [see Use in Specific Populations (8.1, 8.3)]. Advise male patients with female partners of reproductive potential to use condoms during treatment with XALKORI and for at least 90 days after the final dose [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Interstitial Lung Disease (Pneumonitis) [see Warnings and Precautions (5.2)]
- QT Interval Prolongation [see Warnings and Precautions (5.3)]
- Bradycardia [see Warnings and Precautions (5.4)]
- Severe Visual Loss [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data in the Warnings and Precautions section reflect exposure to XALKORI in 1719 patients who received XALKORI 250 mg twice daily enrolled on Studies 1 (including an additional 109 patients who crossed over from the control arm), 2, 3, a single arm trial (n=1063) of ALK-positive NSCLC, and an additional ALK-positive NSCLC expansion cohort of a dose finding study (n=154) [see Warnings and Precautions (5)].
The data described below is based primarily on 343 patients with ALK-positive metastatic NSCLC who received XALKORI 250 mg twice daily from 2 open-label, randomized, active-controlled trials (Studies 1 and 2). The safety of XALKORI was also evaluated in 50 patients with ROS1-positive metastatic NSCLC from a single-arm study (Study 3).
The most common adverse reactions (≥25%) of XALKORI are vision disorders, nausea, diarrhea, vomiting, edema, constipation, elevated transaminases, fatigue, decreased appetite, upper respiratory infection, dizziness, and neuropathy.
Previously Untreated ALK-Positive Metastatic NSCLC - Study 1
The data in Table 3 are derived from 340 patients with ALK-positive metastatic NSCLC who had not received previous systemic treatment for advanced disease who received treatment in a randomized, multicenter, open-label, active-controlled trial (Study 1). Patients in the XALKORI arm (n=171) received XALKORI 250 mg orally twice daily until documented disease progression, intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit. A total of 169 patients in the chemotherapy arm received pemetrexed 500 mg/m2 in combination with cisplatin 75 mg/m2 (n=91) or carboplatin at a dose calculated to produce an area under the concentration-time curve (AUC) of 5 or 6 mg min/mL (n=78). Chemotherapy was given by intravenous infusion every 3 weeks for up to 6 cycles, in the absence of dose-limiting chemotherapy-related toxicities. After 6 cycles, patients remained on study with no additional anticancer treatment, and tumor assessments continued until documented disease progression.
The median duration of study treatment was 10.9 months for patients in the XALKORI arm and 4.1 months for patients in the chemotherapy arm. Median duration of treatment was 5.2 months for patients who received XALKORI after cross over from chemotherapy. Across the 340 patients who were treated in Study 1, the median age was 53 years; 16% of patients were older than 65 years. A total of 62% of patients were female and 46% were Asian.
Serious adverse events were reported in 58 patients (34%) treated with XALKORI. The most frequent serious adverse events reported in patients treated with XALKORI were dyspnea (4.1%) and pulmonary embolism (2.9%). Fatal adverse events in XALKORI-treated patients occurred in 2.3% patients, consisting of septic shock, acute respiratory failure, and diabetic ketoacidosis.
Dose reductions due to adverse reactions were required in 6.4% of XALKORI-treated patients. The most frequent adverse reactions that led to dose reduction in these patients were nausea (1.8%) and elevated transaminases (1.8%).
Permanent discontinuation of XALKORI treatment for adverse reactions was 8.2%. The most frequent adverse reactions that led to permanent discontinuation in XALKORI-treated patients were elevated transaminases (1.2%), hepatotoxicity (1.2%), and ILD (1.2%).
Tables 3 and 4 summarize common adverse reactions and laboratory abnormalities in XALKORI-treated patients.
| Adverse Reaction | XALKORI (N=171) | Chemotherapy (Pemetrexed/Cisplatin or Pemetrexed/Carboplatin) (N=169) |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3–4 (%) | All Grades (%) | Grade 3–4 (%) |
|
|
||||
| Cardiac Disorders | ||||
| Electrocardiogram QT prolonged | 6 | 2 | 2 | 0 |
| Bradycardia† | 14 | 1 | 1 | 0 |
| Eye Disorders | ||||
| Vision disorder‡ | 71 | 1 | 10 | 0 |
| Gastrointestinal Disorders | ||||
| Vomiting | 46 | 2 | 36 | 3 |
| Diarrhea | 61 | 2 | 13 | 1 |
| Constipation | 43 | 2 | 30 | 0 |
| Dyspepsia | 14 | 0 | 2 | 0 |
| Dysphagia | 10 | 1 | 2 | 1 |
| Abdominal pain§ | 26 | 0 | 12 | 0 |
| Esophagitis¶ | 6 | 2 | 1 | 0 |
| General Disorders and Administration Site Conditions | ||||
| Edema# | 49 | 1 | 12 | 1 |
| Pyrexia | 19 | 0 | 11 | 1 |
| Infections and Infestations | ||||
| Upper respiratory infectionÞ | 32 | 0 | 12 | 1 |
| Investigations | ||||
| Increased weight | 8 | 1 | 2 | 0 |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Pain in extremity | 16 | 0 | 7 | 0 |
| Muscle spasm | 8 | 0 | 2 | 1 |
| Nervous System Disorders | ||||
| Dizzinessß | 18 | 0 | 10 | 1 |
| Dysgeusia | 26 | 0 | 5 | 0 |
| Headache | 22 | 1 | 15 | 0 |
Additional adverse reactions occurring at an overall incidence between 1% and 60% in patients treated with XALKORI included nausea (56%), decreased appetite (30%), fatigue (29%), neuropathy (21%; gait disturbance, hypoesthesia, muscular weakness, neuralgia, neuropathy peripheral, paresthesia, peripheral sensory neuropathy, polyneuropathy, sensory disturbance), rash (11%), renal cyst (5%), ILD (1%; ILD, pneumonitis), syncope (1%), and decreased blood testosterone (1%; hypogonadism).
| Laboratory Abnormality | XALKORI | Chemotherapy | ||
|---|---|---|---|---|
| Any Grade (%) | Grade 3–4 (%) | Any Grade (%) | Grade 3–4 (%) |
|
| Additional laboratory test abnormality in patients treated with XALKORI was an increase in creatinine (Any Grade: 99%; Grade 3: 2%; Grade 4: 0%) compared to the chemotherapy arm (Any Grade: 92%; Grade 3: 0%; Grade 4: 1%). | ||||
| Hematology | ||||
| Neutropenia | 52 | 11 | 59 | 16 |
| Lymphopenia | 48 | 7 | 53 | 13 |
| Chemistry | ||||
| ALT elevation | 79 | 15 | 33 | 2 |
| AST elevation | 66 | 8 | 28 | 1 |
| Hypophosphatemia | 32 | 10 | 21 | 6 |
Previously Treated ALK-Positive Metastatic NSCLC - Study 2
The data in Table 5 are derived from 343 patients with ALK-positive metastatic NSCLC enrolled in a randomized, multicenter, active-controlled, open-label trial (Study 2). Patients in the XALKORI arm (n=172) received XALKORI 250 mg orally twice daily until documented disease progression, intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit. A total of 171 patients in the chemotherapy arm received pemetrexed 500 mg/m2 (n=99) or docetaxel 75 mg/m2 (n=72) by intravenous infusion every 3 weeks until documented disease progression, intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit. Patients in the chemotherapy arm received pemetrexed unless they had received pemetrexed as part of first-line or maintenance treatment.
The median duration of study treatment was 7.1 months for patients who received XALKORI and 2.8 months for patients who received chemotherapy. Across the 347 patients who were randomized to study treatment (343 received at least 1 dose of study treatment), the median age was 50 years; 14% of patients were older than 65 years. A total of 56% of patients were female and 45% of patients were Asian.
Serious adverse reactions were reported in 64 patients (37.2%) treated with XALKORI and 40 patients (23.4%) in the chemotherapy arm. The most frequent serious adverse reactions reported in patients treated with XALKORI were pneumonia (4.1%), pulmonary embolism (3.5%), dyspnea (2.3%), and ILD (2.9%). Fatal adverse reactions in XALKORI-treated patients in Study 2 occurred in 9 (5%) patients, consisting of: acute respiratory distress syndrome, arrhythmia, dyspnea, pneumonia, pneumonitis, pulmonary embolism, ILD, respiratory failure, and sepsis.
Dose reductions due to adverse reactions were required in 16% of XALKORI-treated patients. The most frequent adverse reactions that led to dose reduction in the patients treated with XALKORI were ALT elevation (7.6%) including some patients with concurrent AST elevation, QTc prolongation (2.9%), and neutropenia (2.3%).
XALKORI was discontinued for adverse reactions in 15% of patients. The most frequent adverse reactions that led to discontinuation of XALKORI were ILD (1.7%), ALT and AST elevation (1.2%), dyspnea (1.2%), and pulmonary embolism (1.2%).
Tables 5 and 6 summarize common adverse reactions and laboratory abnormalities in XALKORI-treated patients.
| Adverse Reaction | XALKORI (N=172) | Chemotherapy (Pemetrexed or Docetaxel) (N=171) |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3–4 (%) | All Grades (%) | Grade 3–4 (%) |
|
|
||||
| Nervous System Disorders | ||||
| Dizziness† | 22 | 1 | 8 | 0 |
| Dysgeusia | 26 | 0 | 9 | 0 |
| Syncope | 3 | 3 | 0 | 0 |
| Eye Disorders | ||||
| Vision disorder‡ | 60 | 0 | 9 | 0 |
| Cardiac Disorders | ||||
| Electrocardiogram QT prolonged | 5 | 3 | 0 | 0 |
| Bradycardia§ | 5 | 0 | 0 | 0 |
| Investigations | ||||
| Decreased weight | 10 | 1 | 4 | 0 |
| Gastrointestinal Disorders | ||||
| Vomiting | 47 | 1 | 18 | 0 |
| Nausea | 55 | 1 | 37 | 1 |
| Diarrhea | 60 | 0 | 19 | 1 |
| Constipation | 42 | 2 | 23 | 0 |
| Dyspepsia | 8 | 0 | 3 | 0 |
| Infections and Infestations | ||||
| Upper respiratory infection¶ | 26 | 0 | 13 | 1 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Pulmonary embolism# | 6 | 5 | 2 | 2 |
| General Disorders and Administration Site Conditions | ||||
| EdemaÞ | 31 | 0 | 16 | 0 |
Additional adverse reactions occurring at an overall incidence between 1% and 30% in patients treated with XALKORI included decreased appetite (27%), fatigue (27%), neuropathy (19%; dysesthesia, gait disturbance, hypoesthesia, muscular weakness, neuralgia, peripheral neuropathy, paresthesia, peripheral sensory neuropathy, polyneuropathy, burning sensation in skin), rash (9%), ILD (4%; acute respiratory distress syndrome, ILD, pneumonitis), renal cyst (4%), esophagitis (2%), hepatic failure (1%), and decreased blood testosterone (1%; hypogonadism).
| Laboratory Abnormality | XALKORI | Chemotherapy | ||
|---|---|---|---|---|
| Any Grade (%) | Grade 3–4 (%) | Any Grade (%) | Grade 3–4 (%) |
|
| Additional laboratory test abnormality in patients treated with XALKORI was an increase in creatinine (Any Grade: 96%; Grade 3: 1%; Grade 4: 0%) compared to the chemotherapy arm (Any Grade: 72%; Grade 3: 0%; Grade 4: 0%). | ||||
| Hematology | ||||
| Neutropenia | 49 | 12 | 28 | 12 |
| Lymphopenia | 51 | 9 | 60 | 25 |
| Chemistry | ||||
| ALT elevation | 76 | 17 | 38 | 4 |
| AST elevation | 61 | 9 | 33 | 0 |
| Hypokalemia | 18 | 4 | 10 | 1 |
| Hypophosphatemia | 28 | 5 | 25 | 6 |
7. Drug Interactions
7.1 Drugs That May Increase Crizotinib Plasma Concentrations
Coadministration of crizotinib with strong cytochrome P450 (CYP) 3A inhibitors increases crizotinib plasma concentrations [see Clinical Pharmacology (12.3)]. Avoid concomitant use of strong CYP3A inhibitors, including but not limited to clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, troleandomycin, voriconazole, grapefruit, and grapefruit juice. If the use of strong CYP3A inhibitors cannot be avoided, reduce the dose of XALKORI to 250 mg orally once daily [see Clinical Pharmacology (12.3)]. After discontinuation of a strong CYP3A inhibitor, resume the XALKORI dose used prior to initiating the strong CYP3A inhibitor. Exercise caution with concomitant use of moderate CYP3A inhibitors.
7.2 Drugs That May Decrease Crizotinib Plasma Concentrations
Coadministration of crizotinib with strong CYP3A inducers decreases crizotinib plasma concentrations [see Clinical Pharmacology (12.3)]. Avoid concomitant use of strong CYP3A inducers, including but not limited to carbamazepine, phenobarbital, phenytoin, rifabutin, rifampin, and St. John's Wort.
7.3 Drugs Whose Plasma Concentrations May Be Altered By Crizotinib
Crizotinib inhibits CYP3A both in vitro and in vivo [see Clinical Pharmacology (12.3)]. Avoid concomitant use of CYP3A substrates with narrow therapeutic range, including but not limited to alfentanil, cyclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, and tacrolimus in patients taking XALKORI. If concomitant use of these CYP3A substrates with narrow therapeutic range is required in patients taking XALKORI, dose reductions of the CYP3A substrates may be required due to adverse reactions.
8. Use In Specific Populations
8.4 Pediatric Use
The safety and efficacy of XALKORI in pediatric patients has not been established.
8.5 Geriatric Use
Of the total number of patients with ALK-positive metastatic NSCLC in clinical studies of XALKORI (n=1669), 16% were 65 years or older and 3.8% were 75 years or older. No overall differences in safety or effectiveness were observed between these patients and younger patients.
Clinical studies of XALKORI in patients with ROS1-positive metastatic NSCLC did not include sufficient numbers of patients age 65 years and older to determine whether they respond differently from younger patients.
8.6 Hepatic Impairment
Crizotinib concentrations increased in patients with pre-existing moderate (any AST and total bilirubin >1.5 times ULN and ≤3 times ULN) or severe (any AST and total bilirubin >3 times ULN) hepatic impairment [see Clinical Pharmacology (12.3)]. Reduce XALKORI dose in patients with moderate or severe hepatic impairment [see Dosage and Administration (2.2)]. No dose adjustment is recommended in patients with pre-existing mild hepatic impairment.
8.7 Renal Impairment
Increased exposure to crizotinib occurred in patients with pre-existing severe renal impairment (CLcr <30 mL/min) not requiring dialysis. Administer XALKORI at a dose of 250 mg taken orally once daily in patients with severe renal impairment not requiring dialysis [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. No dose adjustment is recommended in patients with mild to moderate renal impairment.
10. Overdosage
There have been no known cases of XALKORI overdose. There is no antidote for XALKORI.
11. Xalkori Description
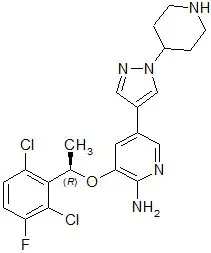
XALKORI (crizotinib) is an oral receptor tyrosine kinase inhibitor. The molecular formula for crizotinib is C21H22Cl2FN5O. The molecular weight is 450.34 daltons. Crizotinib is described chemically as (R)-3-[1-(2,6-Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine.
The chemical structure of crizotinib is shown below:
Crizotinib is a white to pale-yellow powder with a pKa of 9.4 (piperidinium cation) and 5.6 (pyridinium cation). The solubility of crizotinib in aqueous media decreases over the range pH 1.6 to pH 8.2 from greater than 10 mg/mL to less than 0.1 mg/mL. The log of the distribution coefficient (octanol/water) at pH 7.4 is 1.65.
XALKORI capsules are supplied as printed hard-shell capsules containing 250 mg or 200 mg of crizotinib together with colloidal silicon dioxide, microcrystalline cellulose, anhydrous dibasic calcium phosphate, sodium starch glycolate, magnesium stearate, and hard gelatin capsule shells as inactive ingredients.
The pink opaque capsule shell components contain gelatin, titanium dioxide, and red iron oxide. The white opaque capsule shell components contain gelatin and titanium dioxide. The printing ink contains shellac, propylene glycol, strong ammonia solution, potassium hydroxide, and black iron oxide.
12. Xalkori - Clinical Pharmacology
12.1 Mechanism of Action
Crizotinib is an inhibitor of receptor tyrosine kinases including ALK, Hepatocyte Growth Factor Receptor (HGFR, c-Met), ROS1 (c-ros), and Recepteur d'Origine Nantais (RON). Translocations can affect the ALK gene resulting in the expression of oncogenic fusion proteins. The formation of ALK fusion proteins results in activation and dysregulation of the gene's expression and signaling which can contribute to increased cell proliferation and survival in tumors expressing these proteins. Crizotinib demonstrated concentration-dependent inhibition of ALK, ROS1, and c-Met phosphorylation in cell-based assays using tumor cell lines and demonstrated antitumor activity in mice bearing tumor xenografts that expressed echinoderm microtubule-associated protein-like 4 (EML4)- or nucleophosmin (NPM)-ALK fusion proteins or c-Met.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with crizotinib have not been conducted.
Crizotinib was genotoxic in an in vitro micronucleus assay in Chinese Hamster Ovary cultures, in an in vitro human lymphocyte chromosome aberration assay, and in in vivo rat bone marrow micronucleus assays. Crizotinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay.
No specific studies with crizotinib have been conducted in animals to evaluate the effect on fertility; however, crizotinib is considered to have the potential to impair reproductive function and fertility in humans based on findings in repeat-dose toxicity studies in the rat. Findings observed in the male reproductive tract included testicular pachytene spermatocyte degeneration in rats given greater than or equal to 50 mg/kg/day for 28 days (greater than 1.7 times the recommended human dose based on AUC). Findings observed in the female reproductive tract included single-cell necrosis of ovarian follicles of a rat given 500 mg/kg/day (approximately 10 times the recommended human dose based on body surface area) for 3 days.
14. Clinical Studies
14.1 ALK-Positive Metastatic NSCLC
Previously Untreated ALK-Positive Metastatic NSCLC - Study 1
The efficacy and safety of XALKORI for the treatment of patients with ALK-positive metastatic NSCLC, who had not received previous systemic treatment for advanced disease, was demonstrated in a randomized, multicenter, open-label, active-controlled study (Study 1). Patients were required to have ALK-positive NSCLC as identified by the FDA-approved assay, Vysis ALK Break-Apart fluorescence in situ hybridization (FISH) Probe Kit, prior to randomization. The major efficacy outcome measure was progression-free survival (PFS) according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 as assessed by independent radiology review (IRR) committee. Additional efficacy outcome measures included objective response rate (ORR) as assessed by IRR, duration of response (DOR), and overall survival (OS). Patient-reported lung cancer symptoms were assessed at baseline and periodically during treatment.
Patients were randomized to receive XALKORI (n=172) or chemotherapy (n=171). Randomization was stratified by Eastern Cooperative Oncology Group (ECOG) performance status (0–1, 2), race (Asian, non-Asian), and brain metastases (present, absent). Patients in the XALKORI arm received XALKORI 250 mg orally twice daily until documented disease progression, intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit. Chemotherapy consisted of pemetrexed 500 mg/m2 with cisplatin 75 mg/m2 or carboplatin AUC of 5 or 6 mg∙min/mL by intravenous infusion every 3 weeks for up to 6 cycles. Patients in the chemotherapy arm were not permitted to receive maintenance chemotherapy. At the time of documented disease progression, as per independent radiology review, patients randomized to chemotherapy were offered XALKORI.
The demographic characteristics of the overall study population were 62% female, median age of 53 years, baseline ECOG performance status 0 or 1 (95%), 51% White and 46% Asian, 4% current smokers, 32% past smokers, and 64% never smokers. The disease characteristics of the overall study population were metastatic disease in 98% of patients, 92% of patients' tumors were classified as adenocarcinoma histology, 27% of patients had brain metastases, and 7% received systemic chemotherapy as adjuvant or neoadjuvant therapy. Of those randomized to chemotherapy, 70% received XALKORI after IRR documented progression.
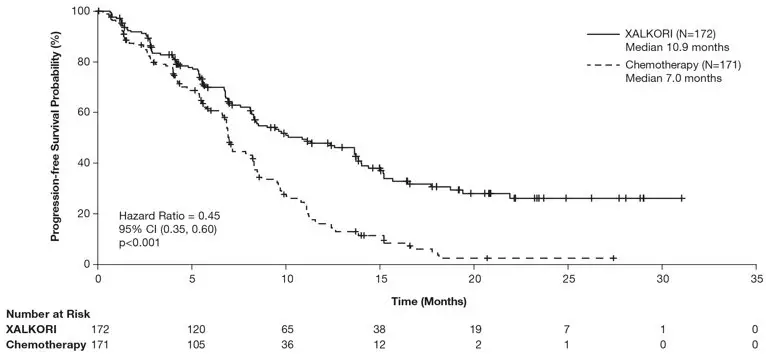
Study 1 demonstrated a statistically significant improvement in PFS in the patients treated with XALKORI. The OS analysis conducted at the time of the PFS analysis did not suggest a difference in survival between arms. Table 7 and Figure 1 summarize the efficacy results. Exploratory patient-reported symptom measures of baseline and post-treatment dyspnea, cough, and chest pain suggested a delay in time to development of or worsening of dyspnea, but not cough or chest pain, in patients treated with XALKORI as compared to chemotherapy. The patient-reported delay in onset or worsening of dyspnea may be an overestimation, because patients were not blinded to treatment assignment.
| XALKORI (N=172) | Chemotherapy (N=171) |
|
|---|---|---|
| HR=hazard ratio; CI=confidence interval; IRR=independent radiology review; NR=not reached; CR=complete response; PR=partial response. | ||
|
||
| Progression-Free Survival (Based on IRR) | ||
| Number of Events (%) | 100 (58%) | 137 (80%) |
| Progressive Disease | 89 (52%) | 132 (77%) |
| Death | 11 (6%) | 5 (3%) |
| Median, Months (95% CI) | 10.9 (8.3, 13.9) | 7.0 (6.8, 8.2) |
| HR (95% CI)* | 0.45 (0.35, 0.60) | |
| p-value† | <0.001 | |
| Overall Survival | ||
| Number of Events (%) | 44 (26%) | 46 (27%) |
| Median, Months (95% CI) | NR | NR |
| HR (95% CI)* | 0.82 (0.54, 1.26) | |
| p-value† | 0.36 | |
| Tumor Responses (Based on IRR) | ||
| Objective Response Rate % (95% CI) | 74% (67, 81) | 45% (37, 53) |
| CR, n (%) | 3 (1.7%) | 2 (1.2%) |
| PR, n (%) | 125 (73%) | 75 (44%) |
| p-value‡ | <0.001 | |
| Duration of Response | ||
| Median, Months (95% CI) | 11.3 (8.1, 13.8) | 5.3 (4.1, 5.8) |
| Figure 1. Kaplan-Meier Curves of Progression-Free Survival as Assessed by IRR in Study 1 |
|---|
|
|
Previously Treated ALK-Positive Metastatic NSCLC - Study 2
The efficacy and safety of XALKORI as monotherapy for the treatment of 347 patients with ALK-positive metastatic NSCLC, previously treated with 1 platinum-based chemotherapy regimen, were demonstrated in a randomized, multicenter, open-label, active-controlled study (Study 2). The major efficacy outcome was PFS according to RECIST version 1.1 as assessed by IRR. Additional efficacy outcomes included ORR as assessed by IRR, DOR, and OS.
Patients were randomized to receive XALKORI 250 mg orally twice daily (n=173) or chemotherapy (n=174). Chemotherapy consisted of pemetrexed 500 mg/m2 (if pemetrexed-naïve; n=99) or docetaxel 75 mg/m2 (n=72) intravenously (IV) every 21 days. Patients in both treatment arms continued treatment until documented disease progression, intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit. Randomization was stratified by ECOG performance status (0–1, 2), brain metastases (present, absent), and prior EGFR tyrosine kinase inhibitor treatment (yes, no). Patients were required to have ALK-positive NSCLC as identified by the FDA-approved assay, Vysis ALK Break-Apart FISH Probe Kit, prior to randomization. At the time of the final analysis of overall survival, 154 (89%) patients randomized to the chemotherapy arm subsequently received XALKORI.
The demographic characteristics of the overall study population were 56% female, median age of 50 years, baseline ECOG performance status 0 or 1 (90%), 52% White and 45% Asian, 4% current smokers, 33% past smokers, and 63% never smokers. The disease characteristics of the overall study population were metastatic disease in at least 95% of patients and at least 93% of patients' tumors were classified as adenocarcinoma histology.
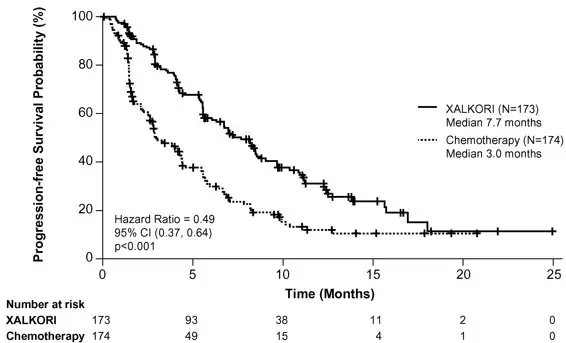
Study 2 demonstrated a statistically significant improvement in PFS in the patients treated with XALKORI. Table 8 and Figure 2 summarize the efficacy results.
| XALKORI (N=173) | Chemotherapy (N=174) |
|
|---|---|---|
| HR=hazard ratio; CI=confidence interval; IRR=independent radiology review; CR=complete response; PR=partial response. | ||
|
||
| Progression-Free Survival (Based on IRR) | ||
| Number of Events (%) | 100 (58%) | 127 (73%) |
| Progressive Disease | 84 (49%) | 119 (68%) |
| Death | 16 (9%) | 8 (5%) |
| Median, Months (95% CI) | 7.7 (6.0, 8.8) | 3.0* (2.6, 4.3) |
| HR (95% CI)† | 0.49 (0.37, 0.64) | |
| p-value‡ | <0.001 | |
| Overall Survival | ||
| Number of Events (%) | 116 (67%) | 126 (72%) |
| Median, Months (95% CI) | 21.7 (18.9,30.5) | 21.9 (16.8,26.0) |
| HR (95% CI)† | 0.85 (0.66, 1.10) | |
| p-value‡ | 0.229 | |
| Tumor Responses (Based on IRR) | ||
| Objective Response Rate % (95% CI) | 65% (58, 72) | 20% (14, 26) |
| CR, n (%) | 1 (0.6%) | 0 |
| PR, n (%) | 112 (65%) | 34 (20%) |
| p-value§ | <0.001 | |
| Duration of Response | ||
| Median, Months (95% CI) | 7.4 (6.1, 9.7) | 5.6 (3.4, 8.3) |
| Figure 2. Kaplan-Meier Curves of Progression-Free Survival as Assessed by IRR in Study 2 |
|---|
 |
14.2 ROS1-Positive Metastatic NSCLC
The efficacy and safety of XALKORI was investigated in a multicenter, single-arm study (Study 3), in which patients with ROS1-positive metastatic NSCLC received XALKORI 250 mg orally twice daily. Patients were required to have histologically-confirmed advanced NSCLC with a ROS1 rearrangement, age 18 years or older, ECOG performance status of 0, 1, or 2, adequate organ function, and measurable disease. The efficacy outcome measures were ORR and DOR according to RECIST version 1.0 as assessed by IRR and investigator, with imaging performed every 8 weeks for the first 60 weeks.
Baseline demographic and disease characteristics were female (56%), median age of 53 years, baseline ECOG performance status of 0 or 1 (98%), White (54%), Asian (42%), past smokers (22%), never smokers (78%), metastatic disease (92%), adenocarcinoma (96%), no prior systemic therapy for metastatic disease (14%), and prior platinum-based chemotherapy for metastatic disease (80%). The ROS1 status of NSCLC tissue samples was determined by laboratory-developed break-apart FISH (96%) or RT-PCR (4%) clinical trial assays. For assessment by FISH, ROS1 positivity required that ≥15% of a minimum of 50 evaluated nuclei contained a ROS1 gene rearrangement.
Efficacy results are summarized in Table 9.
| Efficacy Parameters | IRR (N=50) | Investigator-Assessed (N=50) |
|---|---|---|
| IRR=independent radiology review; CI=confidence interval; NR=not reached. | ||
|
||
| Objective Response Rate (95% CI) | 66% (51, 79) | 72% (58, 84) |
| Complete Response, n | 1 | 5 |
| Partial Response, n | 32 | 31 |
| Duration of Response | ||
| Median, Months (95% CI) | 18.3 (12.7, NR) | NR (14.5, NR) |
16. How is Xalkori supplied
- 250 mg capsules
Hard gelatin capsule with pink opaque cap and body, printed with black ink "Pfizer" on the cap, "CRZ 250" on the body; available in:Bottles of 60 capsules: NDC 0069-8140-20 - 200 mg capsules
Hard gelatin capsule with pink opaque cap and white opaque body, printed with black ink "Pfizer" on the cap, "CRZ 200" on the body; available in:Bottles of 60 capsules: NDC 0069-8141-20
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).

| PATIENT INFORMATION XALKORI® (zal-KOR-ee) (crizotinib) capsules |
||
|---|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: March 2016 | |
|
What is the most important information I should know about XALKORI? |
||
| XALKORI may cause serious side effects, including: | ||
|
||
|
|
|
|
||
|
|
|
|
||
| What is XALKORI? | ||
| XALKORI is a prescription medicine that is used to treat people with non-small cell lung cancer (NSCLC) that has spread to other parts of the body and is caused by a defect in either a gene called ALK (anaplastic lymphoma kinase) or a gene called ROS1. | ||
| It is not known if XALKORI is safe and effective in children. | ||
| What should I tell my healthcare provider before taking XALKORI? | ||
| Before you take XALKORI, tell your healthcare provider if you: | ||
|
||
| Tell your healthcare provider about the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. | ||
| How should I take XALKORI? | ||
|
||
| What should I avoid while taking XALKORI? | ||
|
||
| XALKORI may cause serious side effects, including: | ||
|
||
The most common side effects of XALKORI include:
|
||
| XALKORI may cause decreased fertility in females and males. In females, this could affect your ability to become pregnant. In males, this could affect your ability to father a child. Talk to your healthcare provider if you have concerns about fertility. | ||
| Tell your healthcare provider if you have any side effect that bothers you or that does not go away. | ||
| These are not all of the possible side effects of XALKORI. For more information, ask your healthcare provider or pharmacist. | ||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||
| How should I store XALKORI? | ||
|
||
| Keep XALKORI and all medicines out of the reach of children. | ||
| General information about XALKORI | ||
| Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use XALKORI for a condition for which it was not prescribed. Do not give XALKORI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about XALKORI that is written for health professionals. | ||
| What are the ingredients in XALKORI? | ||
| Active ingredient: crizotinib | ||
| Inactive ingredients: colloidal silicon dioxide, microcrystalline cellulose, anhydrous dibasic calcium phosphate, sodium starch glycolate, and magnesium stearate. | ||
| Pink opaque capsule shell contains: gelatin, titanium dioxide, and red iron oxide. | ||
| White opaque capsule shell contains: gelatin and titanium dioxide. | ||
| Printing ink contains: shellac, propylene glycol, strong ammonia solution, potassium hydroxide, and black iron oxide. | ||
|
|
||
| LAB-0441-8.0 | ||
| For more information, go to www.XALKORI.com. | ||


| XALKORI
crizotinib capsule |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| XALKORI
crizotinib capsule |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) |
| Registrant - Pfizer Inc (113480771) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Pharmaceuticals LLC | 829084552 | REPACK(0069-8141, 0069-8140) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Ireland Pharmaceuticals | 989811526 | API MANUFACTURE(0069-8140, 0069-8141) | |