Drug Detail:Lupkynis (Voclosporin [ voe-kloe-spor-in ])
Drug Class: Calcineurin inhibitors
Highlights of Prescribing Information
LUPKYNIS (voclosporin) capsules, for oral use
Initial U.S. Approval: 2021
WARNING: MALIGNANCIES AND SERIOUS INFECTIONS
See full prescribing information for complete boxed warning.
Increased risk for developing serious infections and malignancies with LUPKYNIS or other immunosuppressants that may lead to hospitalization or death. (
5.1,
5.2)
Indications and Usage for Lupkynis
LUPKYNIS is a calcineurin-inhibitor immunosuppressant indicated in combination with a background immunosuppressive therapy regimen for the treatment of adult patients with active lupus nephritis (LN). ( 1, 14)
Limitations of Use: Safety and efficacy of LUPKYNIS have not been established in combination with cyclophosphamide. Use of LUPKYNIS is not recommended in this situation.
Lupkynis Dosage and Administration
Administration:
- LUPKYNIS must be swallowed whole on an empty stomach. ( 2.1)
- Administer consistently as close to a 12-hour schedule as possible, and with at least 8 hours between doses. ( 2.1)
- If a dose is missed, instruct the patient to take it as soon as possible within 4 hours after missing the dose. Beyond the 4-hour time frame, instruct the patient to wait until the usual scheduled time to take the next regular dose. Instruct the patient not to double the next dose. ( 2.1)
- Instruct patients to avoid eating grapefruit or drinking grapefruit juice while taking LUPKYNIS. ( 2.1, 7.1)
Dosage Recommendations:
- Before initiating LUPKYNIS, establish an accurate baseline estimated glomerular filtration rate (eGFR) and check blood pressure (BP).
- Use of LUPKYNIS is not recommended in patients with a baseline eGFR ≤45 mL/min/1.73 m 2 unless the benefit exceeds the risk; these patients may be at increased risk for acute and/or chronic nephrotoxicity. ( 2.2, 5.3)
- Do not initiate LUPKYNIS in patients with baseline BP >165/105 mmHg or with hypertensive emergency. ( 2.2, 5.4)
- Recommended starting dose: 23.7 mg orally, twice a day. ( 2.3)
- Use LUPKYNIS in combination with mycophenolate mofetil (MMF) and corticosteroids. ( 2.3)
- Modify the LUPKYNIS dose based on eGFR (
2.3,
5.3):
- Assess eGFR every two weeks for the first month, and every four weeks thereafter.
- If eGFR <60 mL/min/1.73 m 2 and reduced from baseline by >20% and <30%, reduce the dose by 7.9 mg twice a day. Re-assess eGFR within two weeks; if eGFR is still reduced from baseline by >20%, reduce the dose again by 7.9 mg twice a day.
- If eGFR <60 mL/min/1.73 m 2 and reduced from baseline by ≥30%, discontinue LUPKYNIS. Re-assess eGFR within two weeks; consider re-initiating LUPKYNIS at a lower dose (7.9 mg twice a day) only if eGFR has returned to ≥80% of baseline.
- For patients that had a decrease in dose due to eGFR, consider increasing the dose by 7.9 mg twice a day for each eGFR measurement that is ≥80% of baseline; do not exceed the starting dose.
- Monitor blood pressure every two weeks for the first month after initiating LUPKYNIS, and as clinically indicated thereafter. For patients with BP >165/105 mmHg or with hypertensive emergency, discontinue LUPKYNIS and initiate antihypertensive therapy. ( 2.3, 5.4)
- If the patient has not experienced therapeutic benefit by 24 weeks, consider discontinuation of LUPKYNIS. ( 2.3)
- Consider the risks and benefits of LUPKYNIS treatment beyond one year in light of the patient’s treatment response and risk of worsening nephrotoxicity. ( 2.3, 5.3)
Dosage Adjustments:
- Patients with severe renal impairment: the recommended dose is 15.8 mg twice daily. ( 2.4, 8.6)
- Patients with mild and moderate hepatic impairment: the recommended dose is 15.8 mg twice daily. ( 2.4, 8.7)
Dosage Forms and Strengths
Capsules: 7.9 mg ( 3)
Contraindications
- Patients concomitantly using strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin). ( 4)
- Known serious or severe hypersensitivity reaction to LUPKYNIS or any of its excipients. ( 4)
Warnings and Precautions
- Nephrotoxicity (acute and/or chronic): May occur due to LUPKYNIS or concomitant nephrotoxic drugs. Monitor renal function; consider dosage reduction. ( 5.3)
- Hypertension: May require antihypertensive therapy; monitor relevant drug interactions. ( 5.4)
- Neurotoxicity: Including risk of posterior reversible encephalopathy syndrome (PRES); monitor for neurologic abnormalities; reduce dosage or discontinue LUPKYNIS. ( 5.5)
- Hyperkalemia: Risk may be increased with other agents associated with hyperkalemia; monitor serum potassium levels. ( 5.6)
- QT Prolongation: Consider obtaining electrocardiograms and monitoring electrolytes in patients at high risk. ( 5.7)
- Immunizations: Avoid live vaccines. ( 5.8)
- Pure Red Cell Aplasia: Consider discontinuation. ( 5.9)
Adverse Reactions/Side Effects
The most commonly reported adverse reactions (≥3%) were: glomerular filtration rate decreased, hypertension, diarrhea, headache, anemia, cough, urinary tract infection, abdominal pain upper, dyspepsia, alopecia, renal impairment, abdominal pain, mouth ulceration, fatigue, tremor, acute kidney injury, and decreased appetite. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Aurinia Pharmaceuticals at 1-833-672-0028 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Moderate CYP3A4 inhibitors: Reduce LUPKYNIS daily dosage to 15.8 mg in the morning and 7.9 mg in the evening. ( 2.5, 7.1, 12.3)
- Strong and moderate CYP3A4 inducers: Avoid co-administration. ( 7.1, 12.3)
- Certain P-gp substrates: Reduce dosage of certain P-gp substrates with a narrow therapeutic window when co-administered with LUPKYNIS. ( 7.2, 12.3)
Use In Specific Populations
- Pregnancy: May cause fetal harm. ( 8.1)
- Lactation: Advise not to breastfeed. ( 8.2)
- Renal Impairment: Use of LUPKYNIS is not recommended in patients with a baseline eGFR ≤45 mL/min/1.73 m 2 unless the benefit exceeds the risk. If used in patients with severe renal impairment at baseline, LUPKYNIS should be used at a reduced dose. ( 2.4, 8.6)
- Hepatic Impairment:
- Mild and moderate hepatic impairment: Dose reduction is required.
- Severe hepatic impairment: Avoid LUPKYNIS use. ( 2.4, 8.7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2021
Full Prescribing Information
WARNING: MALIGNANCIES AND SERIOUS INFECTIONS
Increased risk for developing malignancies and serious infections with LUPKYNIS or other immunosuppressants that may lead to hospitalization or death [see Warnings and Precautions ( 5.1, 5.2)].
1. Indications and Usage for Lupkynis
LUPKYNIS is indicated in combination with a background immunosuppressive therapy regimen [see Clinical Studies ( 14)] for the treatment of adult patients with active lupus nephritis (LN).
Limitations of Use: Safety and efficacy of LUPKYNIS have not been established in combination with cyclophosphamide. Use of LUPKYNIS is not recommended in this situation.
2. Lupkynis Dosage and Administration
2.1 Important Administration Instructions
- LUPKYNIS capsules must be swallowed whole and must not be opened, crushed, or divided.
- LUPKYNIS should be taken on an empty stomach consistently as close to a 12-hour schedule as possible, and with a minimum of 8 hours between doses.
- If a dose is missed, instruct the patient to take it as soon as possible within 4 hours after missing the dose. Beyond the 4-hour time frame, instruct the patient to wait until the usual scheduled time to take the next regular dose. Instruct the patient not to double the next dose.
- Instruct patients to avoid eating grapefruit or drinking grapefruit juice while taking LUPKYNIS [see Drug Interactions ( 7.1)] .
2.2 Prior to Initiating LUPKYNIS Therapy
Establish an accurate baseline estimated glomerular filtration rate (eGFR). Use of LUPKYNIS is not recommended in patients with a baseline eGFR ≤45 mL/min/1.73 m 2 unless the benefit exceeds the risk; these patients may be at increased risk for acute and/or chronic nephrotoxicity [see Warnings and Precautions ( 5.3)] .
Check blood pressure (BP) at baseline. Do not initiate LUPKYNIS in patients with BP >165/105 mmHg or with hypertensive emergency [see Warnings and Precautions ( 5.4)] .
2.3 Dosage Recommendations
The recommended starting dose of LUPKYNIS is 23.7 mg twice a day.
Use LUPKYNIS in combination with mycophenolate mofetil (MMF) and corticosteroids [see Clinical Studies ( 14)] .
Safety and efficacy of LUPKYNIS have not been established in combination with cyclophosphamide. Use of LUPKYNIS is not recommended in this situation.
Dosage of LUPKYNIS is based on the patient’s eGFR. Modify LUPKYNIS dosage based on eGFR [see Warnings and Precautions ( 5.3)] :
- Assess eGFR every two weeks for the first month, and every four weeks thereafter.
- If eGFR <60 mL/min/1.73 m 2 and reduced from baseline by >20% and <30%, reduce the dose by 7.9 mg twice a day. Re-assess eGFR within two weeks; if eGFR is still reduced from baseline by >20%, reduce the dose again by 7.9 mg twice a day.
- If eGFR <60 mL/min/1.73 m 2 and reduced from baseline by ≥30%, discontinue LUPKYNIS. Re-assess eGFR within two weeks; consider re-initiating LUPKYNIS at a lower dose (7.9 mg twice a day) only if eGFR has returned to ≥80% of baseline.
- For patients that had a decrease in dose due to eGFR, consider increasing the dose by 7.9 mg twice a day for each eGFR measurement that is ≥80% of baseline; do not exceed the starting dose.
Monitor blood pressure every two weeks for the first month after initiating LUPKYNIS, and as clinically indicated thereafter [see Warnings and Precautions ( 5.4)] . For patients with BP >165/105 mmHg or with hypertensive emergency, discontinue LUPKYNIS and initiate antihypertensive therapy.
If the patient does not experience therapeutic benefit by 24 weeks, consider discontinuation of LUPKYNIS.
Safety and efficacy have not been established beyond one year [see Clinical Trials Experience ( 6.1) and Clinical Studies ( 14)] . Consider the risks and benefits of longer durations of treatment in light of the patient’s treatment response and risk of worsening nephrotoxicity [see Warnings and Precautions ( 5.3)] .
2.4 Dosage Recommendations in Patients with Renal and Hepatic Impairment
Use of LUPKYNIS is not recommended in patients with a baseline eGFR ≤45 mL/min/1.73 m 2 unless the benefit exceeds the risk; LUPKYNIS has not been studied in patients with a baseline eGFR ≤45 mL/min/1.73 m 2. If used in patients with severe renal impairment at baseline, the recommended starting dose is 15.8 mg twice a day [see Use in Specific Populations ( 8.6) and Clinical Pharmacology ( 12.3)] .
In patients with mild and moderate hepatic impairment (Child-Pugh A and Child-Pugh B), the recommended dose is 15.8 mg twice daily. LUPKYNIS is not recommended to be used in patients with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations ( 8.7) and Clinical Pharmacology ( 12.3)] .
2.5 Dosage Adjustments due to Drug Interactions
When co-administering LUPKYNIS with moderate CYP3A4 inhibitors (e.g., verapamil, fluconazole, diltiazem), reduce LUPKYNIS daily dosage to 15.8 mg in the morning and 7.9 mg in the evening. No dose adjustment of LUPKYNIS is recommended when LUPKYNIS is co-administered with mild CYP3A4 inhibitors [see Drug Interactions ( 7.1) and Clinical Pharmacology ( 12.3)] .
3. Dosage Forms and Strengths
Capsules: 7.9 mg (voclosporin) oval, pink/orange in color, imprinted on one side with VCS in white ink.
4. Contraindications
LUPKYNIS is contraindicated in:
- Patients concomitantly using strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin) because these medications can significantly increase exposure to LUPKYNIS which may increase the risk of acute and/or chronic nephrotoxicity [see Warnings and Precautions ( 5.3), Drug Interactions ( 7.1), and Pharmacokinetics ( 12.3)] .
- Patients who have had a known serious or severe hypersensitivity reaction to LUPKYNIS or any of its excipients.
5. Warnings and Precautions
5.1 Lymphoma and Other Malignancies
Immunosuppressants, including LUPKYNIS, increase the risk of developing lymphomas and other malignancies, particularly of the skin. The risk appears to be related to the intensity and duration of immunosuppression rather than to the use of any specific agent. Examine patients for skin changes and advise to avoid or limit sun exposure and to avoid artificial UV light (tanning beds, sun lamps) by wearing protective clothing and using a broad spectrum sunscreen with a high protection factor (SPF 30 or higher).
5.2 Serious Infections
Immunosuppressants, including LUPKYNIS, increase the risk of developing bacterial, viral, fungal, and protozoal infections, including opportunistic infections. These infections may lead to serious, including fatal, outcomes. Viral infections reported include cytomegalovirus and herpes zoster infections.
Monitor for the development of infection. Consider the benefits and risks for the individual patient; use the lowest effective dose needed to maintain response.
5.3 Nephrotoxicity
LUPKYNIS, like other calcineurin-inhibitors, can cause acute and/or chronic nephrotoxicity. Nephrotoxicity was reported in clinical trials [see Adverse Reactions ( 6.1)] . Monitor eGFR regularly during treatment, and consider dose reduction or discontinuation in patients with decreases in eGFR from baseline [see Dosage and Administration ( 2.3)] ; persistent decrease of eGFR should be evaluated for chronic calcineurin-inhibitor nephrotoxicity.
Consider the risks and benefits of LUPKYNIS treatment in light of the patient’s treatment response and risk of worsening nephrotoxicity, including in the following situations:
- Longer treatment duration beyond one year. Safety and efficacy of LUPKYNIS have not been established beyond one year [see Adverse Reactions ( 6.1) and Clinical Studies ( 14)] .
- Co-administration with drugs associated with nephrotoxicity. The risk for acute and/or chronic nephrotoxicity is increased when LUPKYNIS is concomitantly administered with drugs associated with nephrotoxicity.
5.4 Hypertension
Hypertension is a common adverse reaction of LUPKYNIS therapy and may require antihypertensive therapy [see Adverse Reactions ( 6.1)] . Some antihypertensive drugs can increase the risk for hyperkalemia [see Warnings and Precautions ( 5.7)] . Certain calcium-channel blocking agents (verapamil and diltiazem) may increase voclosporin blood concentrations and require dosage reduction of LUPKYNIS [see Dosage and Administration ( 2.5) and Drug Interactions ( 7.2)] .
Monitor blood pressure regularly during treatment and treat new-onset hypertension and exacerbations of pre-existing hypertension. If a patient experiences increases in blood pressure that cannot be managed with dose reduction of LUPKYNIS or other appropriate medical intervention, consider discontinuation of LUPKYNIS [see Dosage and Administration ( 2.3)] .
5.5 Neurotoxicity
LUPKYNIS, like other calcineurin-inhibitors, may cause a spectrum of neurotoxicities [see Adverse Reactions ( 6.1)] . The most severe neurotoxicities include posterior reversible encephalopathy syndrome (PRES), delirium, seizure, and coma; others include tremors, paresthesias, headache, mental status changes, and changes in motor and sensory functions. Monitor for neurologic symptoms and consider dosage reduction or discontinuation of LUPKYNIS if neurotoxicity occurs.
5.6 Hyperkalemia
Hyperkalemia, which may be serious and require treatment, has been reported with calcineurin-inhibitors including LUPKYNIS [see Adverse Reactions ( 6.1)] . Concomitant use of agents associated with hyperkalemia (e.g., potassium-sparing diuretics, ACE inhibitors, angiotensin receptor blockers) may increase the risk for hyperkalemia. Monitor serum potassium levels periodically during treatment.
5.7 QTc Prolongation
LUPKYNIS prolongs the QTc interval in a dose-dependent manner after single dose administration at a dose higher than the recommended lupus nephritis therapeutic dose [see Clinical Pharmacology ( 12.2)] . The use of LUPKYNIS in combination with other drugs that are known to prolong QTc may result in clinically significant QT prolongation.
Certain circumstances may increase the risk of the occurrence of torsade de pointes and/or sudden death in association with the use of drugs that prolong the QTc interval, including (1) bradycardia; (2) hypokalemia or hypomagnesemia; (3) concomitant use of other drugs that prolong the QTc interval; and (4) presence of congenital prolongation of the QT interval.
5.8 Immunizations
Avoid the use of live attenuated vaccines during treatment with LUPKYNIS (e.g., intranasal influenza, measles, mumps, rubella, oral polio, BCG, yellow fever, varicella, and TY21a typhoid vaccines).
Inactivated vaccines noted to be safe for administration may not be sufficiently immunogenic during treatment with LUPKYNIS.
5.9 Pure Red Cell Aplasia
Cases of pure red cell aplasia (PRCA) have been reported in patients treated with another calcineurin-inhibitor immunosuppressant. All of these patients reported risk factors for PRCA such as parvovirus B19 infection, underlying disease, or concomitant medications associated with PRCA. A mechanism for calcineurin-inhibitor-induced PRCA has not been elucidated. If PRCA is diagnosed, consider discontinuation of LUPKYNIS.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Lymphoma and Other Malignancies [see Warnings and Precautions ( 5.1)]
- Serious Infections [see Warnings and Precautions ( 5.2)]
- Nephrotoxicity due to LUPKYNIS and Drug Interactions [see Warnings and Precautions ( 5.3)]
- Hypertension [see Warnings and Precautions ( 5.4)]
- Neurotoxicity [see Warnings and Precautions ( 5.5)]
- Hyperkalemia [see Warnings and Precautions ( 5.6)]
- QTc Prolongation [see Warnings and Precautions ( 5.7)]
- Pure Red Cell Aplasia [see Warnings and Precautions ( 5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 355 patients with LN were treated with voclosporin in the Phase 2 and 3 clinical studies with 224 exposed for at least 48 weeks.
Patients in Study 1 were randomized to LUPKYNIS 23.7 mg twice a day or placebo [see Clinical Studies ( 14)] . Patients in Study 2 were randomized to LUPKYNIS 23.7 mg twice a day, voclosporin 39.5 mg twice a day, or placebo.
Patients received background treatment with MMF 2 g daily and an IV bolus of corticosteroids followed by a pre-specified oral corticosteroid taper dosing schedule; LUPKYNIS dosing was adjusted based on eGFR and BP.
A total of 267 patients received at least 1 dose of LUPKYNIS 23.7 mg twice a day with 184 exposed for at least 48 weeks. A total of 88 patients received at least 1 dose of voclosporin 39.5 mg twice a day with 40 exposed for 48 weeks.
Table 1 lists common adverse reactions occurring in at least 3% of patients receiving LUPKYNIS and at an incidence at least 2% greater than placebo in Studies 1 and 2.
|
||
| Adverse Reaction | LUPKYNIS
23.7 mg twice a day (n=267) | Placebo
(n=266) |
|---|---|---|
| Glomerular filtration rate decreased * | 26% | 9% |
| Hypertension | 19% | 9% |
| Diarrhea | 19% | 13% |
| Headache | 15% | 8% |
| Anemia | 12% | 6% |
| Cough | 11% | 2% |
| Urinary tract infection | 10% | 6% |
| Abdominal pain upper | 7% | 2% |
| Dyspepsia | 6% | 3% |
| Alopecia | 6% | 3% |
| Renal Impairment | 6% | 3% |
| Abdominal pain | 5% | 2% |
| Mouth ulceration | 4% | 1% |
| Fatigue | 4% | 1% |
| Tremor | 3% | 1% |
| Acute kidney injury | 3% | 1% |
| Decreased appetite | 3% | 1% |
Other adverse reactions reported in less than 3% of patients in the LUPKYNIS 23.7 mg group and at a 2% higher rate than in the placebo group through 48/52 weeks included gingivitis and hypertrichosis.
The integrated LN dataset is presented in the Specific Adverse Reactions section:
Placebo-controlled Studies: Studies 1 and 2 were integrated to represent safety through 48/52 weeks for placebo (n=266), LUPKYNIS 23.7 mg twice a day (n=267), and voclosporin 39.5 mg twice a day (n=88).
Exposure adjusted incidence rates were adjusted by study for all the adverse events reported in this section.
Specific Adverse Reactions
Infections
Infections were reported in 146 patients (107.4 per 100 patient-years) treated with placebo, 166 patients (135.2 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 58 patients (167.5 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. The most frequent infections were upper respiratory tract infections, urinary tract infections, viral upper respiratory tract infections, and herpes zoster.
Serious infections were reported in 27 patients (12.0 per 100 patient-years) treated with placebo, 27 patients (11.9 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 10 patients (14.4 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. The most frequent serious infections were pneumonia, gastroenteritis, and urinary tract infections.
Opportunistic infections were reported in 2 patients (0.9 per 100 patient-years) treated with placebo, 3 patients (1.3 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 1 patient (1.4 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. The most frequent opportunistic infections were cytomegalovirus chorioretinitis, cytomegalovirus infection, and herpes zoster cutaneous disseminated.
Nephrotoxicity
Glomerular filtration rate decreased was the most frequently reported adverse reaction, reported in 25 patients (11.3 per 100 patient-years) treated with placebo, 70 patients (37.1 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 27 patients (48.7 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. In patients treated with LUPKYNIS 23.7 mg twice a day, decreases in glomerular filtration rate occurred within the first 3 months of LUPKYNIS treatment in 50/70 (71%), with 39/50 (78%) resolved or improved following dose modification, and of those 25/39 (64%) resolved or improved within 1 month [see Dosage and Administration ( 2.3)] . Decreases in glomerular filtration rate resulted in permanent discontinuation of LUPKYNIS in 10/70 (14%), and resolved in 4/10 (40%) 3 months after treatment discontinuation.
Renal adverse reactions (defined as renal impairment, acute kidney injury, blood creatinine increased, azotemia, renal failure, oliguria, and proteinuria) were reported in 22 patients (9.5 per 100 patient-years) treated with placebo, 26 patients (11.3 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 11 patients (16.5 per 100 patient-years) treated with voclosporin 39.5 mg twice a day.
Serious renal adverse reactions were reported in 9 patients (3.7 per 100 patient-years) treated with placebo, 13 patients (5.6 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 0 patients (0 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. The most frequent serious renal adverse reactions were acute kidney injury and renal impairment.
Hypertension
Hypertension was reported in 23 patients (10.3 per 100 patient-years) treated with placebo, 51 patients (25.2 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 16 patients (26.0 per 100 patient-years) treated with voclosporin 39.5 mg twice a day.
Serious hypertension was reported in 1 patient (0.4 per 100 patient-years) treated with placebo, 5 patients (2.1 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 2 patients (2.8 per 100 patient-years) treated with voclosporin 39.5 mg twice a day.
Neurotoxicity
Nervous system disorders were reported in 44 patients (21.6 per 100 patient-years) treated with placebo, 74 patients (38.9 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 24 patients (42.5 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. The most frequent neurological adverse reactions were headache, tremor, dizziness, post herpetic neuralgia, migraine, paresthesia, hypoaesthesia, seizure, tension headache, and disturbance in attention.
Serious nervous system disorders were reported in 2 patients (0.9 per 100 patient-years) treated with placebo, 9 patients (3.9 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 3 patients (4.3 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. The most frequent serious neurological adverse reactions were headache, migraine, seizure, and posterior reversible encephalopathy syndrome.
Malignancy
Malignancies were reported in 0 patients (0 per 100 patient-years) treated with placebo, 4 patients (1.7 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 0 patients (0 per 100 patient-years) treated with voclosporin 39.5 mg twice a day. These consisted of single occurrences of stage 0 cervical carcinoma, skin neoplasm, pyoderma gangrenosum, and breast tumor excision.
Hyperkalemia
Hyperkalemia was reported in 2 patients (0.8 per 100 patient-years) treated with placebo, 5 patients (2.1 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 1 patient (1.4 per 100 patient-years) treated with voclosporin 39.5 mg twice a day.
QT Prolongation
QT prolongation was reported in 0 patients (0 per 100 patient-years) treated with placebo, 2 patients (0.9 per 100 patient-years) treated with LUPKYNIS 23.7 mg, and 1 patient (1.4 per 100 patient-years) treated with voclosporin 39.5 mg twice a day.
7. Drug Interactions
7.1 Effect of Other Drugs on LUPKYNIS
Strong and Moderate CYP3A4 Inhibitors
Voclosporin is a sensitive CYP3A4 substrate. Co-administration with strong or moderate CYP3A4 inhibitors increases voclosporin exposure [see Clinical Pharmacology ( 12.3)] , which may increase the risk of LUPKYNIS adverse reactions. Co-administration of LUPKYNIS with strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin) is contraindicated [see Contraindications ( 4)] . Reduce LUPKYNIS dosage when co-administered with moderate CYP3A4 inhibitors (e.g., verapamil, fluconazole, diltiazem) [see Dosage and Administration ( 2.4)] . Avoid food or drink containing grapefruit when taking LUPKYNIS.
Strong and Moderate CYP3A4 Inducers
Voclosporin is a sensitive CYP3A4 substrate. Co-administration with strong or moderate CYP3A4 inducers decreases voclosporin exposure [see Clinical Pharmacology ( 12.3)] , which may decrease the efficacy of LUPKYNIS. Avoid co-administration of LUPKYNIS with strong or moderate CYP3A4 inducers.
7.2 Effect of LUPKYNIS on Other Drugs
Certain P-gp Substrates
Voclosporin is a P-gp inhibitor. Co-administration of voclosporin increases exposure of P-gp substrates [see Clinical Pharmacology ( 12.3)] , which may increase the risk of adverse reactions of these substrates. For certain P-gp substrates with a narrow therapeutic window, reduce the dosage of the substrate as recommended in its prescribing information, if needed.
OATP1B1 Substrates
The effect of LUPKYNIS on OATP1B1 substrates (e.g., statins) has not been studied clinically. However, voclosporin is an OATP1B1 inhibitor in vitro, and information suggests an increase in the concentration of these substrates is possible [see Clinical Pharmacology ( 12.3)] . Monitor for adverse reactions of OATP1B1 substrates when used concomitantly with LUPKYNIS.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Avoid use of LUPKYNIS in pregnant women due to the alcohol content of the drug formulation. The available data on the use of LUPKYNIS in pregnant patients are insufficient to determine whether there is a drug-associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with systemic lupus erythematosus (SLE) (see Clinical Considerations) .
LUPKYNIS may be used in combination with a background immunosuppressive therapy regimen that includes MMF. MMF used in pregnant women and men whose female partners are pregnant can cause fetal harm (major birth defects and miscarriage). Refer to the MMF prescribing information for more information on its use during pregnancy.
In animal reproductive studies, oral administration of either voclosporin or a 50:50 mixture of voclosporin and its cis-isomer was embryocidal and fetocidal in rats and rabbits at doses 15- and 1-times, respectively, the maximum recommended human dose (MRHD) of 23.7 mg twice a day, based on drug exposure AUC. There were no treatment-related fetal malformations or variations. Additional findings of reduced placental and fetal body weights occurred in rabbits at 0.1 to 0.3-times the MRHD and in rats at higher drug exposures. Voclosporin was transferred across the placenta in pregnant rats. For rats, but not all doses in rabbits, these effects were associated with maternal toxicity consisting of reductions in body weight gain. Dystocia was evident in a pre- and postnatal study in rats, but there were no effects of voclosporin on postnatal growth and development (see Data) .
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnant women with SLE are at increased risk of adverse pregnancy outcomes, including worsening of the underlying disease, premature birth, miscarriage, and intrauterine growth restriction. Maternal LN increases the risk of hypertension and preeclampsia/eclampsia. Passage of maternal autoantibodies across the placenta may result in adverse neonatal outcomes, including neonatal lupus and congenital heart block.
Fetal/Neonatal adverse reactions
The formulation of LUPKYNIS contains alcohol (21.6 mg of dehydrated ethanol per capsule for a total daily dose of 129.4 mg/day). Published studies have demonstrated that alcohol is associated with fetal harm including central nervous system abnormalities, behavioral disorders and impaired intellectual development. There is no safe level of alcohol exposure in pregnancy; therefore, avoid use of LUPKYNIS in pregnant women.
Data
Animal Data
Voclosporin (90 to 95% trans-isomer) is the active ingredient in LUPKYNIS. Animal reproductive studies were primarily conducted with an approximate 50:50 mixture of voclosporin and its cis-isomer. Similarity of the toxicity effects of the 50:50 mixture and voclosporin was demonstrated in comparative toxicity studies with adult rats. Interconversion between cis and trans isomers was not detected with in vitro or in vivo studies.
In an embryofetal developmental study, pregnant rats were dosed orally, during the period of organogenesis from gestation days 6-17, with the 50:50 mixture of voclosporin and its cis-isomer, litter size was reduced due to increased fetal resorptions and deaths at drug exposures approximately 15-times the MRHD (on an AUC basis with a maternal oral dose of 25 mg/kg/day). Surviving fetuses had reduced placental weights and slightly reduced fetal weights. There were no treatment-related fetal malformations or variations with doses up to 15-times the MRHD, although reductions in ossification sites were observed in the metatarsal bones. This dose was associated with maternal toxicity based on decreased body weight gain. The no effect dose for both fetal and maternal effects occurred at a drug exposure approximately 7-times the MRHD (on an AUC basis with a maternal oral dose of 10 mg/kg/day).
Two embryofetal developmental studies were conducted in pregnant rabbits that received either the 50:50 mixture of voclosporin and its cis-isomer or voclosporin during the period of organogenesis from gestation days 6-18. Litter size was reduced due to increased fetal resorptions and deaths with 50:50 mixture at drug exposures approximately the MRHD (on an AUC basis with a maternal oral dose of 20 mg/kg/day). Increased resorptions were observed with voclosporin at 0.1-times the MRHD (on an AUC basis with a maternal dose of 20 mg/kg/day); however, litter size was not significantly affected. Decreased placental weights and fetal body weights were observed with the 50:50 mixture at doses 0.3-times the MRHD and higher (on an AUC basis with maternal oral doses of 10 mg/kg/day and higher). Decreased fetal body weights were observed with voclosporin at doses 0.1-times the MRHD and higher (on an AUC basis with maternal oral doses of 5 mg/kg/day and higher). There were no treatment-related malformations or variations. Both studies had reductions of ossification sites in the metacarpal bones with the 50:50 mixture at doses 2-times the MRHD, and the sternabrae and hyoid body and/or arches with voclosporin at doses 0.1-times the MRHD and higher. The high dose of 20 mg/kg/day 50:50 mixture or voclosporin was associated with maternal toxicity based on decreased body weight gains. These rabbit studies indicated that the toxicity of 50:50 mixture of voclosporin and its cis-isomer and voclosporin were qualitatively similar; however, voclosporin was more potent than the 50:50 mixture, consistent with its pharmacological potency. The no effect dose for the fetal effects of voclosporin occurred at a drug exposure approximately 0.01-times the MRHD (on an AUC basis with a maternal oral dose of 1 mg/kg/day).
In a pre-and post-natal developmental study, rats were dosed from gestation day 7 through lactation day 20 with a 50:50 mixture of voclosporin and its cis-isomer. Dystocia (delayed parturition) occurred at a dose 12-times the MRHD (on an AUC basis with a maternal oral dose of 25 mg/kg/day) that resulted in reductions of the mean number of total pups delivered and surviving pups per litter. This dose was associated with maternal toxicity based on decreased body weight gain. No adverse effects on dams or their pups were observed at doses 3-times the MRHD and lower (on an AUC basis with a maternal oral dose of 10 mg/kg/day). There were no effects on behavioral and physical development, or the reproductive performance of male or female pups. The no effect dose for delivery and pup survival was 10 mg/kg/day.
8.2 Lactation
Risk Summary
There are no available data on the presence of voclosporin in human milk, the effects on the breastfed infant, or the effects on milk production. Voclosporin is present in milk of lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Given the serious adverse reactions seen in adult patients treated with LUPKYNIS such as increased risk of serious infections, advise patients that breastfeeding is not recommended during treatment and for at least 7 days after the last dose of LUPKYNIS (approximately 6 elimination half-lives).
8.3 Females and Males of Reproductive Potential
LUPKYNIS may be used in combination with a background immunosuppressive therapy regimen that includes MMF. If LUPKYNIS is administered with MMF, the information for MMF regarding pregnancy testing, contraception, and infertility also applies to this combination regimen. Refer to MMF prescribing information for additional information.
8.4 Pediatric Use
The safety and efficacy of LUPKYNIS in pediatric patients has not been established.
8.5 Geriatric Use
Clinical studies of LUPKYNIS did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Use of LUPKYNIS is not recommended in patients with a baseline eGFR ≤45 mL/min/1.73 m 2 unless the benefit exceeds the risk. If used in patients with severe renal impairment at baseline, LUPKYNIS should be used at a reduced dose [see Dosage and Administration ( 2.4)] . No dosage adjustment is recommended in patients with mild or moderate renal impairment at baseline [see Clinical Pharmacology ( 12.3)] . Monitor eGFR closely.
After initiating therapy, dosing adjustments should be made based on eGFR [see Dosage and Administration ( 2.3)] .
10. Overdosage
Cases of accidental overdose have been reported with LUPKYNIS; symptoms may include tremor, headache, nausea and vomiting, infections, tachycardia, urticaria, lethargy, and increases in blood urea nitrogen, serum creatinine, and alanine aminotransferase levels.
No specific antidote to LUPKYNIS therapy is available. If overdose occurs, general supportive measures and symptomatic treatment should be conducted, including stopping treatment with LUPKYNIS and assessing blood urea nitrogen, serum creatinine, eGFR and alanine aminotransferase levels.
Consider contacting a poison center (1-800-222-1222) or medical toxicologist for advice and review of overdosage management recommendations.
11. Lupkynis Description
LUPKYNIS (voclosporin) capsules, a calcineurin-inhibitor immunosuppressant, is available for administration as soft gelatin capsules containing 7.9 mg voclosporin per capsule. Inactive ingredients include alcohol, Vitamin E polyethylene glycol succinate (NF), polysorbate 40 (NF), medium-chain triglycerides (NF), gelatin, sorbitol, glycerin, iron oxide yellow, iron oxide red, titanium dioxide, and water.
Voclosporin (90 to 95% trans-isomer) is the active ingredient in LUPKYNIS. Chemically, voclosporin is named: Cyclo{{(6E)-(2S,3R,4R)-3-hydroxy-4-methyl-2-(methylamino)-6,8-nonadienoyl}-L-2-aminobutyryl-N-methyl-glycyl-N-methyl-L-leucyl-L-valyl-N-methyl-L-leucyl-L-alanyl-D-alanyl-N-methyl-L-leucyl-N-methyl-L-leucyl-N-methyl-L-valyl}. The chemical structure of voclosporin is:
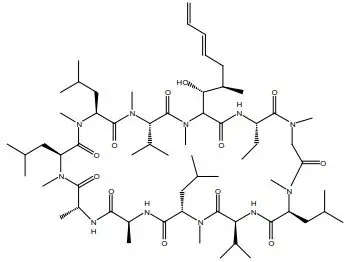
Voclosporin has an empirical formula of C 63H 111N 11O 12 and a molecular weight of 1214.6 g/mole. It appears as white to off-white solid matter. At ambient temperature, voclosporin is freely soluble in acetone, acetonitrile, ethanol, and methanol, and practically insoluble in heptanes (USP). Voclosporin is practically insoluble (less than 0.1 g/L at 20ºC) in water and melts above 144ºC with decomposition.
12. Lupkynis - Clinical Pharmacology
12.1 Mechanism of Action
LUPKYNIS is a calcineurin-inhibitor immunosuppressant. The mechanism of voclosporin suppression of calcineurin has not been fully established. Activation of lymphocytes involves an increase in intracellular calcium concentrations that bind to the calcineurin regulatory site and activate calmodulin binding catalytic subunit and through dephosphorylation activates the transcription factor, Nuclear Factor of Activated T-Cell Cytoplasmic (NFATc). The immunosuppressant activity results in inhibition of lymphocyte proliferation, T-cell cytokine production, and expression of T-cell activation surface antigens.
Studies in animal models also support a non-immunological role for calcineurin inhibition in kidney function to stabilize actin cytoskeleton and stress fibers in podocytes leading to increased podocyte integrity in glomeruli.
12.2 Pharmacodynamics
Calcineurin inhibition
Concentration-dependent calcineurin inhibition, measured as the percent of maximum calcineurin inhibition, was observed after oral administration of voclosporin twice daily in healthy volunteers. There is little or no lag time between the time to maximum drug concentration and the time to maximum calcineurin inhibition. Voclosporin inhibits calcineurin in a dose-dependent manner up to a maximum dose of 1.0 mg/kg.
Cardiac Electrophysiology
In a randomized, placebo- and active-controlled (moxifloxacin 400 mg), single dose study with parallel study design, dose-dependent QT prolonging effect was detected with LUPKYNIS in the dose range of 0.5–4.5 mg/kg (up to 9-fold coverage of the therapeutic exposure). Dose-dependent QT prolongation effect was observed with a time to maximum QTc increase occurring at 4–6-hour postdose across different dose levels. The maximum mean placebo-adjusted changes of QTcF from baseline after LUPKYNIS 0.5 mg/kg, 1.5 mg/kg, 3.0 mg/kg, and 4.5 mg/kg dose were 6.4 msec, 17.5 msec, 25.7 msec, and 34.6 msec, respectively.
In a separate, randomized, placebo-controlled, crossover study in 31 healthy subjects, an absence of large mean increases (i.e., >20 msec) was observed following 7 days of treatment with LUPKYNIS at 0.3, 0.5 and 1.5 mg/kg twice daily (approximately 6-fold coverage of the therapeutic exposure).
The mechanism for the QT prolonging effect as observed in the single-dose and multiple-dose studies is unknown.
12.3 Pharmacokinetics
The whole blood voclosporin pharmacokinetics increase in a greater than dose-proportional manner over the therapeutic range. With a twice daily dosing regimen, voclosporin achieves steady-state after 6 days and the accumulation is approximately 2-fold.
Absorption
The median T max of voclosporin is 1.5 hours (1 to 4 hours) when administered on an empty stomach.
Effect of Food
Co-administration of voclosporin with food decreased both the rate and extent of absorption: with either low- or high-fat meals, C max and AUC of voclosporin were reduced by 29% to 53% and 15% to 25%, respectively.
Distribution
The apparent volume of distribution (V ss/F) of voclosporin is 2,154 L.
Protein binding of voclosporin is 97%. Voclosporin partitions extensively into red blood cells and distribution between whole blood and plasma is concentration- and temperature-dependent.
Elimination
The mean apparent clearance at steady-state (CL ss/F) of voclosporin is 63.6 L/h, and mean terminal half-life (t 1/2) is approximately 30 hours (24.9 to 36.5 hours).
Metabolism
Voclosporin is predominantly metabolized by CYP3A4. Voclosporin is the major circulating component and the pharmacologic activity is mainly attributed to the parent molecule. A major metabolite in human whole blood represented 16.7% of total exposure and is about 8-fold less potent than the parent molecule.
Excretion
Following single oral administration of radiolabeled voclosporin 70 mg, 92.7% of the radioactivity was recovered in feces (including 5% as unchanged voclosporin), and 2.1% was recovered in urine (including 0.25% as unchanged voclosporin).
Specific Populations
There were no clinically significant differences in the pharmacokinetics of voclosporin based on age (18 to 66 years), sex, race (Caucasian, Black, Asian, other), or body weight (37 to 133 kg).
Patients with Renal Impairment
Voclosporin C max and AUC were similar in volunteers with mild (CL Cr 60 to 89 mL/min as estimated by Cockcroft-Gault) and moderate (CL Cr 30 to 59 mL/min) renal impairment compared to volunteers with normal renal function (CL Cr ≥90 mL/min). The C max and AUC increased 1.5- and 1.7-fold, respectively, in volunteers with severe renal impairment (CL Cr <30 mL/min). The effect of end-stage renal disease (ESRD) with or without hemodialysis on the pharmacokinetics of voclosporin is unknown.
Patients with Hepatic Impairment
Voclosporin C max and AUC increased approximately 1.5- to 2.0-fold in volunteers with mild hepatic impairment (Child-Pugh A) or moderate hepatic impairment (Child-Pugh B). The effect of severe hepatic impairment (Child-Pugh C) on the pharmacokinetics of voclosporin is unknown.
Drug Interaction Studies
Effect of Other Drugs on LUPKYNIS
The effect of co‑administered drugs on the exposure of voclosporin is shown in Table 2.
|
Notes: CI = Confidence interval; CYP = Cytochrome P450; P-gp = P-glycoprotein; QD = once daily; TID = every 8 hours. 1 Ratios for C max and AUC compare co-administration of the medication with voclosporin vs. administration of voclosporin alone. |
|||
| Co-administered Drug | Regimen of Co-administered
Drug | Ratio
(90% CI) 1 |
|
|---|---|---|---|
| C max | AUC | ||
| Ketoconazole (strong CYP3A4 inhibitor) | 400 mg QD for 9 days | 6.45
(5.02, 8.29) | 18.55
(15.89, 21.65) |
| Verapamil (moderate CYP3A4 and
strong P-gp inhibitor) | 80 mg TID for 10 days | 2.08
(1.89, 2.28) | 2.71
(2.56, 2.87) |
| Rifampin (strong CYP3A4 inducer) | 600 mg QD for 10 days | 0.32
(0.28, 0.37) | 0.13
(0.11, 0.15) |
-
Moderate CYP3A inhibitors: Co-administration of multiple doses of fluconazole or diltiazem is predicted to increase voclosporin C max and AUC 0-12 approximately 2- and 3-fold, respectively.
-
Weak CYP3A inhibitors: Co-administration of multiple doses of fluvoxamine and cimetidine is predicted to have minimal effects on voclosporin C max and AUC 0-12.
-
Moderate CYP3A inducers: Co-administration of multiple doses of efavirenz is predicted to decrease voclosporin C max and AUC 0-12 by 61% and 70%, respectively.
In vitro, voclosporin is not a substrate for breast cancer resistance protein (BCRP) or organic anion transporting polypeptides OATP1B1 and OATP1B3.
Effect of LUPKYNIS on other Drugs
Voclosporin was studied on a background therapy that included MMF. Voclosporin 23.7 mg twice a day in patients with SLE (with or without LN) had no effect on MPA exposure. Also, clinical studies indicate that voclosporin is a weak inhibitor of P-gp and has no clinically relevant effects on the pharmacokinetics of the sensitive CYP3A4 substrate midazolam. Summary of the results from clinical studies which evaluated the effect of voclosporin on other drugs is provided in Table 3.
|
Notes: BID = twice daily; CI = Confidence interval; CYP = Cytochrome P450; MMF = mycophenolate mofetil; P-gp = P-glycoprotein. 1 Ratios for C max and AUC compare co-administration of the medication with voclosporin vs administration of the medication alone. 2 Observed effect of voclosporin on mycophenolic acid (MPA). |
|||
| Co-administered Drug | Multiple Dose Regimen of
Voclosporin | Ratio
(90% CI) 1 |
|
|---|---|---|---|
| C max | AUC | ||
| MMF 2 (immunosuppressant) | 23.7 mg BID | 0.94
(0.77, 1.16) | 1.09
(0.94, 1.26) |
| Digoxin (P-gp substrate) | 0.4 mg/kg BID | 1.51
(1.40, 1.63) | 1.25
(1.19, 1.31) |
| Midazolam (sensitive CYP3A4 substrate) | 0.4 mg/kg BID | 0.89
(0.80, 0.99) | 1.02
(0.93, 1.12) |
Based on in vitro studies, voclosporin does not inhibit BCRP, CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or induce CYP1A2, 2B6, 3A4. Voclosporin is an inhibitor of OATP1B1 and OATP1B3.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year mouse carcinogenicity study, administration of voclosporin at oral doses of 3, 10, or 30 mg/kg/day resulted in an increased incidence of malignant lymphoma in high dose females (7.5 times the MRHD on an AUC basis) and a dose-responsive trend for increase in malignant lymphoma in males. Malignant lymphoma was considered drug related in mice. In a 2-year rat carcinogenicity study, oral administration of voclosporin at doses up to 1.25 mg/kg/day in males and 2.5 mg/kg/day in females (doses that result in approximately similar drug exposures in rats) resulted in no statistically significant increases of tumor incidences.
In a 39-week oral toxicology study with monkeys, malignant lymphomas occurred at a dose of 150 mg/kg/day (approximately 4- and 7-times MRHD based on AUC for male and female animals, respectively). [At this dose, monkeys experienced high levels of immunosuppression as indicated by maximum calcineurin inhibition levels (E max) of greater than 80%.]
Voclosporin was not mutagenic or clastogenic in a standard battery of genetic toxicity studies that included the in vitro bacterial reverse mutation assay, in vitro Chinese hamster ovary cell chromosomal aberration assay, and in vivo rat micronucleus assay.
Voclosporin had no effect on fertility at doses up to 25 mg/kg/day in male and female rats (16- and 9-times MRHD based on AUC, respectively).
14. Clinical Studies
The safety and efficacy of LUPKYNIS were investigated in Study 1 (NCT03021499), a 52-week, randomized, double-blind, placebo-controlled trial in patients with a diagnosis of systemic lupus erythematosus and with International Society of Nephrology / Renal Pathology Society (ISN/RPS) biopsy-proven active Class III or IV LN (alone or in combination with Class V LN) or Class V LN. Patients with Class III or IV LN (alone or in combination with Class V LN) were required to have a urine protein to creatinine (UPCR) ratio of ≥1.5 mg/mg; patients with Class V LN were required to have a UPCR of ≥2 mg/mg.
A total of 357 patients with LN were randomized in a 1:1 ratio to receive either LUPKYNIS 23.7 mg twice daily or placebo.
Patients in both arms received background treatment with MMF and corticosteroids as follows:
- Oral MMF at a target dose of 2 g/day (1 g twice a day). (Patients not already receiving MMF were started on MMF 500 mg twice a day with escalation to MMF 1 g twice a day after the first week.) Dose increases up to 3 g/day were allowed.
- Intravenous (IV) methylprednisolone on Day 1 and Day 2 at a dose of 500 mg/day (body weight ≥45 kg) or 250 mg/day (body weight <45 kg) followed by a reducing taper of oral corticosteroids [oral prednisone 25 mg/day (body weight ≥45 kg) or 20 mg/day (body weight <45 kg); tapered to achieve a target dose of 2.5 mg/day by Week 16].
Throughout the study, patients were prohibited from using immunosuppressants (other than MMF and hydroxychloroquine/chloroquine) and from changing/commencing angiotensin II receptor blockers (ARBs) or angiotensin converting enzyme (ACE) inhibitors.
Patients with baseline eGFR ≤45 mL/min/1.73 m 2 were not enrolled in this study.
Dosage was adjusted based on eGFR and BP in a pre-defined dosage adjustment protocol. Dosage adjustments should follow the dosage recommendations [see Dosage and Administration ( 2.3)] .
The median age of patients was 31 years (range 18 to 72). The proportion of women was 88%. Approximately 36.1% were White, 9.5% were Black, 30.5% were Asian, 1.1% were American Indian or Alaska Native, and 22.7% were multiple race or other. Approximately 32.5% were Hispanic or Latino.
The mean (SD) daily dose of voclosporin was 41.3 (±9.7) mg/day. The mean (SD) daily dose of MMF was 1.9 (±0.4) g/day; 9% received >2 but ≤3 g/day of MMF. The mean (SD) daily dose of IV methylprednisolone (on Day 1 was 495 (±90) mg/day and Day 2 was 487 (±55) mg/day). The mean (SD) starting oral corticosteroid dose (Day 3) was 22.8 (±4.8) mg/day; approximately 81% received ≤2.5 mg/day of oral corticosteroids at Week 16.
The distribution by kidney biopsy class was Class III or IV (60.8%), Class III or IV in combination with Class V (24.9%), and Class V (14.3%). Mean (SD) eGFR on entry was 91 (±30) mL/min/1.73 m 2. Mean (SD) UPCR on entry was 4.0 (±2.5) mg/mg.
The primary efficacy endpoint was the proportion of patients achieving complete renal response at Week 52. Complete renal response was defined as follows (both must be met):
- UPCR of ≤0.5 mg/mg, and
- eGFR ≥60 mL/min/1.73 m 2 or no confirmed decrease from baseline in eGFR of >20% or no treatment- or disease-related eGFR-associated event (defined as blood creatinine increased, creatinine renal clearance decreased, glomerular filtration rate decreased, serum creatinine increased, renal impairment, renal failure, or renal failure acute) at time of assessment.
In order to be considered a responder, the patient must not have received more than 10 mg prednisone for ≥3 consecutive days or for ≥7 days in total during Weeks 44 through 52. Patients who received rescue medication or withdrew from the study were considered non-responders.
A higher proportion of patients in the LUPKYNIS arm than the placebo arm achieved complete renal response at Week 52 (Table 4).
|
eGFR = Estimated glomerular filtration rate, UPCR: Urine protein-creatinine ratio CI: Confidence interval, a In order to be considered a responder, the patient must not have received more than 10 mg prednisone for ≥3 consecutive days or for ≥7 days in total during Weeks 44 through 52. Patients who received rescue medication or withdrew from the study were considered non-responders. b Treatment- or disease-related eGFR-associated event is defined as blood creatinine increased, creatinine renal clearance decreased, glomerular filtration rate decreased, serum creatinine increased, renal impairment, renal failure, or renal failure acute. |
|||
| LUPKYNIS
(N=179) | Placebo
(N=178) | Odds Ratio | |
|---|---|---|---|
| Primary Endpoint | |||
| Complete Renal Response at Week 52 [n (%)] a | 73 (40.8) | 40 (22.5) | 2.7 (95% CI: 1.6, 4.3);
p<0.001 |
| Components of Primary Endpoint | |||
| UPCR ≤0.5 mg/mg [n (%)] | 81 (45.3) | 41 (23.0) | 3.1 (95% CI: 1.9, 5.0) |
| eGFR ≥60 mL/min/1.73 m
2 or no confirmed
decrease from baseline in eGFR of >20% or no treatment- or disease-related eGFR-associated adverse event b at time of assessment [n (%)] | 147 (82.1) | 135 (75.8) | 1.5 (95% CI: 0.8, 2.5) |
A higher proportion of patients in the LUPKYNIS arm than the placebo arm achieved complete renal response at Week 24 (32.4% vs. 19.7%; odds ratio: 2.2; 95% CI: 1.3, 3.7). Time to UPCR of ≤0.5 mg/mg was shorter in the LUPKYNIS arm than the placebo arm (median time of 169 days vs. 372 days; hazard ratio: 2.0; 95% CI: 1.5, 2.7).
16. How is Lupkynis supplied
LUPKYNIS (voclosporin) capsules 7.9 mg are oval, pink/orange capsules, imprinted on one side with VCS in white ink, packed in cold-formed aluminum blisters, consisting of laminated backing and lidding materials that are thermo-sealed together. Four individual 3 × 5 blister strips are assembled into a cardboard wallet.
LUPKYNIS is available in:
- NDC 75626-001-01: Wallet containing 60 capsules
- NDC 75626-001-02: Carton containing three wallets (180 capsules)
LUPKYNIS is provided in child-proof packaging to avoid unintentional ingestion of study medication by children.
Store at controlled room temperature 20ºC to 25ºC (68ºF to 77ºF); excursions permitted to 15ºC to 30ºC (59ºF to 86ºF) [See USP Controlled Room Temperature].
Do not put LUPKYNIS in another container. Keep capsules in their original packaging until ready to be taken.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Administration
Advise patients to:
- Swallow LUPKYNIS capsules whole, and not to open, crush, or divide LUPKYNIS capsules.
- Take LUPKYNIS on an empty stomach consistently as close to a 12-hour schedule as possible, and with a minimum of 8 hours between doses.
- If a dose is missed, take it as soon as possible within 4 hours after missing the dose. Beyond the 4-hour time frame, wait until the usual scheduled time to take the next regular dose. Do not double the next dose.
- Avoid eating grapefruit or drinking grapefruit juice while taking LUPKYNIS.
Development of Lymphoma and Other Malignancies
Inform patients that they are at an increased risk of developing lymphomas and other malignancies, particularly of the skin, due to immunosuppression. Advise patients to limit exposure to sunlight and ultraviolet (UV) light by wearing protective clothing and use a sunscreen with a high protection factor [see Boxed Warning and Warnings and Precautions ( 5.1)] .
Increased Risk of Infection
Inform patients that they are at an increased risk of developing a variety of infections, including opportunistic infections, due to immunosuppression and to contact their physician if they develop any symptoms of infection such as fever, sweats or chills, cough or flu-like symptoms, muscle aches, or warm, red, painful areas on the skin [see Boxed Warning and Warnings and Precautions ( 5.2)] .
Nephrotoxicity (Acute and/or Chronic)
Inform patients that LUPKYNIS can have toxic effects on the kidney that should be monitored. Advise patients to attend all visits and complete all blood tests ordered by their medical team [see Warnings and Precautions ( 5.3)] .
Hypertension
Inform patients that LUPKYNIS can cause high blood pressure which may require treatment with antihypertensive therapy. Advise patients to monitor their blood pressure [see Warnings and Precautions ( 5.4)] .
Neurotoxicity
Inform patients that they are at risk of developing adverse neurologic effects including seizure, altered mental status, and tremor. Advise patients to contact their physician should they develop vision changes, delirium, or tremors [see Warnings and Precautions ( 5.5)] .
Hyperkalemia
Inform patients that LUPKYNIS can cause hyperkalemia. Monitoring of potassium levels may be necessary, especially with concomitant use of other drugs known to cause hyperkalemia [see Warnings and Precautions ( 5.6)] .
Drug Interactions
Advise patients to tell their healthcare provider when they start or stop taking any concomitant medications. Strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin) are contraindicated with LUPKYNIS, and other CYP3A4 enzyme modulating drugs can alter LUPKYNIS exposure [see Contraindications ( 4) and Drug Interactions ( 7.1)] .
Pregnancy
Inform female patients of the potential risk to a fetus and to avoid use of LUPKYNIS during pregnancy. When LUPKYNIS is administered in combination with MMF, refer patients to the MMF medication guide. Advise females to inform their healthcare provider if they are pregnant or become pregnant [see Use in Specific Populations ( 8.1, 8.3)] .
Lactation
Advise women not to breastfeed during treatment with LUPKYNIS and for 7 days after the last dose of LUPKYNIS [see Use in Specific Populations ( 8.2)] .
Immunizations
Inform patients that LUPKYNIS can interfere with the usual response to immunizations and that they should avoid live vaccines [see Warnings and Precautions ( 5.8)] .
Manufactured for:
Aurinia Pharmaceuticals Inc.
#1203-4464 Markham Street
Victoria, BC V8Z7X8
Canada
Distributed by:
Aurinia Pharma U.S., Inc.
77 Upper Rock Circle
Suite 700
Rockville, MD 20850
USA
Medication Guide
| Medication Guide
LUPKYNIS™ (loop kye’ nis) (voclosporin) capsules, for oral use |
||
| Read this Medication Guide before you start taking LUPKYNIS and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment. If you have any questions about LUPKYNIS, ask your healthcare provider or pharmacist. | ||
| What is the most important information I should know about LUPKYNIS?
LUPKYNIS can cause serious side effects, including:
|
||
|
|
|
|
|
|
| See “What are the possible side effects of LUPKYNIS?” for more information about side effects. | ||
What is LUPKYNIS?
|
||
Do not take LUPKYNIS:
|
||
Before you take LUPKYNIS, tell your healthcare provider about all your medical conditions, including if you:
|
||
How should I take LUPKYNIS?
|
||
What should I avoid while taking LUPKYNIS?
|
||
| What are the possible side effects of LUPKYNIS?
LUPKYNIS may cause serious side effects, including:
|
||
|
|
|
|
|
|
|
||
|
||
|
|
|
|
|
|
| ||
| These are not all the possible side effects of LUPKYNIS.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to www.fda.gov/medwatch. |
||
How should I store LUPKYNIS?
|
||
| General information about the safe and effective use of LUPKYNIS
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use LUPKYNIS for a condition for which it was not prescribed. Do not give LUPKYNIS to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about LUPKYNIS that is written for healthcare professionals. |
||
| What are the ingredients in LUPKYNIS?
Active ingredient : voclosporin Inactive ingredients: alcohol, Vitamin E polyethylene glycol succinate, polysorbate 40, medium-chain triglycerides, gelatin Manufactured for: Aurinia Pharmaceuticals Inc., Victoria, BC V8Z 7X8 Canada Distributed by: Aurinia Pharma U.S., Inc., Rockville, MD 20850 USA For more information, go to www.LUPKYNIS.com or call 1-833-AURINIA |
||
This Medication Guide has been approved by the U.S. Food and Drug Administration Issued: 01/2021
| LUPKYNIS
voclosporin capsule |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Aurinia Pharma U.S., Inc. (117485579) |
| Registrant - Aurinia Pharma U.S., Inc. (117485579) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| MRIGlobal | 007173453 | analysis(75626-001) | |
