Drug Detail:Sunlenca (Lenacapavir)
Drug Class: Miscellaneous antivirals
Highlights of Prescribing Information
SUNLENCA® (lenacapavir) tablets, for oral use
SUNLENCA® (lenacapavir) injection, for subcutaneous use
Initial U.S. Approval: 2022
Indications and Usage for Sunlenca
SUNLENCA, a human immunodeficiency virus type 1 (HIV-1) capsid inhibitor, in combination with other antiretroviral(s), is indicated for the treatment of HIV-1 infection in heavily treatment-experienced adults with multidrug resistant HIV-1 infection failing their current antiretroviral regimen due to resistance, intolerance, or safety considerations. (1)
Sunlenca Dosage and Administration
- Recommended dosage – Initiation with one of two options followed by once every 6-months maintenance dosing. Tablets may be taken without regard to food. (2.1)
| Initiation Option 1 | |
| Day 1 | 927 mg by subcutaneous injection (2 × 1.5 mL injections) 600 mg orally (2 × 300 mg tablets) |
| Day 2 | 600 mg orally (2 × 300 mg tablets) |
| Initiation Option 2 | |
| Day 1 | 600 mg orally (2 × 300 mg tablets) |
| Day 2 | 600 mg orally (2 × 300 mg tablets) |
| Day 8 | 300 mg orally (1 × 300 mg tablet) |
| Day 15 | 927 mg by subcutaneous injection (2 × 1.5 mL injections) |
| Maintenance | |
| 927 mg by subcutaneous injection (2 × 1.5 mL injections) every 6 months (26 weeks) from the date of the last injection +/-2 weeks. | |
- Missed dose: If more than 28 weeks since last injection and clinically appropriate to continue SUNLENCA, restart initiation from Day 1, using either Option 1 or Option 2. (2.2)
- Two 1.5 mL subcutaneous injections are required for complete dose. (2.3)
Dosage Forms and Strengths
Tablets: 300 mg
Injection: 463.5 mg/1.5 mL (309 mg/mL) in single-dose vials. (3)
Contraindications
Concomitant administration of SUNLENCA is contraindicated with strong CYP3A inducers. (4)
Warnings and Precautions
Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.1)
Residual concentrations of lenacapavir may remain in systemic circulation for up to 12 months or longer. Counsel patients regarding the dosing schedule; non-adherence could lead to loss of virologic response and development of resistance. (5.2)
May increase exposure and risk of adverse reactions to drugs primarily metabolized by CYP3A initiated within 9 months after the last subcutaneous dose of SUNLENCA. (5.2)
If discontinued, initiate an alternative, fully suppressive antiretroviral regimen where possible no later than 28 weeks after the final injection of SUNLENCA. If virologic failure occurs, switch to an alternative regimen if possible. (5.2)
Injection site reactions may occur, and nodules and indurations may be persistent. (5.3)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence greater than or equal to 3%, all grades) are nausea and injection site reactions. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Consult the Full Prescribing Information prior to and during treatment for important drug interactions. (4, 7, 12.3)
Use In Specific Populations
- Lactation: Individuals infected with HIV should be instructed not to breastfeed due to the potential for HIV transmission. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2022
Full Prescribing Information
1. Indications and Usage for Sunlenca
SUNLENCA, in combination with other antiretroviral(s), is indicated for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in heavily treatment-experienced adults with multidrug resistant HIV-1 infection failing their current antiretroviral regimen due to resistance, intolerance, or safety considerations.
2. Sunlenca Dosage and Administration
2.1 Recommended Dosage
SUNLENCA can be initiated using one of two recommended dosage regimens, see Table 1 and Table 2 below. Healthcare providers should determine the appropriate initiation regimen for the patient [see Clinical Pharmacology (12.3)]. SUNLENCA oral tablets may be taken with or without food.
| Treatment Time | |
|---|---|
|
|
| Dosage of SUNLENCA: Initiation | |
| Day 1 | 927 mg by subcutaneous injection (2 × 1.5 mL injections) 600 mg orally (2 × 300 mg tablets) |
| Day 2 | 600 mg orally (2 × 300 mg tablets) |
| Dosage of SUNLENCA: Maintenance | |
| Every 6 months (26 weeks) * +/-2 weeks | 927 mg by subcutaneous injection (2 × 1.5 mL injections) |
| Treatment Time | |
|---|---|
|
|
| Dosage of SUNLENCA: Initiation | |
| Day 1 | 600 mg orally (2 × 300 mg tablets) |
| Day 2 | 600 mg orally (2 × 300 mg tablets) |
| Day 8 | 300 mg orally (1 × 300 mg tablet) |
| Day 15 | 927 mg by subcutaneous injection (2 × 1.5 mL injections) |
| Dosage of SUNLENCA: Maintenance | |
| Every 6 months (26 weeks) * +/-2 weeks | 927 mg by subcutaneous injection (2 × 1.5 mL injections) |
2.2 Missed Dose
During the maintenance period, if more than 28 weeks have elapsed since the last injection and if clinically appropriate to continue SUNLENCA treatment, restart the initiation dosage regimen from Day 1, using either Option 1 or Option 2 [see Dosage and Administration (2.1)].
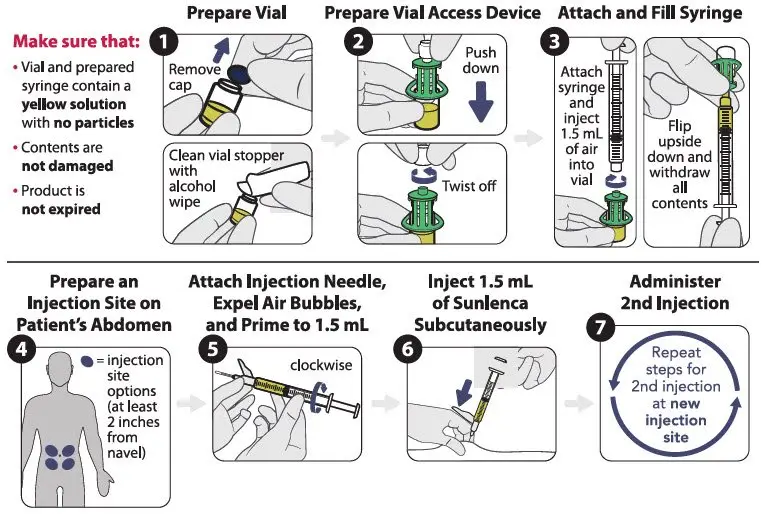
2.3 Preparation and Administration of Subcutaneous Injection
SUNLENCA injection is for administration into the abdomen by a healthcare provider.
Use aseptic technique. Visually inspect the solution in the vials and prepared syringe for particulate matter and discoloration prior to administration. SUNLENCA injection is a yellow solution. Do not use SUNLENCA injection if the solution is discolored or if it contains particulate matter. Once the solution is withdrawn from the vials, the subcutaneous injections should be administered as soon as possible [see How Supplied/Storage and Handling (16)].
Refer to Figure 1 to identify the components for use in the administration steps. The administration steps are provided in Figure 2.
The injection kit components are for single use only. Use of a vial access device is required. Two 1.5 mL injections are required for a complete dose.
Figure 1 SUNLENCA Injection Kit Components

Figure 2 SUNLENCA Injection Steps
4. Contraindications
Concomitant administration of SUNLENCA with strong CYP3A inducers is contraindicated due to decreased lenacapavir plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to SUNLENCA [see Drug Interactions (7.1)].
5. Warnings and Precautions
5.1 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections [such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia (PCP), or tuberculosis], which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.2 Long-Acting Properties and Potential Associated Risks with SUNLENCA
Residual concentrations of lenacapavir may remain in the systemic circulation of patients for prolonged periods (up to 12 months or longer after the last subcutaneous dose). It is important to counsel patients that maintenance dosing by injection is required every 6 months, because missed doses or non-adherence to injections could lead to loss of virologic response and development of resistance [see Dosage and Administration (2.1)].
Lenacapavir, a moderate CYP3A inhibitor, may increase the exposure to, and therefore potential risk of adverse reactions from, drugs primarily metabolized by CYP3A initiated within 9 months after the last subcutaneous dose of SUNLENCA [see Drug Interactions and Clinical Pharmacology (7.2, 12.3)].
If SUNLENCA is discontinued, to minimize the potential risk of developing viral resistance, it is essential to initiate an alternative, fully suppressive antiretroviral regimen where possible no later than 28 weeks after the final injection of SUNLENCA. If virologic failure occurs during treatment, switch the patient to an alternative regimen if possible [see Dosage and Administration (2.2)].
5.3 Injection Site Reactions
Administration of SUNLENCA may result in local injection site reactions (ISRs). If clinically significant ISRs occur, evaluate and institute appropriate therapy and follow-up.
Manifestations of ISRs may include swelling, pain, erythema, nodule, induration, pruritus, extravasation or mass. Nodules and indurations at the injection site may take longer to resolve than other ISRs. In clinical studies, after a median follow-up of 553 days, 30% of nodules and 13% of indurations (in 10% and 1% of subjects, respectively) associated with the first injections of SUNLENCA had not fully resolved. Measurements and qualitative assessments of ISRs were not routinely reported. Where described, the majority of the injection site nodules and indurations were palpable but not visible, and had a maximum size of approximately 1 to 4 cm [see Adverse Reactions (6.1)].
The mechanism driving the persistence of injection site nodules and indurations in some patients is not fully understood, but based on available data, they may be related to the presence of the subcutaneous drug depot. In some patients who had a skin biopsy performed of an injection site nodule or induration, dermatopathology revealed foreign body inflammation or granulomatous response.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in other sections of the labeling:
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.1)]
- Injection Site Reactions [see Warnings and Precautions (5.3)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The primary safety assessment of SUNLENCA was based on data from heavily treatment-experienced adult subjects with HIV who received SUNLENCA in a Phase 2/3 trial (CAPELLA; N=72) through Week 52 (median duration on study of 71 weeks) [see Clinical Studies (14)], as well as supportive data in treatment-naïve adult subjects with HIV who received SUNLENCA in a Phase 2 trial (CALIBRATE; N=157) through Week 54 (median duration of exposure of 66 weeks).
The most common adverse reactions (all Grades) reported in at least 3% of subjects in CAPELLA were nausea and injection site reactions. The proportion of subjects in CAPELLA who discontinued treatment with SUNLENCA due to adverse events, regardless of severity, was 1% (Grade 1 injection site nodule in 1 subject). Table 3 displays the frequency of adverse reactions (all Grades) greater than or equal to 3% in the SUNLENCA group.
| Adverse Reactions | SUNLENCA + Background Regimen (N=72) |
|---|---|
|
|
| Injection Site Reactions | 65% |
| Nausea | 4% |
The majority (96%) of all adverse reactions associated with SUNLENCA were mild or moderate in severity.
Laboratory Abnormalities
The frequency of laboratory abnormalities (Grades 3 to 4) occurring in at least 2% of subjects in CAPELLA are presented in Table 4. A causal association between SUNLENCA and these laboratory abnormalities has not been established.
| Laboratory Parameter Abnormality | SUNLENCA + Background Regimen (N=72) * |
|---|---|
| ALT= alanine aminotransferase; AST= aspartate aminotransferase; ULN = upper limit of normal | |
|
|
| Creatinine ( >1.8 × ULN or ≥1.5 × baseline) | 13% |
| Glycosuria (>2+) † | 6% |
| Hyperglycemia (fasting) (>250 mg/dL) | 5% |
| Proteinuria (>2+) † | 4% |
| ALT (≥5 × ULN) † | 3% |
| AST (≥5 × ULN) | 3% |
| Direct Bilirubin (>ULN) † | 3% |
7. Drug Interactions
7.2 Effect of SUNLENCA on Other Drugs
Lenacapavir is a moderate inhibitor of CYP3A. Due to the long half-life of lenacapavir following subcutaneous administration, SUNLENCA may increase the exposure of drugs primarily metabolized by CYP3A [see Clinical Pharmacology (12.3)] initiated within 9 months after the last subcutaneous dose of SUNLENCA, which may increase the potential risk of adverse reactions. See the prescribing information of the sensitive CYP3A substrate for dosing recommendations with moderate inhibitors of CYP3A.
7.3 Established and Other Potentially Significant Drug Interactions
Table 5 provides a listing of clinically significant drug interactions with recommended prevention or management strategies, but is not all inclusive. The drug interactions described are based on studies conducted with SUNLENCA or are drug interactions that may occur with SUNLENCA [see Contraindications (4) and Clinical Pharmacology (12.3)].
| Concomitant Drug Class: Drug Name | Effect on Concentration * | Clinical Comment |
|---|---|---|
|
||
| Antiarrhythmics:
digoxin | ↑ digoxin | Use with caution and monitor digoxin therapeutic concentration. |
| Anticoagulants:
Direct Oral Anticoagulants (DOACs) rivaroxaban dabigatran edoxaban | ↑ DOAC | Refer to the DOAC prescribing information for concomitant administration with combined moderate CYP3A and P-gp inhibitors. |
| Anticonvulsants:
carbamazepine oxcarbazepine phenobarbital phenytoin | ↓ lenacapavir | Concomitant administration of carbamazepine, oxcarbazepine, phenobarbital, or phenytoin may result in loss of therapeutic effect and development of resistance. Concomitant administration of SUNLENCA with carbamazepine or phenytoin is contraindicated. Concomitant administration of SUNLENCA with oxcarbazepine or phenobarbital is not recommended. Consider use of alternative anticonvulsants. |
| Antiretroviral Agents:
atazanavir/cobicistat † atazanavir/ritonavir | ↑ lenacapavir (atazanavir/cobicistat, atazanavir/ritonavir) | Concomitant administration of efavirenz, nevirapine, or tipranavir/ritonavir may result in loss of therapeutic effect and development of resistance. |
| efavirenz †
nevirapine tipranavir/ritonavir | ↓ lenacapavir (efavirenz, nevirapine, tipranavir/ritonavir) | Concomitant administration with atazanavir/cobicistat, atazanavir/ritonavir, efavirenz, nevirapine, or tipranavir/ritonavir is not recommended. |
| Antimycobacterials:
rifabutin rifampin † rifapentine | ↓ lenacapavir | Concomitant administration of rifabutin, rifampin and rifapentine may result in loss of therapeutic effect and development of resistance. Concomitant administration of SUNLENCA with rifampin is contraindicated [see Contraindications (4)]. Concomitant administration of SUNLENCA with rifabutin or rifapentine is not recommended. |
| Corticosteroids (systemic):
Dexamethasone Hydrocortisone/cortisone | ↑ corticosteroids (systemic) | Concomitant administration with corticosteroids whose exposures are significantly increased by CYP3A inhibitors can increase the risk for Cushing's syndrome and adrenal suppression. Initiate with the lowest starting dose and titrate carefully while monitoring for safety. |
| Ergot derivatives:
dihydroergotamine ergotamine methylergonovine | ↑ dihydroergotamine ↑ ergotamine ↑ methylergonovine | Concomitant administration of SUNLENCA with dihydroergotamine, ergotamine or methylergonovine is not recommended. |
| Herbal Products:
St. John's wort ‡ (Hypericum perforatum) | ↓ lenacapavir | Concomitant administration of St. John's wort may result in loss of therapeutic effect and development of resistance. Concomitant administration of SUNLENCA with St. John's wort is contraindicated. |
| HMG-CoA Reductase Inhibitors:
lovastatin simvastatin | ↑ lovastatin ↑ simvastatin | Initiate lovastatin and simvastatin with the lowest starting dose and titrate carefully while monitoring for safety (e.g., myopathy). |
| Narcotic analgesics metabolized by CYP3A:
e.g., fentanyl, oxycodone | ↑ fentanyl ↑ oxycodone | Careful monitoring of therapeutic effects and adverse reactions associated with CYP3A-metabolized narcotic analgesics (including potentially fatal respiratory depression) is recommended with co-administration. |
| tramadol | ↑ tramadol | A decrease in dose may be needed for tramadol with concomitant use. |
| Narcotic analgesic for treatment of opioid dependence:
buprenorphine, methadone | buprenorphine: effects unknown methadone: effects unknown | Initiation of buprenorphine or methadone in patients taking SUNLENCA: Carefully titrate the dose of buprenorphine or methadone to the desired effect; use the lowest feasible initial or maintenance dose. Initiation of SUNLENCA in patients taking buprenorphine or methadone: A dose adjustment for buprenorphine or methadone may be needed. Monitor clinical signs and symptoms. |
| Opioid Antagonist:
naloxegol | ↑ naloxegol | Avoid use with SUNLENCA; if unavoidable, decrease the dosage of naloxegol and monitor for adverse reactions. |
| Phosphodiesterase-5 (PDE-5) Inhibitors:
sildenafil tadalafil vardenafil | ↑ PDE-5 inhibitors | Use of PDE-5 inhibitors for pulmonary arterial hypertension (PAH): Concomitant administration of SUNLENCA with tadalafil for the treatment of PAH is not recommended. Use of PDE-5 inhibitors for erectile dysfunction (ED): Refer to the prescribing information of PDE-5 inhibitors for dose recommendations. |
| Sedatives/Hypnotics:
midazolam (oral) † triazolam | ↑ midazolam (oral) ↑ triazolam | Use with caution when midazolam or triazolam is concomitantly administered with SUNLENCA |
7.4 Drugs without Clinically Significant Interactions with SUNLENCA
Based on drug interaction studies conducted with SUNLENCA, no clinically significant drug interactions have been observed with: darunavir/cobicistat, cobicistat, famotidine, pitavastatin, rosuvastatin, tenofovir alafenamide, and voriconazole.
8. Use In Specific Populations
8.4 Pediatric Use
The safety and effectiveness of SUNLENCA have not been established in pediatric patients.
8.5 Geriatric Use
Clinical studies of SUNLENCA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
No dosage adjustment of SUNLENCA is recommended in patients with mild, moderate or severe renal impairment (estimated creatinine clearance greater than or equal to 15 mL per minute). SUNLENCA has not been studied in patients with ESRD (estimated creatinine clearance less than 15 mL per minute) [see Clinical Pharmacology (12.3)].
10. Overdosage
No data are available on overdose of SUNLENCA in patients. If overdose occurs, monitor the patient for evidence of toxicity. Treatment of overdose with SUNLENCA consists of general supportive measures including monitoring of vital signs as well as observation of the clinical status of the patient. As lenacapavir is highly bound to plasma proteins, it is unlikely to be significantly removed by dialysis.
11. Sunlenca Description
SUNLENCA tablets and SUNLENCA injection contain lenacapavir sodium, a capsid inhibitor.
The chemical name of lenacapavir sodium is: Sodium (4-chloro-7-(2-((S)-1-(2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamido)-2-(3,5-difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide.
Lenacapavir sodium has a molecular formula of C39H31ClF10N7NaO5S2, a molecular weight of 990.3, and the following structural formula:
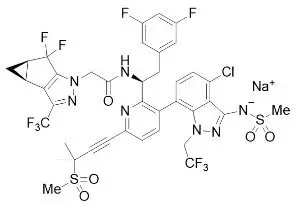
Lenacapavir sodium is a light yellow to yellow solid and is practically insoluble in water.
12. Sunlenca - Clinical Pharmacology
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of lenacapavir are provided in Table 6 and Table 7. The estimated lenacapavir exposures are comparable between the two recommended dosing regimens.
| Oral | Subcutaneous | ||
|---|---|---|---|
|
|||
| Absorption | |||
| % Absolute bioavailability | 6 to 10 | 100 * | |
| Tmax† | 4 hours | 77 to 84 days ‡ | |
| Effect of Food | |||
| Effect of low-fat meal (relative to fasting) § | AUCinf ratio | 98.6 (58.2,167.2) | - |
| Cmax ratio | 115.8 (55.4, 242.1) | - | |
| Effect of high-fat meal (relative to fasting) ¶ | AUCinf ratio | 115.2 (72.0, 184.5) | - |
| Cmax ratio | 145.2 (77.9, 270.5) | - | |
| Distribution | |||
| Apparent volume of distribution (Vd/F, L) | 19240 | 9500 to 11700 | |
| % bound to human plasma proteins | >98.5 | ||
| Blood-to-plasma ratio | 0.5 to 0.7 # | ||
| Elimination | |||
| t1/2 | 10 to 12 days | 8 to 12 weeks | |
| Clearance (mean apparent clearance, L/h) | 55 | 4.2 | |
| % of dose of unchanged drug in plasma Þ | 69 | ||
| Metabolism | |||
| Metabolic pathway(s) | CYP3A (minor) UGT1A1 (minor) |
||
| Excretion | |||
| Major routes of elimination | Excretion of unchanged drug into feces ß | ||
| % of dose excreted in urine Þ | <1 | ||
| % of dose excreted in feces (% unchanged) ß | 76 (33) | ||
| Parameter Mean (%CV) | Recommended Dosing Regimen, Option 1 * | Recommended Dosing Regimen, Option 2 † |
|
|---|---|---|---|
| Day 1: 600 mg (oral) + 927 mg (SC) Day 2: 600 mg (oral) | Days 1 and 2: 600 mg (oral), Day 8: 300 mg (oral), Day 15: 927 mg (SC) |
||
| Day 1 to end of Month 6 | Days 1 to 15 | Day 15 to end of Month 6 | |
| CV = coefficient of variation; NA = not applicable; SC = subcutaneous | |||
|
|||
| Cmax
(ng/mL) | 97.1 (61.6) | 124.4 (85.1) | 87.3 (49.4) |
| AUCtau
(h∙ng/mL) | 234294.8 (65.1) | 25962.9 (67.8) | 251907.2 (48.2) |
| Ctrough
(ng/mL) | 29.2 (90.8) | 48.6 (52.1) | 35.1 (59.2) |
The estimated exposures of lenacapavir were 43% to 100% higher in subjects with HIV-1 infection compared to subjects without HIV-1 infection.
Drug Interaction Studies
Clinical Studies
Clinical drug-drug interaction study indicated that lenacapavir is a substrate of CYP3A, P-gp, and UGT1A1. Table 8 summarizes the pharmacokinetic effects of other drugs on lenacapavir.
Lenacapavir is a moderate inhibitor of CYP3A. Lenacapavir is an inhibitor of P-gp and BCRP but does not inhibit OATP. Table 9 summarizes the pharmacokinetic effects of lenacapavir on other drugs.
| Coadministered Drug | Dose of Coadministered Drug (mg) | Mean Ratio of Lenacapavir Pharmacokinetic Parameters (90% CI); No effect = 1.00 | |
|---|---|---|---|
| Cmax | AUC | ||
|
|||
| Cobicistat (fed) (Inhibitor of CYP3A [strong] and P-gp) | 150 once daily | 2.10 (1.62, 2.72) | 2.28 (1.75, 2.96) |
| Darunavir / cobicistat (fed) (Inhibitor of CYP3A [strong] and inhibitor and inducer of P-gp) | 800/150 once daily | 2.30 (1.79, 2.95) | 1.94 (1.50, 2.52) |
| Voriconazole (fasted) (Inhibitor of CYP3A [strong]) | 400 twice daily, 200 twice daily ‡ | 1.09 (0.81, 1.47) | 1.41 (1.10, 1.81) |
| Atazanavir / cobicistat (fed) (Inhibitor of CYP3A [strong] and UGT1A1 and P-gp) | 300/150 once daily | 6.60 (4.99, 8.73) | 4.21 (3.19, 5.57) |
| Rifampin (fasted) (Inducer of CYP3A [strong] and P-gp and UGT) | 600 once daily | 0.45 (0.34, 0.60) | 0.16 (0.12, 0.20) |
| Efavirenz (fasted) (Inducer of CYP3A [moderate] and P-gp) | 600 once daily | 0.64 (0.45, 0.92) | 0.44 (0.32, 0.59) |
| Famotidine (2 hours before, fasted) | 40 once daily | 1.01 (0.75, 1.34) | 1.28 (1.00, 1.63) |
| Coadministered Drug | Dose of Coadministered Drug (mg) | Mean Ratio of Coadministered Drug Pharmacokinetic Parameters (90% CI) ‡; No effect = 1.00 | |
|---|---|---|---|
| Cmax | AUC | ||
|
|||
| Tenofovir alafenamide (fed) (substrate of P-gp) | 25 single dose | 1.24 (0.98, 1.58) | 1.32 (1.09, 1.59) |
| Tenofovir §
(substrate of P-gp) | 1.23 (1.05, 1.44) | 1.47 (1.27, 1.71) |
|
| Pitavastatin (simultaneous administration, fed) (substrate of OATP) | 2 single dose | 1.00 (0.84, 1.19) | 1.11 (1.00, 1.25) |
| Pitavastatin (3 days after lenacapavir, fed) (substrate of OATP) | 2 single dose | 0.85 (0.69, 1.05) | 0.96 (0.87, 1.07) |
| Rosuvastatin (fed) (substrate of BCRP and OATP) | 5 single dose | 1.57 (1.38, 1.80) | 1.31 (1.19, 1.43) |
| Midazolam (simultaneous administration, fed) (substrate of CYP3A) | 2.5 single dose | 1.94 (1.81, 2.08) | 3.59 (3.30, 3.91) |
| 1-hydroxymidazolam ¶
(substrate of CYP3A) | 0.54 (0.50, 0.59) | 0.76 (0.72, 0.80) |
|
| Midazolam (1 day after lenacapavir, fed) (substrate of CYP3A) | 2.5 single dose | 2.16 (2.02, 2.30) | 4.08 (3.77, 4.41) |
| 1-hydroxymidazolam ¶
(substrate of CYP3A) | 0.52 (0.48, 0.57) | 0.84 (0.80, 0.88) |
|
14. Clinical Studies
The efficacy and safety of SUNLENCA in HIV-1 infected, heavily treatment-experienced subjects with multidrug resistance is based on 52-week data from CAPELLA, a randomized, placebo-controlled, double-blind, multicenter trial (NCT 04150068).
CAPELLA was conducted in 72 heavily treatment-experienced subjects with multiclass resistant HIV-1. Subjects were required to have a viral load ≥ 400 copies/mL, documented resistance to at least two antiretroviral medications from each of at least 3 of the 4 classes of antiretroviral medications (NRTI, NNRTI, PI and INSTI), and ≤ 2 fully active antiretroviral medications from the 4 classes of antiretroviral medications remaining at baseline due to resistance, intolerability, drug access, contraindication, or other safety concerns.
The trial was composed of two cohorts. Subjects were enrolled into the randomized cohort (cohort 1, N=36) if they had a < 0.5 log10 HIV-1 RNA decline compared to the screening visit. Subjects were enrolled into the non-randomized cohort (cohort 2, N=36) if they had a ≥ 0.5 log10 HIV-1 RNA decline compared to the screening visit or after cohort 1 reached its planned sample size.
In the 14-day functional monotherapy period, subjects in cohort 1 were randomized in a 2:1 ratio in a blinded fashion to receive either SUNLENCA or placebo, while continuing their failing regimen. This period was to establish the virologic activity of SUNLENCA. After the functional monotherapy period, subjects who had received SUNLENCA continued on SUNLENCA along with an optimized background regimen (OBR); subjects who had received placebo during this period initiated SUNLENCA along with an OBR.
Subjects in cohort 1 had a mean age of 52 years (range: 24 to 71), 72% were male, 46% were White, 46% were Black, and 9% were Asian. 29% percent of subjects identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.3 log10 copies/mL (range: 2.3 to 5.4). 19% of subjects had baseline viral loads greater than 100,000 copies/mL. The mean baseline CD4+ cell count was 161 cells/mm3 (range: 6 to 827). 75% of subjects had CD4+ cell counts below 200 cells/mm3. The mean number of years since subjects first started HIV treatment was 24 years (range: 7 to 33); the mean number of antiretroviral agents in failing regimens at baseline was 4 (range: 1 to 7). The percentage of subjects in the randomized cohort with known resistance to at least 2 agents from the NRTI, NNRTI, PI and INSTI classes was 97%, 94%, 78% and 75%, respectively. In cohort 1, 53% of subjects had no fully active agents, 31% had 1 fully active agent, and 17% had 2 or more fully active agents within their initial failing regimen, including 6% of subjects were who were receiving fostemsavir, which was an investigational agent at the start of the CAPELLA trial.
Subjects in cohort 2 initiated SUNLENCA and an OBR on Day 1.
Subjects in cohort 2 had a mean age of 48 years (range: 23 to 78), 78% were male, 36% were White, 31% were Black, 33% were Asian, and 14% of subjects identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.1 log10 copies/mL (range: 1.3 to 5.7). 19% of subjects had baseline viral loads greater than 100,000 copies/mL. The mean baseline CD4+ cell count was 258 cells/mm3 (range: 3 to 1296). 53% of subjects had CD4+ cell counts below 200 cells/mm3. The mean number of years since subjects first started HIV treatment was 19 years (range: 3 to 35); the mean number of antiretroviral agents in failing regimens at baseline was 4 (range: 2 to 7). The percentage of subjects in the non-randomized cohort with known resistance to at least 2 agents from the NRTI, NNRTI, PI and INSTI classes was 100%, 100%, 83% and 64%, respectively. In cohort 2, 31% of subjects had no fully active agents, 42% had 1 fully active agent, and 28% had 2 or more fully active agents within their initial failing regimen, including 6% of subjects who were receiving fostemsavir, which was an investigational agent at the start of the CAPELLA trial.
The primary efficacy endpoint was the proportion of subjects in cohort 1 achieving ≥ 0.5 log10 copies/mL reduction from baseline in HIV-1 RNA at the end of the functional monotherapy period. The results of the primary endpoint analysis are shown in Table 10.
| SUNLENCA (N=24) | Placebo (N=12) |
|
|---|---|---|
|
||
| Proportion of Subjects Achieving a ≥ 0.5 log10 Decrease in Viral Load | 87.5% | 16.7% |
| Treatment Difference (95% CI) | 70.8% (34.9% to 90.0%) * | |
The results at Weeks 26 and 52 are provided in Table 11 and Table 12.
| SUNLENCA plus OBR (N=36) |
||
|---|---|---|
| Week 26 | Week 52 | |
| OBR = optimized background regimen | ||
|
||
| HIV-1 RNA < 50 copies/mL | 81% | 83% |
| HIV-1 RNA ≥ 50 copies/mL‡ | 19% | 14% |
| No virologic data in Week 26 or 52 Window | 0 | 3% |
| Discontinued Study Drug Due to AE or Death § | 0 | 0 |
| Discontinued Study Drug Due to Other Reasons ¶ and Last Available HIV-1 RNA < 50 copies/mL | 0 | 3% |
| Missing Data During Window but on Study Drug | 0 | 0 |
| SUNLENCA plus OBR (N=36) |
||
|---|---|---|
| Week 26 | Week 52 | |
| ARV = antiretroviral; DRV=darunavir; DTG=dolutegravir; INSTI = integrase strand-transfer inhibitor; OBR = optimized background regimen; | ||
|
||
| Age (Years) | ||
| < 50 | 100% (9/9) | 89% (8/9) |
| ≥ 50 | 74% (20/27) | 81% (22/27) |
| Gender | ||
| Male | 77% (20/26) | 77% (20/26) |
| Female | 90% (9/10) | 100% (10/10) |
| Race | ||
| Black | 81% (13/16) | 75% (12/16) |
| Non-Black | 84% (16/19) | 89% (17/19) |
| Baseline plasma viral load (copies/mL) | ||
| ≤ 100,000 | 86% (25/29) | 86% (25/29) |
| > 100,000 | 57% (4/7) | 71% (5/7) |
| Baseline CD4+ (cells/mm3) | ||
| < 200 | 78% (21/27) | 78% (21/27) |
| ≥ 200 | 89% (8/9) | 100% (9/9) |
| Baseline INSTI resistance profile | ||
| With INSTI resistance | 85% (23/27) | 81% (22/27) |
| Without INSTI resistance | 63% (5/8) | 88% (7/8) |
| Number of fully active ARV agents in the OBR | ||
| 0 | 67% (4/6) | 67% (4/6) |
| 1 | 86% (12/14) | 79% (11/14) |
| ≥ 2 | 81% (13/16) | 94% (15/16) |
| Use of DTG and/or DRV in the OBR | ||
| With DTG and DRV | 83% (10/12) | 83% (10/12) |
| With DTG, without DRV | 83% (5/6) | 83% (5/6) |
| Without DTG, with DRV | 78% (7/9) | 89% (8/9) |
| Without DTG or DRV | 78% (7/9) | 78% (7/9) |
In cohort 1, at Weeks 26 and 52, the mean change from baseline in CD4+ cell count was 81 cells/mm3 (range: -101 to 522) and 82 cells/mm3 (range: -194 to 467), respectively.
In cohort 2, at Week 26 and 52, 81% (29/36) and 72% (26/36) of patients achieved HIV-1 RNA < 50 copies/mL, respectively, and the mean change from baseline in CD4+ cell count was 97 cells/mm3 (range: -103 to 459) and 113 cells/mm3 (range: -124 to 405), respectively.
16. How is Sunlenca supplied



| SUNLENCA
lenacapavir sodium tablet, film coated |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| SUNLENCA
lenacapavir sodium kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Gilead Sciences (185049848) |
