Drug Detail:Kineret (Anakinra [ an-na-kin-rah ])
Drug Class: Antirheumatics Interleukin inhibitors
Highlights of Prescribing Information
KINERET® (anakinra) injection, for subcutaneous use
Initial U.S. Approval: 2001
Recent Major Changes
| Indications and Usage, DIRA (1.3) | 12/2020 |
| Dosage and Administration, DIRA (2.3) | 12/2020 |
| Warning and Precautions, Serious Infections (5.1) | 12/2020 |
| Warning and Precautions, Hypersensitivity Reactions (5.3) | 12/2020 |
Indications and Usage for Kineret
KINERET is an interleukin-1 receptor antagonist indicated for:
Rheumatoid Arthritis (RA)
- Reduction in signs and symptoms and slowing the progression of structural damage in moderately to severely active rheumatoid arthritis, in patients 18 years of age or older who have failed 1 or more disease modifying antirheumatic drugs (DMARDs) (1.1)
Cryopyrin-Associated Periodic Syndromes (CAPS)
- Treatment of Neonatal-Onset Multisystem Inflammatory Disease (NOMID) (1.2)
Deficiency of Interleukin-1 Receptor Antagonist (DIRA)
- Treatment of Deficiency of Interleukin-1 Receptor Antagonist (DIRA) (1.3)
Kineret Dosage and Administration
Rheumatoid Arthritis (RA)
- The recommended dose of KINERET for the treatment of patients with rheumatoid arthritis is 100 mg/day administered daily by subcutaneous injection. The dose should be administered at approximately the same time every day (2.1)
- Physicians should consider a dose of 100 mg of KINERET administered every other day for RA patients who have severe renal insufficiency or end stage renal disease (defined as creatinine clearance < 30 mL/min, as estimated from serum creatinine levels) (2.4)
Cryopyrin-Associated Periodic Syndromes (CAPS)
- The recommended starting dose of KINERET is 1-2 mg/kg daily for NOMID patients. The dose can be individually adjusted to a maximum of 8 mg/kg daily to control active inflammation. (2.2)
- Physicians should consider administration of the prescribed KINERET dose every other day for NOMID patients who have severe renal insufficiency or end stage renal disease (defined as creatinine clearance < 30 mL/min, as estimated from serum creatinine levels) (2.4)
Deficiency of Interleukin-1 Receptor Antagonist (DIRA)
- The recommended starting dose of KINERET is 1-2 mg/kg daily for patients with DIRA. The dose can be individually adjusted to a maximum of 8 mg/kg daily to control active inflammation. (2.3)
- Physicians should consider administration of the prescribed KINERET dose every other day for patients with DIRA who have severe renal insufficiency or end stage renal disease (defined as creatinine clearance < 30 mL/min, as estimated from serum creatinine levels) (2.4)
See full prescribing information for administration instructions (2.4)
Dosage Forms and Strengths
Injection: 100 mg/0.67 mL solution in a single-use prefilled syringe for subcutaneous injection. Graduated syringe allows for doses between 20 mg and 100 mg. (3)
Contraindications
Known hypersensitivity to E coli-derived proteins, Kineret, or to any component of the product. (4)
Warnings and Precautions
- In RA, discontinue use if serious infection develops. In KINERET-treated NOMID or DIRA patients, the risk of a disease flare when discontinuing KINERET treatment should be weighed against the potential risk of continued treatment. Do not initiate KINERET in patients with active infections. (5.1)
- Use in combination with Tumor Necrosis Factor (TNF) blocking agents is not recommended (5.2)
- Hypersensitivity reactions, including anaphylactic reactions and angioedema, have been reported. Patients with DIRA may have an increased risk of allergic reactions, particularly in the first several weeks after starting KINERET treatment (5.3)
- The impact of treatment with KINERET on active and/or chronic infections and the development of malignancies is not known (5.4)
- Live vaccines should not be given concurrently with KINERET (5.5)
- Neutrophil counts should be assessed prior to initiating KINERET treatment, and while receiving KINERET, monthly for 3 months, and thereafter quarterly for a period up to 1 year (5.6)
Adverse Reactions/Side Effects
Rheumatoid Arthritis (RA)
Most common adverse reactions (incidence ≥ 5%) are injection site reaction, worsening of rheumatoid arthritis, upper respiratory tract infection, headache, nausea, diarrhea, sinusitis, arthralgia, flu like-symptoms, and abdominal pain (6.1)
NOMID
The most common AEs during the first 6 months of treatment (incidence > 10%) are injection site reaction, headache, vomiting, arthralgia, pyrexia, and nasopharyngitis (6.2)
DIRA
The most common AEs are upper respiratory tract infections, rash, pyrexia, influenza like illness, and gastroenteritis (6.3)
To report SUSPECTED ADVERSE REACTIONS, contact Swedish Orphan Biovitrum at 1-866-547-0644 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- A higher rate of serious infections has been observed in RA patients treated with concurrent KINERET and etanercept therapy than in patients treated with etanercept alone. Use of KINERET in combination with TNF blocking agents is not recommended (7)
Use In Specific Populations
- Pediatric use: KINERET is indicated for use in pediatric patients with NOMID and DIRA (8.4)
- Geriatric use: Because there is a higher incidence of infections in the elderly population in general, caution should be used in treating the elderly (8.5)
- Renal impairment: This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function (8.6)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2020
Full Prescribing Information
1. Indications and Usage for Kineret
1.1 Active Rheumatoid Arthritis
KINERET is indicated for the reduction in signs and symptoms and slowing the progression of structural damage in moderately to severely active rheumatoid arthritis (RA), in patients 18 years of age or older who have failed 1 or more disease modifying antirheumatic drugs (DMARDs). KINERET can be used alone or in combination with DMARDs other than Tumor Necrosis Factor (TNF) blocking agents [see Warnings and Precautions (5.2)].
2. Kineret Dosage and Administration
2.1 Active Rheumatoid Arthritis
The recommended dose of KINERET for the treatment of patients with rheumatoid arthritis is 100 mg/day administered daily by subcutaneous injection. Higher doses did not result in a higher response. The dose should be administered at approximately the same time every day.
2.2 Cryopyrin-Associated Periodic Syndromes (CAPS)
The recommended starting dose of KINERET is 1-2 mg/kg for NOMID patients. The dose can be individually adjusted to a maximum of 8 mg/kg daily to control active inflammation.
Adjust doses in 0.5 to 1 mg/kg increments. Once daily administration is generally recommended, but the dose may be split into twice daily administrations. Each syringe is intended for a single use. A new syringe must be used for each dose. Any unused portion after each dose should be discarded.
2.3 Deficiency of Interleukin-1 Receptor Antagonist (DIRA)
The recommended starting dose of KINERET is 1-2 mg/kg daily for patients with DIRA. The dose can be individually adjusted to a maximum of 8 mg/kg daily to control active inflammation. Adjust doses in 0.5 to 1 mg/kg increments.
Each syringe is intended for a single use. A new syringe must be used for each dose. Any unused portion after each dose should be discarded.
2.4 Renal Impairment
Physicians should consider administration of the prescribed dose of KINERET every other day for patients who have severe renal insufficiency or end stage renal disease (defined as creatinine clearance < 30 mL/min, as estimated from serum creatinine levels) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.5 Administration
Instructions on appropriate use should be given by the healthcare provider to the patient or caregiver. Patients or caregivers should not be allowed to administer KINERET until the patient or caregiver has demonstrated a thorough understanding of procedures and an ability to inject the product correctly. The prescribed dose of KINERET should be administered according to the instructions for use and any unused portions discarded. After administration of KINERET it is essential to follow the proper procedure for disposal of syringes and any residual drug. See the “Information for Patients” insert for detailed instructions on the handling and injection of KINERET.
Do not use KINERET beyond the expiration date shown on the carton. Visually inspect the solution for particulate matter and discoloration before administration. There may be trace amounts of small, translucent-to-white amorphous particles of protein in the solution. The prefilled syringe should not be used if the solution is discolored or cloudy, or if foreign particulate matter is present. If the number of translucent-to-white amorphous particles in a given syringe appears excessive, do not use this syringe.
3. Dosage Forms and Strengths
Injection: 100 mg/0.67 mL solution in a single-use prefilled syringe for subcutaneous injection. Graduated syringe allows for doses between 20 and 100 mg.
4. Contraindications
KINERET is contraindicated in patients with known hypersensitivity to E coli-derived proteins, KINERET, or any components of the product [see Hypersensitivity Reactions (5.3)].
5. Warnings and Precautions
5.1 Serious Infections
KINERET has been associated with an increased incidence of serious infections (2%) vs. Placebo (< 1%) in clinical trials in RA. Administration of KINERET in RA should be discontinued if a patient develops a serious infection. In KINERET treated NOMID and DIRA patients the risk of a disease flare when discontinuing KINERET treatment should be weighed against the potential risk of continued treatment. Treatment with KINERET should not be initiated in patients with active infections. The safety and efficacy of KINERET in immunosuppressed patients or in patients with chronic infections have not been evaluated.
Drugs that affect the immune system by blocking tumor necrosis factor (TNF) have been associated with an increased risk of reactivation of latent tuberculosis (TB). It is possible that taking drugs such as KINERET that blocks IL-1 increases the risk of TB or other atypical or opportunistic infections. Health care providers should follow current CDC guidelines both to evaluate for and to treat possible latent tuberculosis infections before initiating therapy with KINERET.
5.2 Use with TNF Blocking Agents
In a 24-week study of concurrent KINERET and etanercept therapy in RA patients, the rate of serious infections in the combination arm (7%) was higher than with etanercept alone (0%). The combination of KINERET and etanercept did not result in higher ACR response rates compared to etanercept alone [see Clinical Studies (14)]. Use of KINERET in combination with TNF blocking agents is not recommended.
5.3 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylactic reactions and angioedema, have been reported with KINERET. If a severe hypersensitivity reaction occurs, administration of KINERET should be discontinued and appropriate therapy initiated.
KINERET is the recombinant form of IL-1Ra that DIRA patients are lacking. Patients with DIRA may have an increased risk of allergic reactions, particularly in the first several weeks after starting KINERET treatment. Patients should be closely monitored during this time period. If a severe allergic reaction occurs, appropriate treatment should be initiated and discontinuation of KINERET should be considered.
5.4 Immunosuppression
The impact of treatment with KINERET on active and/or chronic infections and the development of malignancies is not known [see Adverse Reactions (6)].
5.5 Immunizations
In a placebo-controlled clinical trial (n = 126), no difference was detected in anti-tetanus antibody response between the KINERET and placebo treatment groups when the tetanus/diphtheria toxoids vaccine was administered concurrently with KINERET. No data are available on the effects of vaccination with other inactivated antigens in patients receiving KINERET. No data are available on either the effects of live vaccination or the secondary transmission of infection by live vaccines in patients receiving KINERET. Therefore, live vaccines should not be given concurrently with KINERET.
5.6 Neutrophil Count
Patients receiving KINERET may experience a decrease in neutrophil counts. Neutrophil counts should therefore be assessed prior to initiating KINERET treatment, and while receiving KINERET, monthly for 3 months, and thereafter quarterly for a period up to 1 year.
In the placebo-controlled studies, 8% of RA patients receiving KINERET had decreases in neutrophil counts of at least one World Health Organization (WHO) toxicity grade compared with 2% in the placebo control group. Nine KINERET-treated patients (0.4%) experienced neutropenia (ANC < 1 x 109/L). This is discussed in more detail in the Adverse Reactions (6): Hematologic Events (6.1) section.
In 43 NOMID patients followed for up to 60 months 2 patients experienced neutropenia that resolved over time during continued KINERET treatment. [see Adverse Reactions (6.2)]
6. Adverse Reactions/Side Effects
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
6.1 Clinical Studies Experience in RA
The most serious adverse reactions were:
- Serious Infections – [see Warnings and Precautions (5.1)]
- Neutropenia, particularly when used in combination with TNF blocking agents
The most common adverse reaction with KINERET is injection-site reactions. These reactions were the most common reason for withdrawing from studies.
The data described herein reflect exposure to KINERET in 3025 patients, including 2124 exposed for at least 6 months and 884 exposed for at least one year. Studies 1 and 4 used the recommended dose of 100 mg per day. The patients studied were representative of the general population of patients with rheumatoid arthritis.
Injection-site Reactions
The most common and consistently reported treatment-related adverse event associated with KINERET is injection-site reaction (ISR). In Studies 1 and 4, 71% of patients developed an ISR, which was typically reported within the first 4 weeks of therapy. The majority of ISRs were reported as mild (72.6% mild, 24.1% moderate and 3.2% severe). The ISRs typically lasted for 14 to 28 days and were characterized by 1 or more of the following: erythema, ecchymosis, inflammation, and pain.
Infections
In Studies 1 and 4 combined, the incidence of infection was 39% in the KINERET-treated patients and 37% in placebo-treated patients during the first 6 months of blinded treatment. The incidence of serious infections in Studies 1 and 4 was 2% in KINERET-treated patients and 1% in patients receiving placebo over 6 months. The incidence of serious infection over 1 year was 3% in KINERET-treated patients and 2% in patients receiving placebo. These infections consisted primarily of bacterial events such as cellulitis, pneumonia, and bone and joint infections. Majority of patients (73%) continued on study drug after the infection resolved. No serious opportunistic infections were reported. Patients with asthma appeared to be at higher risk of developing serious infections when treated with KINERET (8 of 177 patients, 4.5%) compared to placebo (0 of 50 patients, 0%).
In open-label extension studies, the overall rate of serious infections was stable over time and comparable to that observed in controlled trials. In clinical studies and postmarketing experience, cases of opportunistic infections have been observed and included fungal, mycobacterial and bacterial pathogens. Infections have been noted in all organ systems and have been reported in patients receiving KINERET alone or in combination with immunosuppressive agents.
In patients who received both KINERET and etanercept for up to 24 weeks, the incidence of serious infections was 7%. The most common infections consisted of bacterial pneumonia (4 cases) and cellulitis (4 cases). One patient with pulmonary fibrosis and pneumonia died due to respiratory failure.
Malignancies
Among 5300 RA patients treated with KINERET in clinical trials for a mean of 15 months (approximately 6400 patient years of treatment), 8 lymphomas were observed for a rate of 0.12 cases/100 patient years. This is 3.6 fold higher than the rate of lymphomas expected in the general population, based on the National Cancer Institute's Surveillance, Epidemiology and End Results (SEER) database.3 An increased rate of lymphoma, up to several fold, has been reported in the RA population, and may be further increased in patients with more severe disease activity. Thirty-seven malignancies other than lymphoma were observed. Of these, the most common were breast, respiratory system, and digestive system. There were 3 melanomas observed in Study 4 and its long-term open-label extension, greater than the 1 expected case. The significance of this finding is not known. While patients with RA, particularly those with highly active disease, may be at a higher risk (up to several fold) for the development of lymphoma, the role of IL-1 blockers in the development of malignancy is not known.
Hematologic Events
In placebo-controlled studies with KINERET, 8% of patients receiving KINERET had decreases in total white blood counts of at least one WHO toxicity grade, compared with 2% of placebo patients. Nine KINERET-treated patients (0.4%) developed neutropenia (ANC < 1 x 109/L). 9 % of patients receiving KINERET had increases in eosinophil differential percentage of at least one WHO toxicity grade, compared with 3 % of placebo patients. Of patients treated concurrently with KINERET and etanercept 2% developed neutropenia (ANC < 1 x 109/L). While neutropenic, one patient developed cellulitis which recovered with antibiotic therapy. 2% of patients receiving KINERET had decreases in platelets, all of WHO toxicity grade one, compared to 0% of placebo patients.
Hypersensitivity Reactions
Hypersensitivity reactions including anaphylactic reactions, angioedema, urticaria, rash, and pruritus have been reported with KINERET.
Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. In Studies 1 and 4, from which data is available for up to 36 months, 49% of patients tested positive for anti-anakinra binding antibodies at one or more time points using a biosensor assay. Of the 1615 patients with available data at Week 12 or later, 30 (2%) tested positive for neutralizing antibodies in a cell-based bioassay. Of the 13 patients with available follow-up data, 5 patients remained positive for neutralizing antibodies at the end of the studies. No correlation between antibody development and adverse events was observed.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assays. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including sample handling, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to KINERET with the incidence of antibodies to other products may be misleading.
Other Adverse Events
Table 1 reflects adverse events in Studies 1 and 4, that occurred with a frequency of ≥ 5% in KINERET-treated patients over a 6-month period.
| Placebo | KINERET 100 mg/day |
||||
| Preferred term | (n = 733) | (n = 1565) | |||
| Injection Site Reaction | 29% | 71% | |||
| Worsening of RA | 29% | 19% | |||
| Upper Respiratory Tract Infections | 17% | 14% | |||
| Headache | 9% | 12% | |||
| Nausea | 7% | 8% | |||
| Diarrhea | 5% | 7% | |||
| Sinusitis | 7% | 7% | |||
| Arthralgia | 6% | 6% | |||
| Flu Like Symptoms | 6% | 6% | |||
| Abdominal Pain | 5% | 5% | |||
6.2 Clinical Study Experience in NOMID
The data described herein reflect an open-label study in 43 NOMID patients exposed to KINERET for up to 60 months adding up to a total exposure of 159.8 patient years.
Patients were treated with a starting dose of 1 to 2 mg/kg/day and an average maintenance dose of 3-4 mg/kg/day adjusted depending on the severity of disease. Among pediatric NOMID patients, doses up to 7.6 mg/kg/day have been maintained for up to 15 months.
There were 24 serious adverse events (SAEs) reported in 14 of the 43 treated patients. The most common type of SAEs reported were infections [see Warnings and Precautions (5.1)]. Five SAEs were related to lumbar puncture, which was part of the study procedure.
There were no permanent discontinuations of study drug treatment due to AEs. Doses were adjusted in 5 patients because of AEs; all were dose increases in connection with disease flares.
The reporting frequency of AEs was highest during the first 6 months of treatment. The incidence of AEs did not increase over time, and no new types of AEs emerged.
The most commonly reported AEs during the first 6 months of treatment (incidence >10%) were injection site reaction (ISR), headache, vomiting, arthralgia, pyrexia, and nasopharyngitis (Table 2).
The most commonly reported AEs during the 60-month study period, calculated as the number of events/patient years of exposure, were arthralgia, headache, pyrexia, upper respiratory tract infection, nasopharyngitis, and rash.
The AE profiles for different age groups <2 years, 2-11 years, and 12-17 years corresponded to the AE profile for patients ≥18 years, with the exception of infections and related symptoms being more frequent in patients <2 years.
Infections
The reporting rate for infections was higher during the first 6 months of treatment (2.3 infections/patient-year) compared to after the first 6 months (1.7 infections/patient year). The most common infections were upper respiratory tract infection, sinusitis, ear infections, and nasopharyngitis.
There were no deaths or permanent treatment discontinuations due to infections. In one patient KINERET administration was temporarily stopped during an infection and in 5 patients the dose of KINERET was increased due to disease flares in connection with infections. Thirteen infections in 7 patients were classified as serious, the most common being pneumonia and gastroenteritis occurring in 3 and 2 patients, respectively. No serious opportunistic infections were reported.
The reporting frequency for infections was highest in patients <12 years of age.
Hematologic Events
After start of KINERET treatment neutropenia was reported in 2 patients. One of these patients experienced an upper respiratory tract infection and an otitis media infection. Both episodes of neutropenia resolved over time with continued KINERET treatment.
Injection Site Reactions
In total, 17 injection site reactions (ISRs) were reported in 10 patients during the 60-month study period. Out of the 17 ISRs, 11 (65%) occurred during the first month and 13 (76%) were reported during the first 6 months. No ISR was reported after Year 2 of treatment. The majority of ISRs were reported as mild (76% mild, 24% moderate). No patient permanently or temporarily discontinued KINERET treatment due to injection site reactions.
Immunogenicity
The immunogenicity of KINERET in NOMID patients was not evaluated.
| Safety population | ||
| (N=43) Total exposure in patient years= 20.8 | ||
| Preferred term | N (%) | Number of events /patient year |
| Injection site reaction | 7 (16.3%) | 0.5 |
| Headache | 6 (14.0%) | 0.7 |
| Vomiting | 6 (14.0%) | 0.6 |
| Arthralgia | 5 (11.6%) | 0.6 |
| Pyrexia | 5 (11.6%) | 0.4 |
| Nasopharyngitis | 5 (11.6%) | 0.3 |
The most common adverse reactions occurring after the first 6-month period of treatment with KINERET (up to 60 months of treatment) included: arthralgia, headache, pyrexia, upper respiratory tract infection, nasopharyngitis, and rash.
6.3 Clinical Study Experience in DIRA
The safety data described in this section reflect exposure to KINERET in 9 patients with DIRA treated for up to 10 years in a natural history study (study 17-I-0016). Most patients received a starting dose of 1 to 2 mg/kg/day and thereafter doses were individually adjusted to reach a stable efficacious dose. The highest dose given was 7.5 mg/kg/day. Overall, the safety profile observed in patients with DIRA treated with KINERET was consistent with the safety profile in NOMID patients.
There were 16 serious adverse events (SAEs) reported in 4 out of 9 treated patients. The most common type of SAEs reported (5 events in 2 patients) were infections [see Warnings and Precautions (5.1)].
The most common adverse events in patients with DIRA were upper respiratory tract infections, rash, pyrexia, influenza like illness and gastroenteritis.
There were no permanent discontinuations of KINERET treatment due to AEs.
Infections
There were 16 infections in 5 patients (reporting rate: 0.28 infections / patient year). The most common infections were upper respiratory tract infections, cellulitis and gastroenteritis.
6.4 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of KINERET. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hepato-biliary disorders:
- elevations of transaminases,
- non-infectious hepatitis
Hematologic events:
- thrombocytopenia, including severe thrombocytopenia (i.e platelet counts <10x109/L)
7. Drug Interactions
No drug-drug interaction studies in human subjects have been conducted. Toxicologic and toxicokinetic studies in rats did not demonstrate any alterations in the clearance or toxicologic profile of either methotrexate or KINERET when the two agents were administered together.
7.1 TNF Blocking Agents
A higher rate of serious infections has been observed in patients treated with concurrent KINERET and etanercept therapy than in patients treated with etanercept alone [see Warnings and Precautions (5.2)]. Two percent of patients treated concurrently with KINERET and etanercept developed neutropenia (ANC < 1 x 109/L). Use of KINERET in combination with TNF blocking agents is not recommended.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Available data from retrospective studies and case reports on KINERET use in pregnant women are insufficient to identify a drug associated risk of major birth defects, miscarriage, or maternal and fetal adverse events. There are risks to the mother and fetus associated with active rheumatoid arthritis or Cryopyrin-Associated Periodic Syndromes (CAPS). In animal reproduction studies, subcutaneous administration of anakinra to pregnant rats and rabbits during organogenesis demonstrated no evidence of fetal harm at doses up to 25 times the maximum recommended human dose (MRHD).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Disease-associated maternal and/or embryo/fetal risk
Published data suggest the risk of adverse pregnancy outcomes in women with rheumatoid arthritis or CAPS is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (<2500 grams), and small for gestational age at birth.
Human Data
The available data from retrospective studies and case reports of anakinra-exposed pregnancies have not identified an increased frequency or pattern of birth defects, miscarriage, or adverse maternal or fetal outcomes. An international multi-center retrospective study of pregnancy outcomes with interleukin-1 inhibitors reported on 23 anakinra-exposed pregnancies. There were 21 live births of healthy infants, 1 miscarriage, and 1 infant with left renal agenesis. The estimated background rate of detected renal malformations is 0.2-2% of all newborns. Another retrospective study reported on 10 anakinra-exposed pregnancies in women with CAPS. There were 9 live births, 1 miscarriage, and 1 fetal demise in a twin pregnancy. The surviving twin was born healthy. Overall, these data cannot definitively establish or exclude any anakinra-associated risks during pregnancy. Methodological limitations of these data include small sample size and the inability to control for confounders such as the timing of drug exposure, underlying maternal disease, and concomitant medication use.
Animal Data
Animal reproduction studies were conducted in rats and rabbits. In embryo-fetal development studies, anakinra was administered throughout the period of organogenesis at the subcutaneous doses of 12.5, 50, and 200 mg/kg/day to pregnant rats from gestation days (GD) 7 to 17 and pregnant rabbits from GD 6 to 18. In these studies, anakinra at doses up to 25 times the MRHD (on a mg/kg basis at maternal subcutaneous doses up to 200 mg/kg/day) revealed no evidence of harm to the fetus.
8.2 Lactation
Risk Summary
There are no data on the presence of anakinra in either human or animal milk or the effects on milk production. Available published data from a small retrospective study and postmarketing case reports do not establish an association between maternal anakinra use during lactation and adverse effects on breastfed infants. The limited clinical data during lactation precludes a clear determination of the risk of KINERET to an infant during lactation. Therefore, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KINERET and any potential adverse effects on the breastfed infant from KINERET or from the underlying maternal condition.
8.4 Pediatric Use
Neonatal-Onset Multisystem Inflammatory Disease (NOMID)
The NOMID study included 36 pediatric patients: 13 below 2 years, 18 between 2 and 11 years, and 5 between 12 and 17 years of age. A subcutaneous KINERET starting dose of 1–2 mg/kg/day was administered in all age groups. An average maintenance dose of 3–4 mg/kg/day was adequate to maintain clinical response throughout the study irrespective of age but a higher dose was, on occasion, required in severely affected patients. The prefilled syringe does not allow doses lower than 20 mg to be administered.
Deficiency of Interleukin-1 Receptor Antagonist (DIRA)
The study in DIRA patients included 9 pediatric patients (ages 1 month to 9 years at start of KINERET treatment). Most patients received a starting dose of 1 to 2 mg/kg/day. Doses up to 7.5 mg/kg/day were given. The prefilled syringe does not allow doses lower than 20 mg to be administered.
Juvenile Rheumatoid Arthritis (JRA)
The safety and effectiveness of KINERET in the treatment of pediatric patients with Juvenile Rheumatoid Arthritis (JRA) have not been established. KINERET was studied in a single randomized, blinded multi-center trial in 86 patients with polyarticular course JRA; ages 2-17 years receiving a dose of 1 mg/kg subcutaneously daily, up to a maximum dose of 100 mg. The 50 patients who achieved a clinical response after a 12-week open-label run-in were randomized to KINERET (25 patients) or placebo (25 patients), administered daily for an additional 16 weeks. A subset of these patients continued open-label treatment with KINERET for up to 1 year in a companion extension study. An adverse event profile similar to that seen in adult RA patients was observed in these studies. These study data are insufficient to demonstrate efficacy and, therefore, KINERET is not recommended for pediatric use in JRA.
8.5 Geriatric Use
A total of 752 RA patients ≥ 65 years of age, including 163 patients ≥ 75 years of age, were studied in clinical trials. No differences in safety or effectiveness were observed between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Because there is a higher incidence of infections in the elderly population in general, caution should be used in treating the elderly.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function.
10. Overdosage
There have been no cases of overdose reported with KINERET in clinical trials of RA or NOMID. In sepsis trials no serious toxicities attributed to KINERET were seen when administered at mean calculated doses of up to 35 times those given patients with RA over a 72-hour treatment period.
11. Kineret Description
KINERET (anakinra) is a recombinant, nonglycosylated form of the human interleukin-1 receptor antagonist (IL-1Ra). KINERET differs from native human IL-1Ra in that it has the addition of a single methionine residue at its amino terminus. KINERET consists of 153 amino acids and has a molecular weight of 17.3 kilodaltons. It is produced by recombinant DNA technology using an E coli bacterial expression system.
KINERET is supplied in single use prefilled glass syringes with 29 gauge needles as a sterile, clear, colorless-to-white, preservative free solution for daily subcutaneous (SC) administration. The solution may contain trace amounts of small, translucent-to-white amorphous proteinaceous particles. Each prefilled glass syringe contains: 0.67 mL (100 mg) of anakinra in a solution (pH 6.5) containing anhydrous citric acid (1.29 mg), disodium EDTA (0.12 mg), polysorbate 80 (0.70 mg), and sodium chloride (5.48 mg) in Water for Injection, USP.
The prefilled syringe contains an outer rigid plastic needle shield attached to an inner needle cover. The syringe or needle shield components are not made with natural rubber latex.
12. Kineret - Clinical Pharmacology
12.1 Mechanism of Action
KINERET blocks the biologic activity of IL-1 alpha and beta by competitively inhibiting IL-1 binding to the interleukin-1 type I receptor (IL-1RI), which is expressed in a wide variety of tissues and organs.
IL-1 production is induced in response to inflammatory stimuli and mediates various physiologic responses including inflammatory and immunological responses. IL-1 has a broad range of activities including cartilage degradation by its induction of the rapid loss of proteoglycans, as well as stimulation of bone resorption. The levels of the naturally occurring IL-1Ra in synovium and synovial fluid from RA patients are not sufficient to compete with the elevated amount of locally produced IL-1.
Spontaneous mutations in the CIAS1/NLRP3 gene have been identified in a majority of patients with cryopyrin-associated periodic syndromes such as NOMID. CIAS1/NLRP3 encodes for cryopyrin, a component of the inflammasome. The activated inflammasome results in proteolytic maturation and secretion of IL-1β, which has an important role in the systemic inflammation and manifestations of NOMID.
DIRA is an autosomal recessive monogenic autoinflammatory disease caused by mutations in the IL1RN gene leading to loss of secretion of the interleukin-1 receptor antagonist (IL-1Ra). The deficiency of IL-1Ra results in unopposed IL-1α and IL-1β pro-inflammatory signaling causing systemic inflammation with skin and bone involvement.
12.3 Pharmacokinetics
The absolute bioavailability of KINERET after a 70 mg subcutaneous bolus injection in healthy subjects (n = 11) is 95%. In subjects with RA, maximum plasma concentrations of KINERET occurred 3 to 7 hours after subcutaneous administration of KINERET at clinically relevant doses (1 to 2 mg/kg; n = 18); the terminal half-life ranged from 4 to 6 hours. In RA patients, no unexpected accumulation of KINERET was observed after daily subcutaneous doses for up to 24 weeks.
The influence of demographic covariates on the pharmacokinetics of KINERET was studied using population pharmacokinetic analysis encompassing 341 patients receiving daily subcutaneous injection of KINERET at doses of 30, 75, and 150 mg for up to 24 weeks. The estimated KINERET clearance increased with increasing creatinine clearance and body weight. After adjusting for creatinine clearance and body weight, gender and age were not significant factors for mean plasma clearance.
In NOMID patients, at a median SC dose of 3 mg/kg once daily and a median treatment time of 3.5 years, the median (range) steady-state serum exposure of anakinra was Cmax 3628 (655–8511) ng/mL (n=16) and C24h 203 (53–1979) ng/mL (n=16). The median (range) half-life of anakinra was 5.7 (3.1–28.2) hours (n=12). There was no obvious gender difference.
Patients With Renal Impairment: The mean plasma clearance of KINERET in subjects with mild (creatinine clearance 50-80 mL/min) and moderate (creatinine clearance 30-49 mL/min) renal insufficiency was reduced by 16% and 50%, respectively. In severe renal insufficiency and end stage renal disease (creatinine clearance < 30 mL/min1), mean plasma clearance declined by 70% and 75%, respectively. Less than 2.5% of the administered dose of KINERET was removed by hemodialysis or continuous ambulatory peritoneal dialysis. Based on these observations, a dose schedule change should be considered for subjects with severe renal insufficiency or end stage renal disease [see Dosage and Administration (2.2)].
Patients with Hepatic Dysfunction: No formal studies have been conducted examining the pharmacokinetics of KINERET administered subcutaneously in patients with hepatic impairment.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Long-term animal studies to evaluate the carcinogenic potential of KINERET were not conducted. KINERET had no effects on fertility and reproductive performance indices in male and female rats subcutaneous doses up to 200 mg/kg/day (approximately 25 times theMRHD on a mg/kg basis).
14. Clinical Studies
14.1 Clinical Studies in RA
The safety and efficacy of KINERET have been evaluated in three randomized, double-blind, placebo-controlled trials of 1790 patients ≥ 18 years of age with active rheumatoid arthritis (RA). An additional fourth study was conducted to assess safety. In the efficacy trials, KINERET was studied in combination with other disease-modifying antirheumatic drugs (DMARDs) other than Tumor Necrosis Factor (TNF) blocking agents (Studies 1 and 2) or as a monotherapy (Study 3).
Study 1 involved 899 patients with active RA who had been on a stable dose of methotrexate (MTX) (10 to 25 mg/week) for at least 8 weeks. All patients had at least 6 swollen/painful and 9 tender joints and either a C-reactive protein (CRP) of ≥ 1.5 mg/dL or an erythrocyte sedimentation rate (ESR) of ≥ 28 mm/hr. Patients were randomized to KINERET or placebo in addition to their stable doses of MTX. The first 501 patients were evaluated for signs and symptoms of active RA. The total 899 patients were evaluated for progression of structural damage.
Study 2 evaluated 419 patients with active RA who had received MTX for at least 6 months including a stable dose (15 to 25 mg/week) for at least 3 consecutive months prior to enrollment. Patients were randomized to receive placebo or one of five doses of KINERET subcutaneous daily for 12 to 24 weeks in addition to their stable doses of MTX.
Study 3 evaluated 472 patients with active RA and had similar inclusion criteria to Study 1 except that these patients had received no DMARD for the previous 6 weeks or during the study. Patients were randomized to receive either KINERET or placebo. Patients were DMARD-naïve or had failed no more than 3 DMARDs.
Study 4 was a placebo-controlled, randomized trial designed to assess the safety of KINERET in 1414 patients receiving a variety of concurrent medications for their RA including some DMARD therapies, as well as patients who were DMARD-free. The TNF blocking agents etanercept and infliximab were specifically excluded. Concurrent DMARDs included MTX, sulfasalazine, hydroxychloroquine, gold, penicillamine, leflunomide, and azathioprine. Unlike Studies 1, 2 and 3, patients predisposed to infection due to a history of underlying disease such as pneumonia, asthma, controlled diabetes, and chronic obstructive pulmonary disease (COPD) were also enrolled [see Adverse Reactions (6)].
In Studies 1, 2 and 3, the improvement in signs and symptoms of RA was assessed using the American College of Rheumatology (ACR) response criteria (ACR20, ACR50, ACR70). In these studies, patients treated with KINERET were more likely to achieve an ACR20 or higher magnitude of response (ACR50 and ACR70) than patients treated with placebo (Table 3). The treatment response rates did not differ based on gender or ethnic group. The results of the ACR component scores in Study 1 are shown in Table 4.
Most clinical responses, both in patients receiving placebo and patients receiving KINERET, occurred within 12 weeks of enrollment.
|
a p < 0.05, KINERET versus placebo |
|||||
|
b p < 0.01, KINERET versus placebo |
|||||
|
c p < 0.001, KINERET versus placebo |
|||||
| Study 1 (Patients on MTX) | Study 3 (No DMARDs) | ||||
| KINERET | |||||
|
Response | Placebo (n = 251) | KINERET 100 mg/day (n = 250) | Placebo (n = 119) | 75 mg/day (n = 115) | 150 mg/day (n = 115) |
| ACR20
Month 3 Month 6 |
24% 22% |
34%a 38%c |
23% 27% |
33% 34% |
33% 43%a |
| ACR50
Month 3 Month 6 |
6% 8% |
13%b 17%b |
5% 8% |
10% 11% |
8% 19%a |
| ACR70
Month 3 Month 6 |
0% 2% |
3%a 6%a |
0% 1% |
0% 1% |
0% 1% |
|
a Health Assessment Questionnaire; 0 = best, 3 = worst; includes eight categories: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities. |
||||||
|
b Visual analog scale; 0 = best, 100 = worst |
||||||
|
c Scale 0 to 68 |
||||||
|
d Scale 0 to 66 |
||||||
| Placebo/MTX | KINERET/MTX 100 mg/day |
|||||
| (n = 251) | (n = 250) | |||||
| Parameter (median) | Baseline | Month 6 | Baseline | Month 6 | ||
| Patient Reported Outcomes | ||||||
| Disability indexa | 1.38 | 1.13 | 1.38 | 1.00 | ||
| Patient global assessmentb | 51.0 | 41.0 | 51.0 | 29.0 | ||
| Painb | 56.0 | 44.0 | 63.0 | 34.0 | ||
| Objective Measures | ||||||
| ESR (mm/hr) | 35.0 | 32.0 | 36.0 | 19.0 | ||
| CRP (mg/dL) | 2.2 | 1.6 | 2.2 | 0.5 | ||
| Physician's Assessments | ||||||
| Tender/painful jointsc | 20.0 | 11.0 | 23.0 | 9.0 | ||
| Physician global assessmentb | 59.0 | 31.0 | 59.0 | 26.0 | ||
| Swollen jointsd | 18.0 | 10.5 | 17.0 | 9.0 | ||
A 24-week study was conducted in 242 patients with active RA on background methotrexate who were randomized to receive either etanercept alone or the combination of KINERET and etanercept. The ACR50 response rate was 31% for patients treated with the combination of KINERET and etanercept and 41% for patients treated with etanercept alone, indicating no added clinical benefit of the combination over etanercept alone. Serious infections were increased with the combination compared to etanercept alone [see Warnings and Precautions (5.1)].
In Study 1, the effect of KINERET on the progression of structural damage was assessed by measuring the change from baseline at month 12 in the Total Modified Sharp Score (TSS) and its subcomponents, erosion score, and joint space narrowing (JSN) score.2 Radiographs of hands/wrists and forefeet were obtained at baseline, 6 months and 12 months and scored by readers who were unaware of treatment group. A difference between placebo and KINERET for change in TSS, erosion score (ES) and JSN score was observed at 12 months (Table 5).
|
* Differences and 95% confidence intervals for the differences in change scores between Placebo/MTX and KINERET/MTX |
||||||
|
** Based on Wilcoxon rank-sum test |
||||||
| Placebo/MTX (N = 450) | KINERET 100 mg/day /MTX (N = 449) | Placebo/MTX vs. KINERET/MTX |
||||
| Baseline | Change at Month 12 | Baseline | Change at Month 12 | 95% Confidence Interval* | p-value** | |
| TSS | 52 | 2.6 | 50 | 1.7 | 0.9 [0.3, 1.6] | < 0.001 |
| Erosion | 28 | 1.6 | 25 | 1.1 | 0.5 [0.1, 1.0] | 0.024 |
| JSN | 24 | 1.1 | 25 | 0.7 | 0.4 [0.1, 0.7] | < 0.001 |
The disability index of the Health Assessment Questionnaire (HAQ) was administered monthly for the first six months and quarterly thereafter during Study 1. Health outcomes were assessed by the Short Form-36 (SF-36) questionnaire. The 1-year data on HAQ in Study 1 showed more improvement with KINERET than placebo. The physical component summary (PCS) score of the SF-36 also showed more improvement with KINERET than placebo but not the mental component summary (MCS).
14.2 Clinical Studies in NOMID
The efficacy of KINERET was evaluated in a prospective, long-term, open-label and uncontrolled study which incorporated a withdrawal period in a subset of 11 patients. This study included 43 NOMID patients 0.7 to 46 years of age treated for up to 60 months. Patients were given an initial KINERET dose of 1–2.4 mg/kg body weight. During the study, the dose was adjusted by 0.5 to 1 mg/kg increments to a protocol-specified maximum of 10 mg/kg daily, titrated to control signs and symptoms of disease. The maximum dose actually studied was 7.6 mg/kg/day. The average maintenance dose was 3 to 4 mg/kg daily. In general, the dose was given once daily, but for some patients, the dose was split into twice daily administrations for better control of disease activity.
NOMID symptoms were assessed with a disease-specific Diary Symptom Sum Score (DSSS), which included the prominent disease symptoms fever, rash, joint pain, vomiting, and headache. In addition, serum amyloid A (SAA), hsCRP, and ESR levels were monitored. Changes in clinical and laboratory parameters from baseline to Months 3 to 6 and from Month 3 (before withdrawal) to the end of the withdrawal period were assessed in the subset of patients who underwent withdrawal. The estimated changes from baseline in DSSS are summarized through Month 60 in Table 6. Results were consistent across all subgroups, including age, gender, presence of CIAS1 mutation, and disease phenotype. Improvements occurred in all individual disease symptoms comprising the DSSS (Table 7), as well as in the serum markers of inflammation. For the 11 patients who went through a withdrawal phase, disease symptoms and serum markers of inflammation worsened after withdrawal and promptly responded to reinstitution of KINERET therapy. Upon withdrawal of treatment, the median time until disease flare criteria were met was 5 days.
|
*Mean (SD) baseline value was 4.5 (3.2) |
||
| Time point | Estimated mean change
from baseline in DSSS* | 95% confidence interval |
| Month 3-6 | -3.5 | -3.7 to -3.3 |
| Month 12 | -3.6 | -3.9 to -3.3 |
| Month 36 | -3.5 | -3.8 to -3.2 |
| Month 60 | -3.5 | -3.8 to -3.1 |
|
*mean (SD) |
||||||
| Visit (month) | Number of patients | Fever score* | Rash score* | Joint pain score* | Vomiting score* | Headache score* |
| Baseline | 29 | 0.5 (0.8) | 1.9 (1.1) | 1.2 (1.1) | 0.1 (0.2) | 0.9 (1.0) |
| 1 | 28 | 0.1 (0.1) | 0.3 (0.5) | 0.2 (0.3) | 0.0 (0.0) | 0.2 (0.3) |
| 3 | 26 | 0.1 (0.2) | 0.1 (0.2) | 0.2 (0.4) | 0.0 (0.1) | 0.1 (0.2) |
| 6 | 25 | 0.0 (0.1) | 0.1 (0.1) | 0.2 (0.4) | 0.0 (0.1) | 0.2 (0.3) |
| 12 | 24 | 0.1 (0.1) | 0.1 (0.2) | 0.1 (0.2) | 0.0 (0.1) | 0.1 (0.2) |
| 36 | 19 | 0.0 (0.2) | 0.0 (0.2) | 0.1 (0.3) | 0.0 (0.0) | 0.2 (0.6) |
| 60 | 15 | 0.0 (0.0) | 0.1 (0.3) | 0.3 (0.7) | 0.0 (0.0) | 0.1 (0.3) |
KINERET treatment also appeared to be associated with improvement of, or stability in, assessments of other NOMID disease manifestations, such as CNS, audiogram, and visual acuity data, up to Month 60.
14.3 Clinical Studies in DIRA
The safety and efficacy of KINERET were evaluated in a long-term natural history study including 9 DIRA patients (ages 1 month to 9 years at the start of KINERET treatment) treated with KINERET for up to 10 years. All patients had genetically confirmed DIRA. The starting dose of KINERET was 1 to 2 mg/kg/day in the 6 patients for which the dose was reported. The dose was then individually adjusted to reach a stable efficacious dose to control active inflammation. The highest KINERET dose studied was 7.5 mg/kg/day. At the last visit during the first KINERET treatment period, the dose ranged between 2.2 and 6.1 mg/kg/day. Inflammatory remission was defined as achievement of all of the following criteria: CRP ≤ 5 mg/L, no pustulosis, no inflammatory bone disease, and no concomitant glucocorticosteroid use. All 9 patients achieved inflammatory remission while on KINERET treatment.
15. References
- Cockcroft DW and Gault HM. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16:31-41.
- Sharp JT, Young DY, Bluhm GB, et al. How many joints in the hands and wrists should be included in a score of radiologic abnormalities used to assess rheumatoid arthritis? Arthritis Rheum. 1985; 28:1326-1335.
- National Cancer Institute. Surveillance, Epidemiology, and End Results Database (SEER) Program. SEER Incidence Crude Rates, 11 Registries, 1992-1999.
16. How is Kineret supplied
KINERET is supplied in single-use preservative free, prefilled glass syringes with 29 gauge needles. Each prefilled glass syringe contains 100 mg of anakinra per 0.67 mL. The full syringe contains 100 mg anakinra. KINERET is dispensed in a 4 x 7 syringe dispensing pack containing 28 syringes (NDC 66658-234-28). KINERET is also dispensed in a 1 x 7 syringe dispensing pack containing 7 syringes (NDC 66658-234-07).
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Instruct patients and their caregivers on the proper dosage and administration of KINERET and provide all patients with the “Patient information and Instructions for Use” insert. While this Patient Information and Instructions for Use provides information about the product and its use, it is not intended to take the place of regular discussions between the patient and healthcare provider. The ability to inject subcutaneously should be assessed to ensure proper administration of KINERET. Thoroughly instruct patients and their caregivers on the importance of proper disposal and caution against the reuse of needles, syringes, and drug product. A puncture-resistant container for the disposal of used syringes should be available to the patient. The full container should be disposed of according to the directions provided by the healthcare provider.
Infections: Inform patients that KINERET may lower the ability of their immune system to fight infections. Advise patients of the importance of contacting their doctor if they develop any symptoms of infection.
Injection-site reactions: Physicians should explain to patients that almost a quarter of patients in the clinical trial experienced a reaction at the injection site. Injection-site reactions may include pain, erythema, swelling, prurities, bruising, mass, inflammation, dermatitis, edema, urticaria, vesicles, warmth, and hemorrhage. Inform patients or their caregivers that the prefilled syringe should be removed from refrigeration and left at room temperature for 30 minutes before injecting. Patients should be cautioned to avoid injecting into an area that is already swollen or red. Any persistent reaction should be brought to the attention of the prescribing physician.
Allergic or other drug reactions: Inform patients about the signs and symptoms of allergic and other adverse drug reactions and the appropriate actions they should take if they experience any of these signs and symptoms. Inform patients with DIRA and their caregivers, that DIRA patients may have an increased risk of allergic reactions, particularly in the first several weeks of treatment and they should be monitored closely.
sobi
SWEDISH ORPAHN BIOVITRUM
Manufactured by:
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm, Sweden
U.S. License No. 1859
© Swedish Orphan Biovitrum AB (publ). All rights reserved.
The product, its production and/or its use may be covered by one or more US Patents, including US Patent Nos. 6,599,873 and 6,858,409 as well as other patents or patents pending.
|
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: December 2020 |
|
| Patient Information
Kineret® (KIN-eh-ret) (anakinra) injection, for subcutaneous use |
|
| What is Kineret?
Kineret is a prescription medicine called an interleukin-1 receptor antagonist (IL-1Ra) used to:
|
|
Do not use Kineret if you are allergic to:
|
|
Before you use Kineret, tell your healthcare provider about all of your medical conditions, including if you:
Kineret and other medicines may affect each other and cause serious side effects. Especially, tell your healthcare provider if you take certain other medicines that:
Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new prescription. |
|
How should I use Kineret?
|
|
| What are the possible side effects of Kineret?
Kineret may cause serious side effects, including:
The most common side effects of Kineret include:
|
|
|
|
|
|
| |
Most injection site reactions are mild, happen early during treatment, and last about 14 to 28 days. Injection site reactions have been observed less frequently in people with NOMID.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of Kineret. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store KINERET?
|
|
| General information about the safe and effective use of KINERET.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. Do not use Kineret for a condition for which it was not prescribed. Do not give Kineret to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about Kineret that is written for health professionals. |
|
| What are the ingredients in KINERET?
Active ingredient: anakinra Inactive ingredients: anhydrous citric acid, disodium EDTA, polysorbate 80, and sodium chloride in Water for Injection, USP Manufactured by: Swedish Orphan Biovitrum AB (publ) SE-112 76 Stockholm, Sweden License No. 1859 © Swedish Orphan Biovitrum AB (publ). All rights reserved. For more information, go to www.kineretrx.com or call 1-866-547-0644. |
|
Instructions for Use
Kineret® (KIN-eh-ret)
(anakinra)
injection, for subcutaneous use
Read this Instructions for Use before you start using Kineret and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Supplies you will need to give your Kineret injection: See Figure A.
- 1 Kineret prefilled syringe
- 1 alcohol wipe
- 1 dry sterile gauze or tissue
- 1 puncture-resistant sharps disposal container
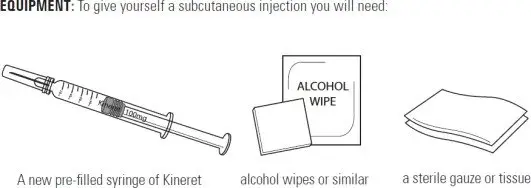 |
| Figure A |
Each Kineret dose comes in a prefilled glass syringe. There are 7 syringes in each new Kineret box, 1 for each day of the week. Use a new Kineret syringe each day. Use the Kineret prefilled syringe that matches the day of the week until all 7 syringes are used.
Setting up for your injection:
Step 1. Take the carton containing the prefilled syringes of Kineret out of the refrigerator. Remove the prefilled syringe from the box that matches the day of the week. Put the carton containing the remaining prefilled syringes back in the refrigerator.
Step 2. Find a clean, flat work surface, such as a table.
Step 3. Check the expiration date (EXP) on the syringe label. See Figure B.
If the expiration date has passed, do not use the syringe. Call your pharmacist or call 1-866-773-5274 for assistance.
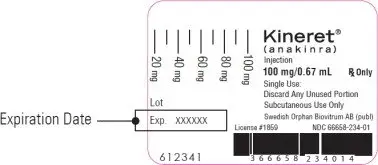 |
| Figure B |
Step 4. Take the Kineret prefilled syringe out of the refrigerator and leave it in room temperature for 30 minutes before your injection.
Make sure the liquid medicine in the prefilled syringe is clear and colorless. It is normal to see a small amount of tiny particles that are white, or that you can see through. Do not inject the medicine if it is cloudy or discolored, or has large or colored particles. Call your healthcare provider or pharmacist if you have any questions about your Kineret prefilled syringe.
Step 5. Gather all the supplies you will need for your injection.
Step 6. Wash your hands with soap and warm water.
Preparing your correct dose of Kineret:
- Preparing a 100mg dose of Kineret:
- Hold the syringe barrel and pull the cover straight off the needle. See Figure C. Do not touch the needle or push the plunger. Throw away the needle cover.
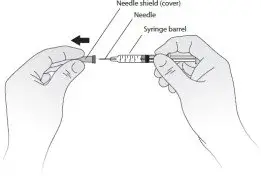
Figure C - You may notice small air bubbles in the prefilled syringe. You do not have to remove the air bubbles before injecting. Injecting the solution with the air bubbles is harmless.
- Carefully place the barrel of the syringe on the table until you are ready to inject. Do not let the needle touch the table. Do not recap the needle.
- Hold the syringe barrel and pull the cover straight off the needle. See Figure C. Do not touch the needle or push the plunger. Throw away the needle cover.
- How to prepare a dose of Kineret less than 100 mg:
- Hold the syringe in 1 hand with the needle pointing straight upwards. See Figure D. Put your thumb on the plunger rod and push slowly until you see a tiny liquid drop at the tip of the needle.
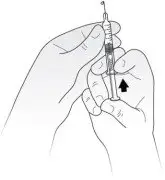
Figure D Turn the syringe so that the needle is now pointing downwards. Place a sterile gauze or tissue on a flat surface and hold the syringe above it with the needle pointing towards the gauze or tissue. See Figure E. Make sure the needle does not touch the gauze or tissue.
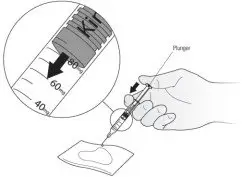
Figure E - Put your thumb on the plunger rod and push slowly until you can see that the top of the plunger has reached the correct number for your Kineret dose your healthcare provider has prescribed. The liquid that was pushed out of the needle will be absorbed by the gauze or tissue. See Figure E.
- If you are not able to select the correct dose of Kineret, throw away the syringe and use a new one.
- Carefully place the barrel of the syringe on the table until you are ready to inject. Do not let the needle touch the table. Do not recap the needle.
- Hold the syringe in 1 hand with the needle pointing straight upwards. See Figure D. Put your thumb on the plunger rod and push slowly until you see a tiny liquid drop at the tip of the needle.
Selecting and preparing the injection site:
Step 7. Choose an injection site. See Figure F.
Recommended injection sites for adults and children include:
- outer area of the upper arms
- abdomen (except the 2-inch area around the belly button)
- front of the middle thighs
- upper outer areas of the buttocks
| Adult | Child |
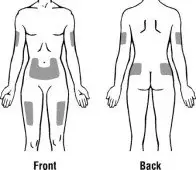 | 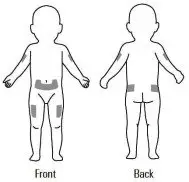 |
| Figure F | |
Choose a new site each time you use Kineret. Choosing a new site may help avoid soreness at 1 site. Do not inject Kineret into an area of skin that is tender, red, bruised, swollen, or hard. Avoid areas of skin with scars or stretch marks. Do not inject Kineret close to a vein that you can see under the surface of your skin.
- Clean your injection site with an alcohol swab. Let the area dry completely.
Giving your injection:
Step 8. Gently pinch a fold of skin at the cleaned injection site.
Step 9. With your other hand, hold the syringe like a pencil at a 45 degree to 90 degree angle to the skin. With a quick, dart-like motion insert the needle into the skin. See Figure G.
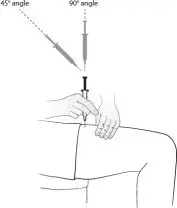 |
| Figure G |
Step 10. After the needle is inserted into the skin, slowly push the plunger all the way down to inject Kineret. See Figure H.
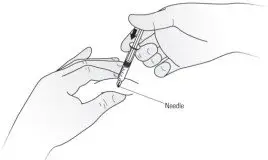 |
| Figure H |
Step 11. When the syringe is empty, pull the needle out of the skin while carefully keeping the needle at the same angle as inserted. See Figure I.
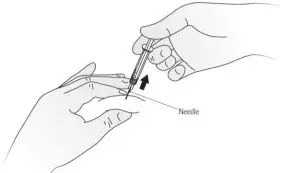 |
| Figure I |
Step 12. Place a dry cotton ball or gauze pad over the injection site and press for several seconds. See Figure J. Do not use an alcohol swab as it may cause stinging. If there is a little bleeding, you may cover the injection site with a small bandage.
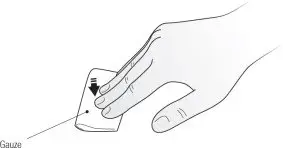 |
| Figure J |
Important Information about your Kineret prefilled syringe:
- Use each Kineret prefilled syringe only 1 time. Do not use a syringe more than 1 time. Do not recap a needle.
- You may not have to use all of the liquid medicine in the prefilled syringe. Your healthcare provider will show you how to find the correct dose of Kineret for you or your child.
- If you notice that some medicine is left in the prefilled syringe, do not inject again with the same prefilled syringe.
- If you drop a prefilled syringe, do not use it. The glass syringe may be broken, or the needle may be bent or dirty. Throw away the prefilled syringe and replace it with a new one. Take a new prefilled syringe from what would be the last day of the week in your current box. For example, if you start on Wednesday, the last day of the week in your series is Tuesday. After using all the remaining prefilled syringes in your current box, start your next box of Kineret prefilled syringes.
Disposal of your Kineret syringes:
- Put your used syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
- Throw away the wet gauze or tissue with your syringe and clean the table surface with a fresh swab.
This Patient Information and Instructions for Use has been approved by the U.S. Food and Drug Administration.
sobi
SWEDISH ORPHAN BIOVITRUM
Manufactured by:
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm, Sweden
License No. 1859
© Swedish Orphan Biovitrum AB (publ). All rights reserved. Revised Date: 12/2020
Principal Display Panel - Carton Label
4 x 7 Syringe Dispensing Packs (28 x 100 mg/0.67 mL Prefilled Glass Syringes) NDC 66658-234-28
sobi
Kineret®
(anakinra)
Injecton
100 mg/0.67 mL
Single Use: Discard Any Unused Portion
Subcutaneous Use Only
Graduated Prefilled Glass Syringes with 29 Gauge Needles
Sterile Solution - No Preservative
Manufactured by Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm, Sweden
License # 1859 Rx Only

Principal Display Panel - Carton Label
(Seven x 100 mg/0.67 mL Prefilled Glass Syringes) NDC 66658-234-07
sobi
Kineret®
(anakinra)
Injecton
100 mg/0.67 mL
Single Use: Discard Any Unused Portion
Subcutaneous Use Only
Graduated Prefilled Glass Syringes with 29 Gauge Needles
Sterile Solution - No Preservative
Refrigerate at 2° to 8°C (36° to 46°F). Do Not Freeze or Shake.
Protect from Light.
Rx Only
Manufactured by Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm, Sweden
License # 1859

| KINERET
anakinra injection, solution |
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
| Labeler - Swedish Orphan Biovitrum AB (publ) (354010589) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Boehringer Ingelheim Pharma GmbH & Co KG (BIP, an affiliate to BI RCV), Germany | 340700520 | ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Boehringer Ingelheim RCV, Austria | 300010883 | API MANUFACTURE(66658-234) , ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Charles River Laboratories Edinburgh Ltd., UK | 226613323 | ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Charles River Laboratories Edinburgh Ltd., UK | 296499353 | ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Charles River Laboratories Inc. USA | 078495006 | ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Eurofins Biolab S.r.l., Italy | 429117112 | ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Hospira Zagreb d.o.o., Croatia | 500625201 | ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon Italia S.p.A., Italy | 338336589 | MANUFACTURE(66658-234) , ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Health AB, Sweden | 354433591 | MANUFACTURE(66658-234) , ANALYSIS(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Rechon Life Science AB, Sweden | 775207769 | LABEL(66658-234) , PACK(66658-234) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Swedish Orphan Biovitrum AB (publ), Sweden | 776024627 | MANUFACTURE(66658-234) | |
