Drug Detail:Crysvita (Burosumab [ bur-oh-sue-mab ])
Drug Class: Miscellaneous metabolic agents
Highlights of Prescribing Information
CRYSVITA® (burosumab-twza) injection, for subcutaneous use
Initial U.S. Approval: 2018
Indications and Usage for Crysvita
CRYSVITA is a fibroblast growth factor 23 (FGF23) blocking antibody indicated for:
- The treatment of X-linked hypophosphatemia (XLH) in adult and pediatric patients 6 months of age and older. (1.1)
- The treatment of FGF23-related hypophosphatemia in tumor-induced osteomalacia (TIO) associated with phosphaturic mesenchymal tumors that cannot be curatively resected or localized in adult and pediatric patients 2 years of age and older. (1.2)
Crysvita Dosage and Administration
For subcutaneous use only (2)
-
Pediatric XLH (6 months and older):
- For patients who weigh less than 10 kg, starting dose regimen is 1 mg/kg of body weight rounded to the nearest 1 mg, administered every two weeks (2.2)
- For patients who weigh 10 kg and greater, starting dose regimen is 0.8 mg/kg of body weight rounded to the nearest 10 mg, administered every two weeks. The minimum starting dose is 10 mg up to a maximum dose of 90 mg. (2.2)
- Adult XLH: Dose regimen is 1 mg/kg body weight rounded to the nearest 10 mg up to a maximum dose of 90 mg administered every four weeks. (2.3)
- Pediatric TIO (2 years and older): Starting dose is 0.4 mg/kg of body weight rounded to the nearest 10 mg every 2 weeks. Dose may be increased up to 2 mg/kg not to exceed 180 mg, administered every two weeks. (2.4)
- Adult TIO: Starting dose is 0.5 mg/kg every four weeks. Dose may be increased up to 2 mg/kg not to exceed 180 mg, administered every two weeks. (2.5)
Dosage Forms and Strengths
Injection: 10 mg/mL, 20 mg/mL, or 30 mg/mL in a single-dose vial (3)
Contraindications
- With oral phosphate and/or active vitamin D analogs. (4)
- When serum phosphorus is within or above the normal range for age. (4)
- In patients with severe renal impairment or end stage renal disease. (4)
Warnings and Precautions
- Hypersensitivity: Discontinue CRYSVITA if serious hypersensitivity reactions occur and initiate appropriate medical treatment. (5.1)
- Hyperphosphatemia and Risk of Nephrocalcinosis: For patients already taking CRYSVITA, dose interruption and/or dose reduction may be required based on a patient's serum phosphorus levels. (5.2, 6.1)
- Injection Site Reactions: Administration of CRYSVITA may result in local injection site reactions. Discontinue CRYSVITA if severe injection site reactions occur and administer appropriate medical treatment. (5.3, 6.1)
Adverse Reactions/Side Effects
- Most common adverse reactions (≥25% in the CRYSVITA group and > Active Control) in pediatric XLH patients are: pyrexia, injection site reaction, cough, vomiting, pain in extremity, headache, tooth abscess, dental caries. (6.1)
- Most common adverse reactions (>5% and in at least 2 patients more than placebo) in adult XLH patients are: back pain, headache, tooth infection, restless legs syndrome, vitamin D decreased, dizziness, constipation, muscle spasms, blood phosphorus increased. (6.1)
- Most common adverse reactions (>10%) in TIO patients are: tooth abscess, muscle spasms, dizziness, constipation, injection site reaction, rash, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Kyowa Kirin, Inc. at 1-844-768-3544 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2023
Full Prescribing Information
1. Indications and Usage for Crysvita
2. Crysvita Dosage and Administration
2.1 Important Dosage and Administration Information
Discontinue oral phosphate and/or active vitamin D analogs (e.g. calcitriol, paricalcitol, doxercalciferol, calcifediol) 1 week prior to initiation of treatment [see Contraindications (4)].
Fasting serum phosphorus concentration should be below the reference range for age prior to initiation of treatment [see Contraindications (4)].
CRYSVITA is administered by subcutaneous injection and should be administered by a healthcare provider.
The maximum volume of CRYSVITA per injection is 1.5 mL. If multiple injections are required, administer at different injection sites.
2.2 Pediatric Patients with X-linked Hypophosphatemia (6 months to less than 18 years of age)
For patients who weigh less than 10 kg, the recommended starting dose is 1 mg/kg of body weight, rounded to the nearest 1 mg, administered every two weeks.
For patients who weigh 10 kg and greater, the recommended starting dose regimen is 0.8 mg/kg of body weight, rounded to the nearest 10 mg, administered every two weeks. The minimum starting dose is 10 mg up to a maximum dose of 90 mg.
After initiation of treatment with CRYSVITA, measure fasting serum phosphorus every 4 weeks for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is above the lower limit of the reference range for age and below 5 mg/dL, continue treatment with the same dose. Follow dose adjustment schedule below to maintain serum phosphorus within the reference range for age.
Dose Adjustment
Reassess fasting serum phosphorus level 4 weeks after dose adjustment.
Do not adjust CRYSVITA more frequently than every 4 weeks.
Dose Increase:
For patients who weigh less than 10 kg, if serum phosphorus is below the reference range for age, the dose may be increased to 1.5 mg/kg, rounded to the nearest 1 mg, administered every two weeks. If additional dose increases are needed, the dose may be increased to the maximum dose of 2 mg/kg, rounded to the nearest 1 mg, administered every two weeks.
For patients who weigh 10 kg or greater, if serum phosphorus is below the reference range for age, the dose may be increased stepwise up to approximately 2 mg/kg, administered every two weeks (maximum dose of 90 mg) according to the dosing schedule shown in Table 1.
| Body Weight (kg) | Starting Dose (mg) | First Dose Increase to (mg) | Second Dose Increase to (mg) |
|---|---|---|---|
| 10 – 14 | 10 | 15 | 20 |
| 15 – 18 | 10 | 20 | 30 |
| 19 – 31 | 20 | 30 | 40 |
| 32 – 43 | 30 | 40 | 60 |
| 44 – 56 | 40 | 60 | 80 |
| 57 – 68 | 50 | 70 | 90 |
| 69 – 80 | 60 | 90 | 90 |
| 81 – 93 | 70 | 90 | 90 |
| 94 – 105 | 80 | 90 | 90 |
| 106 and greater | 90 | 90 | 90 |
Dose Decrease:
If serum phosphorus is above 5 mg/dL, withhold the next dose and reassess the serum phosphorus level in 4 weeks. The patient must have serum phosphorus below the reference range for age to reinitiate CRYSVITA. Once serum phosphorus is below the reference range for age, treatment may be restarted.
For patients who weigh less than 10 kg, restart CRYSVITA at 0.5 mg/kg of body weight, rounded to the nearest 1 mg, administered every two weeks. For patients who weigh 10 kg or more, restart CRYSVITA according to the dose schedule shown in Table 2.
| Previous Dose (mg) | Re-Initiation Dose (mg) |
|---|---|
| 10 | 5 |
| 15 | 10 |
| 20 | 10 |
| 30 | 10 |
| 40 | 20 |
| 50 | 20 |
| 60 | 30 |
| 70 | 30 |
| 80 | 40 |
| 90 | 40 |
After a dose decrease, reassess serum phosphorus level 4 weeks after the dose adjustment. If the level remains below the reference range for age after the re-initiation dose, the dose can be adjusted as outlined under Dose Increase.
2.3 Adult Patients with X-linked Hypophosphatemia (18 years of age and older)
The recommended dose regimen in adults is 1 mg/kg body weight, rounded to the nearest 10 mg up to a maximum dose of 90 mg, administered every four weeks.
After initiation of treatment with CRYSVITA, assess fasting serum phosphorus on a monthly basis, measured 2 weeks post-dose, for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is within the normal range, continue with the same dose.
Dose Decrease
Reassess fasting serum phosphorus level 2 weeks after dose adjustment.
Do not adjust CRYSVITA more frequently than every 4 weeks.
If serum phosphorus is above the normal range, withhold the next dose and reassess the serum phosphorus level after 4 weeks. The patient must have serum phosphorus below the normal range to be able to reinitiate CRYSVITA. Once serum phosphorus is below the normal range, treatment may be restarted at approximately half the initial starting dose up to a maximum dose of 40 mg every 4 weeks according to the dose schedule shown in Table 3. Reassess serum phosphorus 2 weeks after any change in dose.
| Previous Dose (mg) | Re-Initiation Dose (mg) |
|---|---|
| 40 | 20 |
| 50 | 20 |
| 60 | 30 |
| 70 | 30 |
| 80 and greater | 40 |
2.4 Pediatric Patients with Tumor-induced Osteomalacia (2 years to less than 18 years of age)
The recommended starting dose for pediatrics is 0.4 mg/kg body weight administered every 2 weeks, rounded to the nearest 10mg, up to a maximum dose of 2 mg/kg not to exceed 180mg, administered every 2 weeks.
After initiation of treatment with CRYSVITA, assess fasting serum phosphorus on a monthly basis, measured 2 weeks post-dose, for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is within the reference range for age, continue with the same dose. Follow the dose adjustment schedule below to maintain serum phosphorus within the reference range for age.
Dose Adjustment
Reassess fasting serum phosphorus level 4 weeks after dose adjustment.
Do not adjust CRYSVITA more frequently than every 4 weeks.
Dose Increase
If serum phosphorus is below the reference range for age, the dose should be titrated in accordance with Table 4 up to the maximum dose of 2 mg/kg every 2 weeks. The maximum dose should not exceed 180 mg.
| Body Weight (kg) | Starting Dose (mg) | First Dose Increase to (mg) | Second Dose Increase to (mg) | Third Dose* Increase to (mg) |
|---|---|---|---|---|
|
||||
| 10 – 14 | 5 | 10 | 15 | 20 |
| 15 – 18 | 5 | 10 | 20 | 25 |
| 19 – 31 | 10 | 20 | 25 | 30 |
| 32 – 43 | 10 | 30 | 40 | 50 |
| 44 – 56 | 20 | 40 | 50 | 70 |
| 57 – 68 | 20 | 50 | 70 | 90 |
| 69 – 80 | 30 | 60 | 80 | 100 |
| 81 – 93 | 30 | 70 | 100 | 120 |
| 94 – 105 | 40 | 80 | 110 | 140 |
| 106 and greater | 40 | 90 | 130 | 160 |
Dose Decrease
If serum phosphorus is above the reference range for age, withhold the next dose and reassess the serum phosphorus level in 4 weeks. The patient must have serum phosphorus below the reference range for age to reinitiate CRYSVITA. Once serum phosphorus is below the reference range for age, treatment may be restarted at approximately half the initial starting dose, up to a maximum dose of 180 mg administered every 2 weeks for pediatrics. After a dose decrease, reassess serum phosphorus level 4 weeks after the dose adjustment. If the level remains below the reference range for age after the re-initiation dose, the dose can be adjusted as outlined per Table 4.
Dose Interruption
If a patient undergoes treatment of the underlying tumor (i.e., surgical excision or radiation therapy) CRYSVITA treatment should be interrupted and serum phosphorus reassessed after treatment has been completed. CRYSVITA dose should be restarted at the patient's initiation dose if serum phosphorus remains below the lower limit of normal. Follow dose adjustment per Table 4 to maintain serum phosphorus within the reference range for age.
2.5 Adult Patients with Tumor-induced Osteomalacia (18 years of age and older)
The recommended starting dose for adults is 0.5 mg/kg body weight administered every 4 weeks, rounded to the nearest 10 mg, up to a maximum dose of 2 mg/kg not to exceed 180mg, administered every 2 weeks.
After initiation of treatment with CRYSVITA, assess fasting serum phosphorus on a monthly basis, measured 2 weeks post-dose, for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is within the normal range, continue with the same dose. Follow the dose adjustment schedule below to maintain serum phosphorus within the reference range.
Dose Adjustment
Reassess fasting serum phosphorus level 2 weeks after dose adjustment.
Do not adjust CRYSVITA more frequently than every 4 weeks.
Dose Increase
If serum phosphorus is below the normal range, the dose should be titrated in accordance with Table 5 up to the maximum dose of 2 mg/kg not to exceed 180 mg, administered every 2 weeks. For those individuals not reaching a serum phosphorus greater than the lower limit of the normal range, physicians may consider dividing total dose administered every 4 weeks and administering every 2 weeks.
| Starting Dose | First Dose Increase‡ | Second Dose Increase‡ | Third Dose Increase‡ | Fourth Dose Increase | Fifth Dose Increase (maximum dose) |
|
|---|---|---|---|---|---|---|
|
||||||
| If serum phosphorus 2 weeks post-dose adjustment is below lower limit of normal | 0.5 mg/kg every 4 weeks | Increase to: 1 mg/kg every 4 weeks OR 0.5 mg/kg every 2 weeks | Increase to: 1.5 mg/kg every 4 weeks§ OR 0.75 mg/kg every 2 weeks | Increase to: 2 mg/kg every 4 weeks§ OR 1 mg/kg every 2 weeks | Increase to: 1.5 mg/kg not to exceed 180 mg every 2 weeks | Increase to: 2 mg/kg not to exceed 180 mg every 2 weeks |
Dose Decrease
If serum phosphorus is above the normal range, withhold the next dose and reassess the serum phosphorus level in 4 weeks. The patient must have serum phosphorus below the reference range to reinitiate CRYSVITA. Once serum phosphorus is below the reference range, treatment may be restarted at approximately half the initial starting dose, up to a maximum dose of 180 mg administered every 2 weeks for adults. After a dose decrease, reassess serum phosphorus level 2 weeks after the dose adjustment. If the level remains below the reference range after the re-initiation dose, the dose can be adjusted as outlined per Table 5.
Dose Interruption
If a patient undergoes treatment of the underlying tumor (i.e., surgical excision or radiation therapy) CRYSVITA treatment should be interrupted and serum phosphorus reassessed after treatment has been completed. CRYSVITA dose should be restarted at the patient's initiation dose if serum phosphorus remains below the lower limit of normal. Follow dose adjustment per Table 5 to maintain serum phosphorus within the reference range.
2.6 Missed Dose
If a patient misses a dose, resume CRYSVITA as soon as possible at the prescribed dose. To avoid missed doses, treatments may be administered 3 days either side of the scheduled treatment date.
2.7 25-Hydroxy Vitamin D Supplementation
Monitor 25-hydroxy vitamin D levels. Supplement with cholecalciferol or ergocalciferol to maintain 25-hydroxy vitamin D levels in the normal range for age. Do not administer active Vitamin D analogs during CRYSVITA treatment [see Contraindications (4)].
2.8 General Considerations for Subcutaneous Administration
Injection sites should be rotated with each injection administered at a different anatomic location (upper arms, upper thighs, buttocks, or any quadrant of abdomen) than the previous injection. Do not inject into moles, scars, or areas where the skin is tender, bruised, red, hard, or not intact. If a given dose on a dosing day requires multiple vials of CRYSVITA, contents from two vials can be combined for injection. The maximum volume of CRYSVITA per injection is 1.5 mL. If multiple injections are required on a given dosing day, administer at different injection sites. Monitor for signs of reactions [see Warnings and Precautions (5.3)].
Visually inspect CRYSVITA for particulate matter and discoloration prior to administration. CRYSVITA is a sterile, preservative-free, clear to slightly opalescent and colorless to pale brown-yellow solution for subcutaneous injection. Do not use if the solution is discolored or cloudy or if the solution contains any particles or foreign particulate matter.
3. Dosage Forms and Strengths
Injection: 10 mg/mL, 20 mg/mL, or 30 mg/mL clear to slightly opalescent and colorless to pale brown-yellow solution in a single-dose vial.
4. Contraindications
CRYSVITA is contraindicated:
- In concomitant use with oral phosphate and/or active vitamin D analogs (e.g. calcitriol, paricalcitol, doxercalciferol, calcifediol) due to the risk of hyperphosphatemia [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
- When serum phosphorus is within or above the normal range for age [see Warnings and Precautions (5.2)].
- In patients with severe renal impairment or end stage renal disease because these conditions are associated with abnormal mineral metabolism [see Use In Specific Population (8.6)].
5. Warnings and Precautions
5.1 Hypersensitivity
Hypersensitivity reactions (e.g. rash, urticaria) have been reported in patients with CRYSVITA. Discontinue CRYSVITA if serious hypersensitivity reactions occur and initiate appropriate medical treatment [see Adverse Reactions (6.1)].
5.2 Hyperphosphatemia and Risk of Nephrocalcinosis
Increases in serum phosphorus to above the upper limit of normal may be associated with an increased risk of nephrocalcinosis. For patients already taking CRYSVITA, dose interruption and/or dose reduction may be required based on a patient's serum phosphorus levels. Patients with tumor-induced osteomalacia who undergo treatment of the underlying tumor should have dosing interrupted and adjusted to prevent hyperphosphatemia [see Dosage and Administration (2) and Adverse Reactions (6.1)].
6. Adverse Reactions/Side Effects
The following adverse reactions are described below and elsewhere in the labeling:
- Hypersensitivity [see Warnings and Precautions (5.1)]
- Hyperphosphatemia and Risk of Nephrocalcinosis [see Warnings and Precautions (5.2)]
- Injection Site Reactions [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Pediatric Patients with XLH
CRYSVITA was studied in three pediatric XLH studies. Study 1 is a randomized, open-label phase 3 study in XLH patients ages 1 to 12 years, who were randomized to treatment with CRYSVITA or treatment with active control of oral phosphate and active vitamin D (CRYSVITA N = 29, Active Control N = 32). Study 2 is an open-label phase 2 study in XLH patients ages 5 to 12 years (N = 52). Study 3 is an open-label phase 2 study in XLH patients ages 1 to less than 5 years (N = 13). Overall, the patient population was 1-12 years (mean age 7.0 years), 49% male, and 88% white.
In Study 1, patients randomized to CRYSVITA received a mean dose of approximately 0.90 mg/kg (range 0.8-1.2 mg/kg) every 2 weeks. All patients in this group and the active control group completed 64 weeks of treatment.
Adverse reactions occurring in ≥ 10% of subjects in the CRYSVITA group, with higher frequency than in the subjects in the active control group, through the 64-week treatment period in Study 1 are shown in Table 6.
| Adverse Reaction | CRYSVITA (N=29) n (%) | Active Control (N=32) n (%) |
|---|---|---|
| n = number of patients with an event; N = total number of patients who received at least one dose of CRYSVITA or active control | ||
|
||
| Pyrexia | 16 (55) | 6 (19) |
| Injection site reaction* | 15 (52) | 0 (0) |
| Cough† | 15 (52) | 6 (19) |
| Vomiting | 12 (41) | 8 (25) |
| Pain in extremity | 11 (38) | 10 (31) |
| Headache | 10 (34) | 6 (19) |
| Tooth abscess‡ | 10 (34) | 4 (13) |
| Dental caries | 9 (31) | 2 (6) |
| Diarrhea | 7 (24) | 2 (6) |
| Vitamin D decreased§ | 7 (24) | 1 (3) |
| Constipation | 5 (17) | 0 (0) |
| Rash¶ | 4 (14) | 2 (6) |
| Nausea | 3 (10) | 1 (3) |
In Study 2, 26 of the patients received CRYSVITA at a mean dose of 1.05 mg/kg (range 0.4 – 2.0 mg/kg) every 2 weeks at Week 64; the other 26 patients received CRYSVITA every 4 weeks. The mean duration of exposure in Study 2 was 124 weeks. In Study 3, patients received CRYSVITA at a mean dose of 0.90 mg/kg (range 0.8-1.2 mg/kg) every 2 weeks at Week 40. The mean duration of exposure in Study 3 was 45 weeks.
Adverse reactions occurring in more than 10% of CRYSVITA-treated patients from Studies 2 and 3 are shown in Table 7.
| Adverse Reaction | Study 2 (N=52) n (%) | Study 3 (N=13) n (%) | Overall (N=65) n (%) |
|---|---|---|---|
| n = number of patients with an event; N = total number of patients who received at least one dose of CRYSVITA | |||
|
|||
| Headache | 38 (73) | 1 (8) | 39 (60) |
| Injection site reaction* | 35 (67) | 3 (23) | 38 (59) |
| Vomiting | 25 (48) | 6 (46) | 31 (48) |
| Pyrexia | 23 (44) | 8 (62) | 31 (48) |
| Pain in extremity | 24 (46) | 3 (23) | 27 (42) |
| Vitamin D decreased† | 19 (37) | 2 (15) | 21 (32) |
| Rash‡ | 14 (27) | 1 (8) | 15 (23) |
| Toothache | 12 (23) | 2 (15) | 14 (22) |
| Myalgia | 9 (17) | 1 (8) | 10 (15) |
| Tooth abscess | 8 (15) | 3 (23) | 11 (17) |
| Dizziness§ | 8 (15) | 0 (0) | 8 (12) |
Hypersensitivity Reactions
In Study 1 (N=29 for CRYSVITA arm), the most frequent hypersensitivity reactions were rash (10%), injection site rash (10%) and injection site urticaria (7%). In Studies 2 and 3 (N=65), the most frequent hypersensitivity reactions were rash (22%), injection site rash (6%), and urticaria (5%).
Hyperphosphatemia
In pediatric studies, no events of hyperphosphatemia were reported.
Injection Site Reactions (ISR)
In Study 1 (N=29 for CRYSVITA arm), 52% of the patients had a local injection site reaction (e.g. injection site urticaria, erythema, rash, swelling, bruising, pain, pruritus, and hematoma) at the site of CRYSVITA injection. In Studies 2 and 3 (N=65), approximately 58% of the patients had a local injection site reaction at the site of CRYSVITA injection. Injection site reactions were generally mild in severity, occurred within 1 day of injection, lasted approximately 1 to 3 days, required no treatment, and resolved in almost all instances.
Adverse Reactions in Adult Patients with XLH
The safety of CRYSVITA in adult patients with XLH was demonstrated in a randomized, double-blind, placebo-controlled study (Study 4) of 134 patients, age 20-63 years (mean age 41 years), of whom most were white/Caucasian (81%) and female (65%). A total of 68 and 66 patients received at least one dose of CRYSVITA or placebo, respectively. The mean dose of CRYSVITA was 0.95 mg/kg (range 0.3 – 1.2 mg/kg) subcutaneously every 4 weeks. Adverse reactions reported in more than 5% of CRYSVITA-treated patients and 2 patients or more than with placebo from the 24-week placebo-controlled portion of Study 4 are shown in Table 8.
| Adverse Reaction | CRYSVITA (N=68) n (%) | Placebo (N=66) n (%) |
|---|---|---|
| n = number of patients with an event; N = total number of patients who received at least one dose of CRYSVITA or placebo | ||
|
||
| Back pain | 10 (15) | 6 (9) |
| Headache* | 9 (13) | 6 (9) |
| Tooth infection† | 9 (13) | 6 (9) |
| Restless legs syndrome | 8 (12) | 5 (8) |
| Vitamin D decreased‡ | 8 (12) | 3 (5) |
| Dizziness | 7 (10) | 4 (6) |
| Muscle spasms | 5 (7) | 2 (3) |
| Constipation | 6 (9) | 0 (0) |
| Blood phosphorus increased§ | 4 (6) | 0 (0) |
The 24-week placebo controlled study was followed by a 24-week open-label treatment period in which all patients received CRYSVITA subcutaneously every 4 weeks. No new adverse reactions were identified in the open-label extension period.
Hypersensitivity Reactions
In the double-blind period of Study 4, approximately 6% of patients in both the CRYSVITA and placebo treatment groups experienced a hypersensitivity event. The events were mild or moderate and did not require discontinuation.
Hyperphosphatemia
In the double-blind period of Study 4, 7% of patients in the CRYSVITA treatment group experienced hyperphosphatemia meeting the protocol-specified criteria for dose reduction (either a single serum phosphorus greater than 5.0 mg/dL or serum phosphorus greater than 4.5 mg/dL [the upper limit of normal] on two occasions). The hyperphosphatemia was managed with dose reduction. The dose for all patients meeting the protocol-specified criteria was reduced 50 percent. A single patient required a second dose reduction for continued hyperphosphatemia.
Injection Site Reactions (ISR)
In the double-blind period of Study 4, approximately 12% of patients in both the CRYSVITA and placebo treatment groups had a local reaction (e.g. injection site reaction, erythema, rash, bruising, pain, pruritus, and hematoma) at the site of the injection. Injection site reactions were generally mild in severity, occurred within 1 day of injection, lasted approximately 1 to 3 days, required no treatment, and resolved in almost all instances.
Restless Legs Syndrome (RLS)
In the double-blind period of Study 4, approximately 12% of the CRYSVITA treatment group had worsening of baseline restless legs syndrome (RLS) or new onset RLS of mild to moderate severity; these events did not lead to dose discontinuation. Nonserious RLS has also been reported in other repeat dose adult XLH studies; in one case, worsening baseline RLS led to drug discontinuation and subsequent resolution of the event.
Spinal Stenosis
Spinal stenosis is prevalent in adults with XLH and spinal cord compression has been reported. In the CRYSVITA phase 2 and phase 3 studies of adults with XLH (total N=176), a total of 7 patients underwent spinal surgery. Most of these cases appeared to involve progression of a pre-existing spinal stenosis. It is unknown if CRYSVITA therapy exacerbates spinal stenosis or spinal cord compression.
Adverse Reactions in Patients with TIO
The safety of CRYSVITA in patients with TIO was demonstrated in two single-arm clinical studies (Study 6 and Study 7) that enrolled a total of 27 patients. Fourteen patients were male, and patients ranged from 33 to 73 years of age. The mean dose of CRYSVITA was 0.77 mg/kg every 4 weeks and the mean duration of exposure was 121 weeks.
Adverse reactions reported in adult TIO patients in the pooled data from Study 6 and Study 7 are shown in Table 9.
| Adverse Reaction | Overall (N=27) n (%) |
|---|---|
|
|
| Tooth abscess* | 5 (19) |
| Muscle spasms | 5 (19) |
| Dizziness | 4 (15) |
| Constipation | 4 (15) |
| Injection site reaction† | 4 (15) |
| Rash‡ | 4 (15) |
| Headache | 3 (11) |
| Vitamin D deficiency | 2 (7) |
| Hyperphosphatemia | 2 (7) |
| Restless legs syndrome | 2 (7) |
Hypersensitivity reactions
In the pooled data for Studies 6 and 7, 22% of patients experienced a hypersensitivity reaction. The most frequent hypersensitivity reactions were eczema (11%) and rash (11%). The events were mild or moderate in severity.
Hyperphosphatemia
In the pooled data for Studies 6 and 7, 2 patients (7%) experienced hyperphosphatemia which was managed with dose reduction.
Injection site reactions
The frequency of injection site reactions was 15% (injection site reaction, injection site pain, and injection site swelling). The injection site reactions were generally mild in severity, required no treatment and resolved in all cases.
Restless Legs Syndrome
In the pooled data for Studies 6 and 7, 2 patients (7%) experienced symptoms of restless legs syndrome, which were mild and did not require treatment interruption.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to burosumab-twza in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In XLH clinical studies, none (0/13) of the 1- to 4-year-old patients, 19% (10/52) of the 5- to 12-year-old patients, and 15% (20/131) of the adult patients tested positive for anti-drug antibodies (ADA) after receiving CRYSVITA. Among these, three 5- to 12-year-old patients tested positive for neutralizing antibodies. The presence of ADA was not associated with clinically relevant changes in pharmacokinetics, pharmacodynamics, efficacy, and safety of burosumab in patients with XLH.
In one TIO clinical study, 14% (2/14) of the adult patients tested positive for ADA after receiving CRYSVITA. None of the ADA positive patients tested positive for neutralizing antibodies. In another TIO clinical study, none of the 13 adult patients tested positive for ADA after receiving CRYSVITA.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of CRYSVITA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Investigations: Blood phosphorus increased has been reported in pediatric XLH patients receiving CRYSVITA.
7. Drug Interactions
7.1 Oral Phosphate and Active Vitamin D Analogs
Concomitant use of CRYSVITA with oral phosphate and/or active vitamin D analogs will increase phosphate concentrations greater than expected with CRYSVITA alone. This increase may result in hyperphosphatemia which can induce nephrocalcinosis.
Concomitant use of CRYSVITA with oral phosphate and/or active vitamin D analogs is contraindicated.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no available data on CRYSVITA use in pregnant women to inform a drug-associated risk of adverse developmental outcomes. In utero, burosumab-twza exposure in cynomolgus monkeys did not result in teratogenic effects. Adverse effects such as late fetal loss and preterm birth were observed in pregnant cynomolgus monkeys, however, these effects are unlikely to indicate clinical risk because they occurred at a drug exposure that was 15-fold higher, by AUC, than the human exposure at the maximum recommended human dose (MRHD) of 2 mg/kg every 2 weeks and were accompanied by maternal hyperphosphatemia and placental mineralization (see Data). Serum phosphorus levels should be monitored throughout pregnancy [see Dosage and Administration (2.2)]. Report pregnancies to the Kyowa Kirin, Inc. Adverse Event reporting line at 1-844-768-3544.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, the estimated background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
In a reproductive toxicity study in pregnant cynomolgus monkeys, burosumab-twza was administered intravenously once every two weeks from Day 20 of pregnancy to parturition or cesarean section on Day 133, which includes the period of organogenesis, at doses of 0.2-, 2- and 15-fold human exposure at the adult MRHD of 2 mg/kg every 2 weeks. The treatment did not result in teratogenic effects in fetuses or offspring. An increase in late fetal loss, a shortened gestation period, and an increased incidence of preterm births were observed at 15-fold human exposure at the adult MRHD of 2 mg/kg every 2 weeks, concomitant with maternal hyperphosphatemia and placental mineralization. Burosumab-twza was detected in serum from fetuses indicating transport across the placenta. Hyperphosphatemia but no ectopic mineralization was present in fetuses and offspring of dams exposed to 15-fold human exposure at the MRHD of 2 mg/kg dose every 2 weeks. Burosumab-twza did not affect pre- and postnatal growth including survivability of the offspring.
8.2 Lactation
Risk Summary
There is no information regarding the presence of burosumab-twza in human milk, or the effects of burosumab-twza on milk production or the breastfed infant. Maternal IgG is present in breast milk. However, the effects of local gastrointestinal exposure and limited systemic exposure to burosumab-twza in the breastfed infant are unknown. The lack of clinical data during lactation precludes a clear determination of the risk of CRYSVITA to an infant during lactation. Therefore, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CRYSVITA and any potential adverse effects on the breastfed infant from CRYSVITA or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of CRYSVITA have been established in pediatric patients 6 months and older. Safety and effectiveness in pediatric patients 1 year and older with XLH are based on one phase 3, open-label, active control study [61 patients 1-12 years of age (Study 1)] and two open-label studies [52 patients 5 to 12 years of age (Study 2), and 13 patients 1 to 4 years of age (Study 3)] evaluating serum phosphorus and radiographic findings. Safety and effectiveness in patients 6 months to 1 year and adolescents are supported by evidence from the studies in pediatric patients 1 year to less than 13 years of age with additional modeling and simulation of adult and pediatric pharmacokinetic (PK) and pharmacodynamic (PD) data to inform dosing [see Adverse Reactions (6.1) and Clinical Studies (14)].
Safety and effectiveness for CRYSVITA in pediatric patients with XLH below the age of 6 months have not been established.
Safety and effectiveness of CRYSVITA in pediatric patients 2 years and older with TIO are supported by evidence from the studies in adult patients with TIO with additional modeling and simulation of PK data from adult and pediatric XLH patients and adult TIO patients to inform dosing.
Safety and effectiveness for CRYSVITA in pediatric patients with TIO below the age of 2 years have not been established.
8.5 Geriatric Use
Clinical studies of CRYSVITA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
The effect of renal impairment on the pharmacokinetics of burosumab-twza is unknown. However, renal impairment can induce abnormal mineral metabolism which will increase phosphate concentrations greater than expected with CRYSVITA alone. This increase may result in hyperphosphatemia which can induce nephrocalcinosis.
CRYSVITA is contraindicated in patients with severe renal impairment, defined as:
- pediatric patients with estimated glomerular filtration rate (eGFR) 15 mL/min/1.73m2 to 29 mL/min/1.73m2 or end stage renal disease (eGFR < 15 mL/min/1.73m2)
- adult patients with creatinine clearance (CLcr) 15 mL/min to 29 mL/min or end stage renal disease (CLcr < 15 mL/min).
10. Overdosage
There have been no reports of overdose with CRYSVITA. CRYSVITA has been administered in pediatric clinical trials without dose limiting toxicity using doses up to 2 mg/kg body weight with a maximal dose of 90 mg, administered every two weeks. In XLH adult clinical trials, no dose limiting toxicity has been observed using doses up to 1 mg/kg or a maximal total dose of 128 mg every 4 weeks. In non-XLH rabbits and cynomolgus monkeys, ectopic mineralization in multiple tissues and organs was observed at doses of burosumab-twza that resulted in supra-physiologic serum phosphate levels. Adverse effects on bone including reductions in bone mineral density, bone mineralization and bone strength were also observed at exposure greater than human exposure [see Nonclinical Toxicology (13.2)].
In case of overdose, it is recommended that serum phosphorus levels, serum calcium levels and renal function be measured immediately and monitored periodically until resolution to normal/baseline levels. In case of hyperphosphatemia, withhold CRYSVITA and initiate appropriate medical treatment.
11. Crysvita Description
Burosumab-twza is a human immunoglobulin G subclass 1 (IgG1), anti-human fibroblast growth factor 23 (FGF23) antibody produced by recombinant DNA technology using Chinese hamster ovary cells. Burosumab-twza is composed of two heavy chain (γ1-chain) molecules and two light chain (κ-chain) molecules. Each heavy chain has an N-linked carbohydrate moiety at asparagine 297 (Asn297). The molecular weight of burosumab-twza determined by mass spectrometry is approximately 147,000.
CRYSVITA (burosumab-twza) injection for subcutaneous administration is supplied as a sterile, preservative-free, clear to slightly opalescent and colorless to pale brown-yellow solution in a single-dose vial.
Each 1 mL of solution contains 10 mg, 20 mg or 30 mg of burosumab-twza, L-histidine (1.55 mg), L-methionine (1.49 mg), polysorbate 80 (0.5 mg), D-sorbitol (45.91 mg) in Water for Injection, USP. Hydrochloric acid may be used to adjust to a pH of 6.25.
12. Crysvita - Clinical Pharmacology
12.1 Mechanism of Action
X-linked hypophosphatemia is caused by excess fibroblast growth factor 23 (FGF23) which suppresses renal tubular phosphate reabsorption and the renal production of 1,25 dihydroxy vitamin D. Burosumab-twza binds to and inhibits the biological activity of FGF23 restoring renal phosphate reabsorption and increasing the serum concentration of 1,25 dihydroxy vitamin D.
12.2 Pharmacodynamics
Following SC administration in XLH and TIO patients, higher burosumab-twza concentrations were associated with greater increase of serum phosphorus levels. The increase in serum phosphorus was reversible and returned to baseline with elimination of systemic burosumab-twza.
Ratio of renal tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) showed dose-dependent increases from baseline [see Clinical Studies (14)].
Elevation in serum total FGF23 was observed after initiation of burosumab-twza treatment, however, the clinical implication is unknown.
12.3 Pharmacokinetics
The following pharmacokinetic parameters were observed in patients with XLH administered the approved recommended starting dosage based on a 70 kg patient, unless otherwise specified. Based on the population PK analysis, the PK characteristics of burosumab-twza were similar between patients with XLH and TIO.
Burosumab-twza exhibited linear pharmacokinetics following SC injections within the dose range of 0.1 to 1 mg/kg (0.08 to 0.8 times the maximum approved recommended dosage based on a 70 kg patient with XLH).
The steady-state trough mean (± SD) concentration of burosumab-twza was 5.8 (± 3.4) mcg/mL in adult XLH patients.
Absorption
The burosumab-twza mean Tmax values ranged from 8 to 11 days.
Distribution
The apparent volume of distribution of burosumab-twza is 8 L.
Elimination
The apparent clearance is 0.290 L/day. The half-life of burosumab-twza is approximately 19 days.
Metabolism
The exact pathway for burosumab-twza metabolism has not been characterized. Burosumab-twza is expected to be degraded into small peptides and amino acids via catabolic pathways.
Specific Populations
No clinical significant difference in burosumab-twza pharmacokinetics was observed based on age.
The effect of renal or hepatic impairment on the pharmacokinetics of burosumab-twza is unknown.
Pediatric Patients
The steady-state trough concentration was 15.8 (± 9.4) mcg/mL in XLH patients aged 5-12 years, and 11.2 (± 4.6) mcg/mL in XLH patients aged 1-4 years.
Body Weight
Clearance and volume of distribution of burosumab-twza increases with body weight.
Drug Interaction Studies
No drug interaction studies have been conducted with CRYSVITA.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of burosumab-twza has not been evaluated in long term animal studies.
Studies have not been performed to evaluate the mutagenic potential of burosumab-twza.
No specific fertility studies have been performed in animals to evaluate the effects of burosumab-twza.
Toxicology studies with burosumab-twza of up to 40 weeks duration in cynomolgus monkeys did not show significant adverse effects on female reproductive organs at doses up to 16-fold human exposure at the maximum recommended human dose (MRHD) of 2 mg/kg every 2 weeks. In male monkeys, minimal mineralization of the rete testis or seminiferous tubules associated with hyperphosphatemia was observed at 3- to 9-fold human exposure at the MRHD of 2 mg/kg every 2 weeks, but semen analysis did not show any adverse effects.
13.2 Animal Toxicology and/or Pharmacology
In rabbits and cynomolgus monkeys, inhibition of FGF23 signaling by burosumab-twza increased serum phosphate and 1,25 dihydroxy vitamin D. Ectopic mineralization in multiple tissues and organs was observed at doses of burosumab-twza that resulted in supra-physiologic serum phosphate levels. In a study in wild type (WT) and hypophosphatemic Hyp mice, a murine model of XLH, ectopic mineralization was markedly less in Hyp mice.
In adult cynomolgus monkeys, burosumab-twza increased bone turnover, mineral content and/or mineral density and cortical thickness at 9- to 16-fold human exposure at the MRHD of 2 mg/kg every 2 weeks. Adverse effects on bone, including reductions in bone mineral density, bone mineralization and bone strength were observed in adult male monkeys at 9- to 11-fold human exposure at the MRHD of 2 mg/kg every 2 weeks.
In juvenile cynomolgus monkeys, burosumab-twza increased bone turnover, mineral content and/or mineral density and/or cortical thickness at 0.2- to 2-fold clinical pediatric exposure. Bone mineralization was decreased in a male monkey at 2-fold pediatric exposure but there was no effect on bone strength. Burosumab-twza did not affect bone development in juvenile monkeys at doses up to 2-fold pediatric exposure.
14. Clinical Studies
14.1 Pediatric X-linked Hypophosphatemia
CRYSVITA has been evaluated in three studies enrolling a total of 126 pediatric patients with XLH.
Study 1 (NCT 02915705) is a 64-week randomized, open-label study in 61 pediatric XLH patients, 1 to 12 years old that compared treatment with CRYSVITA to active control (oral phosphate and active vitamin D). At time of first dose the mean age of patients was 6.3 years and 44% were male. All patients had radiographic evidence of rickets at baseline, with an RSS score of ≥ 2.0 and had received oral phosphate and active vitamin D analogs for a mean (SD) duration of 4 (3.1) years. Oral phosphate and active vitamin D analogs were discontinued prior to study enrollment for a 7-day washout period and then reinitiated for patients in the active control group. Patients were randomized to receive either CRYSVITA at a starting dose of 0.8 mg/kg every two weeks or oral phosphate (recommended dose 20-60 mg/kg/day) and active vitamin D (recommended doses calcitriol 20-30 ng/kg/day or alfacalcidol 40-60 ng/kg/day). Patients randomized to active control received a mean oral phosphate dose of approximately 41 mg/kg/day (range 18 to 110 mg/kg/day) at Week 40 and approximately 46 mg/kg/day (range 18 mg/kg/day to 166 mg/kg/day) at Week 64. They also received either a mean oral calcitriol dose of 26 ng/kg/day at Week 40 and 27 ng/kg/day at Week 64 or a therapeutically equivalent amount of alfacalcidol. Eight patients in the CRYSVITA arm titrated up to 1.2 mg/kg based on serum phosphorus measurements. All patients completed at least 64 weeks on study.
Serum Phosphorus
In Study 1, CRYSVITA increased mean (SD) serum phosphorus levels from 2.4 (0.24) mg/dL at baseline to 3.3 (0.43) mg/dL at Week 40 and to 3.3 (0.42) mg/dL at Week 64. In the active control group, mean (SD) serum phosphorus concentrations increased from 2.3 (0.26) mg/dL at baseline to 2.5 (0.34) mg/dL at Week 40 and to 2.5 (0.39) mg/dL at Week 64. The renal phosphate reabsorptive capacity as assessed by TmP/GFR increased in the CRYSVITA-treated patients from a mean (SD) of 2.2 (0.37) mg/dL at baseline to 3.4 (0.67) mg/dL and 3.3 (0.65) mg/dL at Week 40 and Week 64, respectively. In the active control group, mean (SD) TmP/GFR decreased from 2.0 (0.33) mg/dL at Baseline to 1.8 (0.35) mg/dL at Week 40, and remained below baseline at Week 64 at 1.9 (0.49) mg/dL.
Figure 1: Serum Phosphorus Concentration and Change from Baseline (mg/dL) (Mean ± SD) by Treatment Group in Children 1-12 Years in Study 1
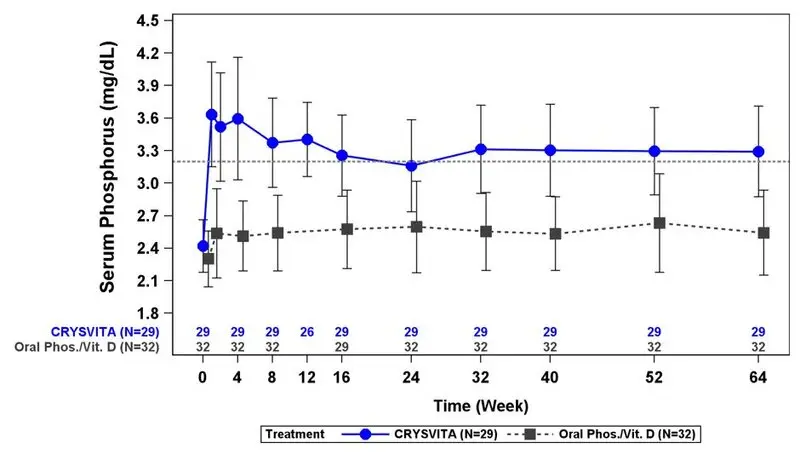
The dotted line represents the lower limit of normal (3.2 mg/dL) for patients in Study 1.
Radiographic Evaluation of Rickets
Radiographs were examined to assess XLH-related rickets using the 10-point Thacher Rickets Severity Score (RSS) and the 7-point Radiographic Global Impression of Change (RGI-C). The RSS score is assigned based on images of the wrist and knee from a single timepoint, with higher scores indicating greater rickets severity. The RGI-C score is assigned based on side-by-side comparisons of wrist and knee radiographs from two timepoints, with higher scores indicating greater improvement in radiographic evidence of rickets. A RGI-C score of +2.0 was defined as radiographic evidence of substantial healing.
In Study 1, baseline mean (SD) total RSS was 3.2 (0.98) in the CRYSVITA group and 3.2 (1.14) in the active control group. After 40 weeks of treatment with CRYSVITA, mean total RSS decreased from 3.2 to 1.1 (0.72) and from 3.2 to 2.5 (1.09) in the active control group. LS mean (SE) RGI-C Global score was +1.9 (0.11) in the CRYSVITA group and +0.8 (0.11) in the active control group at Week 40 (see Table 10). At Week 40, 21 of the 29 patients in the CRYSVITA group and 2 of the 32 patients in the active control arm achieved a RGI-C global score ≥ +2.0. These findings were maintained at Week 64 as shown in Table 10.
| Endpoint Timepoint | CRYSVITA Every 2 Weeks (N=29) | Active Control (N=32) |
|---|---|---|
|
||
| RSS Total Score | ||
| Baseline Mean (SD) | 3.2 (0.98) | 3.2 (1.14) |
| LS Mean change from baseline in total score* (reduction indicates improvement) with 95% CI | ||
| Week 40 | -2.0 (-2.33, -1.75) | -0.7 (-0.98, -0.43) |
| Week 64 | -2.2 (-2.46, -2.00) | -1.0 (-1.31, -0.72) |
| RGI-C Global Score† | ||
| LS Mean score* (positive indicates healing) with 95% CI | ||
| Week 40 | +1.9 (+1.70, +2.14) | +0.8 (+0.56, +0.99) |
| Week 64 | +2.06 (+1.91, +2.20) | +1.03 (+0.77, +1.30) |
Lower Extremity Skeletal Abnormality
In Study 1, lower extremity skeletal abnormalities were assessed by RGI-C in standing long leg radiographs. At Week 64, the CRYSVITA group maintained greater improvement compared with the active control group (LS mean [SE]: +1.25 [0.17] versus +0.29 [0.12]; difference of +0.97 (95% CI: +0.57, +1.37, GEE model)).
Serum Alkaline Phosphatase Activity
For Study 1, mean (SD) serum total alkaline phosphatase activity decreased from 511 (125) at baseline to 337 (86) U/L in the CRYSVITA group (mean change: -33%) and from 523 (154) at baseline to 495 (182) U/L in the active control group (mean change: -5%) at Week 64.
Growth
In Study 1, CRYSVITA treatment for 64 weeks increased standing mean (SD) height Z score from -2.32 (1.17) at baseline to -2.11 (1.11) at Week 64 (LS mean change (SE) of +0.17 (0.07)). In the active control group, mean (SD) height Z score increased from -2.05 (0.87) at baseline to -2.03 (0.83) at Week 64 (LS mean (SE) change of +0.02 (0.04)). The difference between the treatment groups at Week 64 was +0.14 (95% CI: 0.00, +0.29).
Study 2 (NCT 02163577) is a randomized, open-label study in 52 prepubescent XLH patients, 5 to 12 years old, which compared treatment with CRYSVITA administered every 2 weeks versus every 4 weeks. Following an initial 16-week dose titration phase, patients completed 48-weeks of treatment with CRYSVITA every 2 weeks. All 52 patients completed at least 64 weeks on study; no patient discontinued. Burosumab-twza dose was adjusted to target a fasting serum phosphorus concentration of 3.5 to 5.0 mg/dL based on the fasting phosphorus level the day of dosing. Twenty-six of 52 patients received CRYSVITA every two weeks up to a maximum dose of 2 mg/kg. The average dose was 0.73 mg/kg (range: 0.3, 1.5) at Week 16, 0.98 mg/kg (range: 0.4, 2.0) at Week 40 and 1.04 mg/kg (range: 0.4, 2.0) at Week 60. The remaining 26 patients received CRYSVITA every four weeks. At study entry, the mean age of patients was 8.5 years and 46% were male. Ninety-six percent had received oral phosphate and active vitamin D analogs for a mean (SD) duration of 7 (2.4) years. Oral phosphate and active vitamin D analogs were discontinued prior to study enrollment. Ninety-four percent of patients had radiographic evidence of rickets at baseline.
Study 3 (NCT 02750618) is a 64-week open-label study in 13 pediatric XLH patients, 1 to 4 years old. Patients received CRYSVITA at a dose of 0.8 mg/kg every two weeks with 3 patients titrating up to 1.2 mg/kg based on serum phosphorus measurements. All patients completed at least 40 weeks on study; no patients discontinued. At study entry, the mean age of patients was 2.9 years and 69% were male. All patients had radiographic evidence of rickets at baseline and 12 patients had received oral phosphate and active vitamin D analogs for a mean (SD) duration of 16.7 (14.4) months. Oral phosphate and active vitamin D analogs were discontinued prior to study enrollment.
Serum Phosphorus
In Study 2, CRYSVITA increased mean (SD) serum phosphorus levels from 2.4 (0.40) at baseline to 3.3 (0.40) and 3.4 (0.45) mg/dL at Week 40 and Week 64 in the patients who received CRYSVITA every 2 weeks. The ratio of renal tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) increased in these patients from mean (SD) of 2.2 (0.49) at baseline to 3.3 (0.60) and 3.4 (0.53) mg/dL at Week 40 and Week 64.
In Study 3, CRYSVITA increased mean (SD) serum phosphorus levels from 2.5 (0.28) mg/dL at baseline to 3.5 (0.49) mg/dL at Week 40.
Radiographic Evaluation of Rickets
In Study 2, baseline mean (SD) RSS total score was 1.9 (1.17) in patients receiving CRYSVITA every two weeks. After 40 weeks of treatment with CRYSVITA, mean total RSS decreased from 1.9 to 0.8 (see Table 11). After 40 weeks of treatment with CRYSVITA, the mean RGI-C Global score was +1.7 in patients receiving CRYSVITA every two weeks. Eighteen out of 26 patients achieved an RGI-C score of ≥ +2.0. These findings were maintained at Week 64 as shown in Table 11.
In Study 3, baseline mean (SD) total RSS was 2.9 (1.37) in 13 patients. After 40 weeks of treatment with CRYSVITA, mean total RSS decreased from 2.9 to 1.2 and the mean (SE) RGI-C Global score was +2.3 (0.08) (see Table 11). All 13 patients achieved a RGI-C global score ≥ +2.0.
| Endpoint Timepoint | CRYSVITA Every 2 Weeks | |
|---|---|---|
| Study 2*
(N=26) | Study 3†
(N=13) |
|
|
||
| RSS Total Score | ||
| Baseline Mean (SD) | 1.9 (1.17) | 2.9 (1.37) |
| LS Mean change from baseline in total score (reduction indicates improvement) with 95% CI | ||
| Week 40 | -1.1 (-1.28, -0.85) | -1.7 (-2.03, -1.44) |
| Week 64 | -1.0 (-1.2, -0.79) | |
| RGI-C Global Score | ||
| LS Mean score (positive indicates healing) with 95% CI | ||
| Week 40 | +1.7 (+1.48, +1.84) | +2.3 (+2.16, +2.51) |
| Week 64 | +1.6 (+1.34, +1.78) | |
Lower Extremity Skeletal Abnormality
In Study 3, the mean (SE) change in lower limb deformity as assessed by RGI-C, using standing long leg radiographs, was +1.3 (0.14) at Week 40.
Serum Alkaline Phosphatase Activity
For Study 2, mean (SD) serum total alkaline phosphatase activity was 462 (110) U/L at baseline and decreased to 354 (73) U/L at Week 64 (-23%) in the patients who received CRYSVITA every 2 weeks.
For Study 3, mean (SD) serum total alkaline phosphatase activity was 549 (194) U/L at baseline and decreased to 335 (88) U/L at Week 40 (mean change: -36%).
Growth
In Study 2, CRYSVITA treatment for 64 weeks increased standing mean (SD) height Z score from -1.72 (1.03) at baseline to -1.54 (1.13) in the patients who received CRYSVITA every two weeks (LS mean change of +0.19 (95% CI: 0.09 to 0.29).
14.2 Adult X-linked Hypophosphatemia
Study 4 (NCT 02526160) is a randomized, double-blind, placebo-controlled study in 134 adult XLH patients. The study comprises a 24-week placebo-controlled treatment phase followed by a 24-week open-label treatment period in which all patients received CRYSVITA. CRYSVITA was administered at a dose of 1 mg/kg every 4 weeks. At study entry, the mean age of patients was 40 years (range 19 to 66 years) and 35% were male. All patients had skeletal pain associated with XLH/osteomalacia at baseline. The baseline mean (SD) serum phosphorus concentration was below the lower limit of normal at 1.98 (0.31) mg/dL. Oral phosphate and active vitamin D analogs were not allowed during the study. Out of the 134 patients enrolled in the study, one patient in the CRYSVITA group discontinued treatment during the 24-week placebo-controlled treatment period, and 7 patients discontinued CRYSVITA during the open-label treatment period.
Study 5 (NCT 02537431) is a 48-week, open-label, single-arm study in 14 adult XLH patients to assess the effects of CRYSVITA on improvement of osteomalacia as determined by histologic and histomorphometric evaluation of iliac crest bone biopsies. Patients received 1 mg/kg CRYSVITA every four weeks. At study entry, the mean age of patients was 40 years (range 25 to 52 years) and 43% were male. Oral phosphate and active vitamin D analogs were not allowed during the study.
Serum Phosphorus
In Study 4 at baseline, mean (SD) serum phosphorus was 1.9 (0.32) and 2.0 (0.30) mg/dL in the placebo and CRYSVITA groups respectively. During the initial 24-week double-blind, placebo-controlled period, mean (SD) serum phosphorus across the midpoints of dose intervals (2 weeks post dose) was 2.1 (0.30) and 3.2 (0.53) mg/dL in the placebo and CRYSVITA groups, and mean (SD) serum phosphorus across the ends of dose intervals was 2.0 (0.30) and 2.7 (0.45) mg/dL in the placebo and CRYSVITA groups.
A total of 94% of patients treated with CRYSVITA achieved a serum phosphorus level above the lower limit of normal (LLN) compared to 8% in the placebo group through Week 24 (see Table 12).
| Placebo (N = 66) | CRYSVITA (N = 68) |
|
|---|---|---|
| The 95% CIs are calculated using the Wilson score method. | ||
|
||
| Achieved Mean Serum Phosphorus > LLN Across Midpoints of Dose Intervals Through Week 24 - n (%) | 5 (8%) | 64 (94%) |
| 95% CI | (3.3, 16.5) | (85.8, 97.7) |
| p-value* | < 0.0001 | |
During the open-label treatment period, serum phosphorus was maintained during continued CRYSVITA therapy, with no evidence of loss of effect through Week 48.
|
| Figure 2: Mean (± SD) Serum Phosphorus Peak Concentrations (mg/dL) in Study 4*,† |
|
|
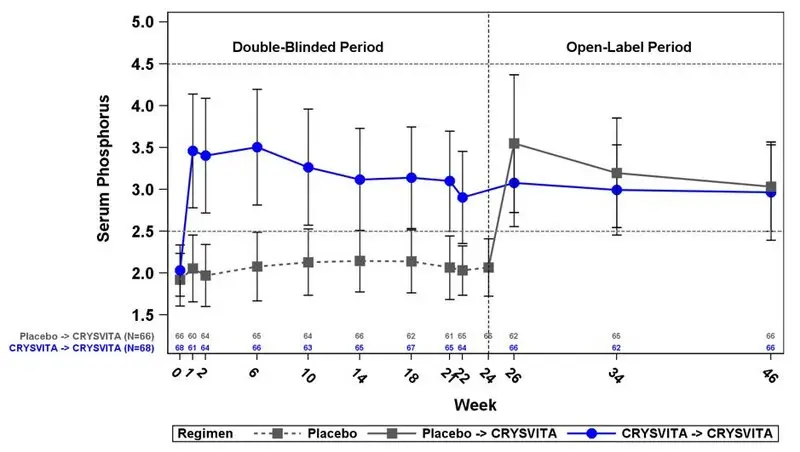
At baseline, the mean (SD) ratio of renal tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) was 1.60 (0.37) and 1.68 (0.40) mg/dL in the placebo and CRYSVITA groups respectively. At Week 22 (midpoint of a dose interval), mean (SD) TmP/GFR was 1.69 (0.37) and 2.73 (0.75) mg/dL in the placebo and CRYSVITA groups. At Week 24 (end of a dose interval), mean (SD) TmP/GFR was 1.73 (0.42) and 2.21 (0.48) mg/dL in the placebo and CRYSVITA groups. During the open-label treatment period, TmP/GFR remained stable during continued CRYSVITA therapy through Week 48.
Radiographic Evaluation of Osteomalacia
In Study 4, a skeletal survey was conducted at baseline to identify osteomalacia-related fractures and pseudofractures. Osteomalacia-related fractures are defined as atraumatic lucencies extending across both bone cortices and pseudofractures are defined as atraumatic lucencies extending across one cortex. There were 52% of patients who had either active (unhealed) fractures (12%) or active pseudofractures (47%) at baseline. The active fractures and pseudofractures were predominantly located in the femurs, tibia/fibula, and metatarsals of the feet. Assessment of these active fracture/pseudofracture sites at Week 24 demonstrated a higher rate of complete healing in the CRYSVITA group compared to placebo as shown in Table 13. During the double-blind, placebo-controlled treatment period through Week 24, a total of 6 new fractures or pseudofractures appeared in 68 patients receiving CRYSVITA, compared to 8 new abnormalities in 66 patients receiving placebo (see Table 13).
| Active Fractures | Active Pseudofractures | Total Fractures | ||||
|---|---|---|---|---|---|---|
| Placebo n (%) | CRYSVITA n (%) | Placebo n (%) | CRYSVITA n (%) | Placebo n (%) | CRYSVITA n (%) |
|
| No. of fractures at baseline | 13 | 14 | 78 | 51 | 91 | 65 |
| Healed at Week 24 | 0 (0%) | 7 (50%) | 7 (9%) | 21 (41%) | 7 (8%) | 28 (43%) |
During the open-label treatment period, the patients who continued receiving CRYSVITA showed continued healing of fractures at Week 48 [active fractures (n = 8, 57%), active pseudofractures (n = 33, 65%)]. In the 'placebo to CRYSVITA' group, fracture healing at Week 48 was observed for active fractures (n = 6, 46%), and active pseudofractures (n = 26, 33%).
Patient Reported Outcomes
Study 4 evaluated patient-reported XLH-related symptoms (pain, joint stiffness, and physical function).
At 24 weeks, the CRYSVITA arm showed a mean improvement from baseline (-7.9) compared to the placebo arm (+0.3) in the stiffness severity score (range 0 to 100; lower scores are reflective of symptom improvement).
At 24 weeks, no significant difference between CRYSVITA and placebo was demonstrated in patient-reported pain intensity or physical function score.
Bone Histomorphometry
In Study 5, after 48 weeks of treatment, healing of osteomalacia was observed in ten patients as demonstrated by decreases in Osteoid volume/Bone volume (OV/BV) from a mean (SD) score of 26% (12.4) at baseline to 11% (6.5), a change of -57%. Osteoid thickness (O.Th) declined in eleven patients from a mean (SD) of 17 (4.1) micrometers to 12 (3.1) micrometers, a change of -33%. Mineralization lag time (MLt) declined in 6 patients from a mean (SD) of 594 (675) days to 156 (77) days, a mean change of -74%.
14.3 Tumor-induced Osteomalacia
CRYSVITA has been evaluated in two studies enrolling a total of 27 patients with TIO.
Study 6 (NCT 02304367) is a single-arm open-label study that enrolled 14 adult patients with a confirmed diagnosis of FGF23-related hypophosphatemia produced by an underlying tumor that was not amenable to surgical excision or could not be located. Of the 14 TIO patients enrolled in Study 6, eight were male, and patients ranged from 33 years to 68 years of age (Median 59.5 years). Oral phosphate and active vitamin D analogs were discontinued two weeks prior to study enrollment. Patients received CRYSVITA every 4 weeks at a weight based starting dose of 0.3 mg/kg that was titrated to achieve a fasting serum phosphorus level of 2.5 to 4.0 mg/dL. The mean dose was 0.83 mg/kg at Week 20, 0.87 mg/kg at Week 48, 0.77 mg/kg at Week 96 and 0.71 mg/kg at Week 144.
Study 7 (NCT 02722798) is a single-arm open-label study. In Study 7, 13 adult patients with a confirmed diagnosis of TIO received CRYSVITA. Of the 13 TIO patients who received treatment in Study 7, six were male, and patients ranged from 41 years to 73 years of age (Median 58.0 years). Oral phosphate and active vitamin D analogs were discontinued two weeks prior to study enrollment. Patients received CRYSVITA every 4 weeks at a weight based starting dose of 0.3 mg/kg that was titrated to achieve a fasting serum phosphorus level of 2.5 to 4.0 mg/dL. The mean (SD) dose was 0.91 (0.59) mg/kg at Week 48, and 0.96 (0.70) mg/kg at Week 88.
Serum Phosphorus
In Study 6, CRYSVITA increased mean (SD) serum phosphorus levels from 1.60 (0.47) mg/dL at baseline to 2.64 (0.76) mg/dL averaged across the midpoint of dose intervals through Week 24 with 50% of patients (7/14) achieving a mean serum phosphorus level above the LLN averaged across the midpoint of dose intervals through Week 24. Increase in the mean serum phosphorus concentrations was sustained near or above the LLN through Week 144 (Figure 3). The ratio of renal tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) increased in these patients from a mean (SD) of 1.12 (0.54) mg/dL at baseline to 2.12 (0.64) mg/dL at Week 48, and remained stable through Week 144.
Figure 3: Serum Phosphorus Concentration and Change from Baseline in Study 6 (mg/dL)
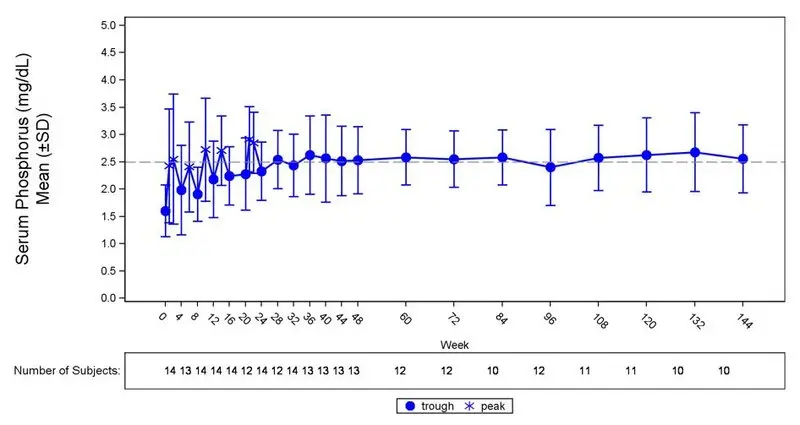
The dotted line represents the lower limit of normal (2.5 mg/dL) for patients in study 6.
In Study 7, CRYSVITA increased mean (SD) serum phosphorus levels from 1.62 (0.49) mg/dL at baseline to 2.63 (0.87) mg/dL averaged across the midpoint of dose intervals through Week 24 with 69% of patients (9/13) achieving a mean serum phosphorus level above the LLN averaged across the midpoint on dose interval through Week 24. Mean serum phosphorus concentrations were sustained above LLN through Week 88. The renal phosphate reabsorptive capacity, as assessed by TmP/GFR, increased from a mean (SD) of 1.15 (0.43) mg/dL at baseline to 2.30 mg/dL (0.48) mg/dL at Week 48.
Bone Histomorphometry
In Study 6, osteomalacia was present at baseline in nine out of 11 patients with paired bone biopsies, and healing was assessed after 48 weeks of treatment. In these 9 patients with osteomalacia at baseline, OV/BV decreased from a mean (SD) score of 21.2% (19.9) at baseline to 13.9% (16.7), a change of -34%. O.Th declined from a mean (SD) of 18.9 (11.9) micrometers to 12.1 (10.1) micrometers, a change of -36%. MLt declined in 3 patients from a mean (SD) of 667 (414) days to 331 (396) days, a change of -50%.
In Study 7, osteomalacia was present at baseline in all 3 patients with paired bone biopsies, and healing was assessed after 48 weeks of treatment. In these 3 patients, OV/BV decreased from a mean (SD) score of 14.0% (15.2) at baseline to 9.2% (5.5), a change of -34%. O.Th declined from a mean (SD) of 16.0 (13.7) micrometers to 13.5 (7.1) micrometers, a change of -16%.
Radiographic Evaluation of Osteomalacia
In Study 6, 99mtechnetium-labelled whole body bone scans were performed at baseline and subsequent timepoints during the study on all 14 patients. Bone scans allow for assessment of sites of increased tracer uptake in a wide range of bone conditions, including osteomalacia. In patients with TIO, increased tracer uptake on bone scan is presumed to be nontraumatic fractures and pseudofractures. At baseline, all patients had areas of tracer uptake with a total of 249 bone abnormalities across 14 patients. The number of areas of tracer uptake decreased from Week 48 through Week 144, suggesting healing of the bone abnormalities.
16. How is Crysvita supplied
CRYSVITA (burosumab-twza) injection for subcutaneous administration is supplied as a sterile, preservative-free, clear to slightly opalescent and colorless to pale brown-yellow solution. The product is available as one single-dose vial per carton in the following strengths:
10 mg/mL (NDC# 42747-102-01)
20 mg/mL (NDC# 42747-203-01)
30 mg/mL (NDC# 42747-304-01)
17. Patient Counseling Information
Drug Interactions
Advise patients not to use any oral phosphate and/or active vitamin D analog products [see Contraindications (4)].
Hypersensitivity Reactions
Advise patients that CRYSVITA may cause hypersensitivity events such as rash, injection site rash and urticaria. Instruct the patients to contact their physician if such reactions occur [see Adverse Reactions (6.1)].
Injection Site Reactions
Inform patients that injection site reactions (e.g. erythema, rash, swelling, bruising, pain, pruritus, urticaria, and hematoma) have occurred at the site of CRYSVITA injection. Instruct the patients to contact their physician if such reactions occur [see Adverse Reactions (6.1)].
Restless Legs Syndrome
Advise patients that CRYSVITA can induce RLS or worsen the symptoms of existing RLS. Instruct the patients to contact their physician if such a reaction occurs [see Adverse Reactions (6.1)].
Pregnancy
Report pregnancies to the Kyowa Kirin, Inc. Adverse Event reporting line at 1-844-768-3544 [see Use in Specific Populations (8.1)].
Manufactured by:
Kyowa Kirin, Inc.
Princeton, NJ 08540
U.S. License No. 2077



| CRYSVITA
burosumab injection |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| CRYSVITA
burosumab injection |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| CRYSVITA
burosumab injection |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Kyowa Kirin, Inc. (014778321) |
