Drug Detail:Ultomiris (Ravulizumab [ rav-ue-liz-ue-mab ])
Drug Class: Selective immunosuppressants
Highlights of Prescribing Information
ULTOMIRIS® (ravulizumab-cwvz) injection, for intravenous or subcutaneous use
Initial U.S. Approval: 2018
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
See full prescribing information for complete boxed warning
Life-threatening meningococcal infections/sepsis have occurred in patients treated with ULTOMIRIS and may become rapidly life-threatening or fatal if not recognized and treated early (5.1).
- Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for meningococcal vaccination in patients with complement deficiencies (5.1).
- Immunize patients with meningococcal vaccines at least 2 weeks prior to administering the first dose of ULTOMIRIS, unless the risks of delaying ULTOMIRIS therapy outweigh the risks of developing a meningococcal infection. See Warnings and Precautions (5.1) for additional guidance on the management of the risk of meningococcal infection.
- Vaccination reduces, but does not eliminate, the risk of meningococcal infection. Monitor patients for early signs of meningococcal infections, and evaluate immediately if infection is suspected.
ULTOMIRIS is available only through a restricted program called the ULTOMIRIS REMS (5.2).
Recent Major Changes
| Indications and Usage (1.3) | 04/2022 |
| Dosing and Administration (2.3, 2.4, 2.5) | 04/2022 |
| Dosage and Administration (2.1, 2.3, 2.4, 2.6) | 07/2022 |
| Warnings and Precautions (5.1, 5.3, 5.6) | 04/2022 |
| Warnings and Precautions (5.2, 5.7) | 07/2022 |
Indications and Usage for Ultomiris
ULTOMIRIS is a complement inhibitor indicated for:
- the treatment of adult and pediatric patients one month of age and older with paroxysmal nocturnal hemoglobinuria (PNH) (1.1).
- the treatment of adult and pediatric patients one month of age and older with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy (TMA) (1.2).
Limitations of Use:
ULTOMIRIS is not indicated for the treatment of patients with Shiga toxin E. coli related hemolytic uremic syndrome (STEC-HUS). - the treatment of adult patients with generalized myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody-positive (1.3).
Ultomiris Dosage and Administration
- See Full Prescribing Information for instructions on dosage, preparation, and administration (2.1, 2.2, 2.3, 2.4, 2.5, 2.6).
- Dilute ULTOMIRIS before use (2.6).
- Only administer as an intravenous infusion through a 0.2 or 0.22 micron filter (2.6).
Dosage Forms and Strengths
Intravenous
Injection: 300 mg/30 mL (10 mg/mL) solution in a single-dose vial (3).
Injection: 300 mg/3 mL (100 mg/mL) solution in a single-dose vial (3).
Injection: 1,100 mg/11 mL (100 mg/mL) solution in a single-dose vial (3).
Subcutaneous
Injection: 245 mg/3.5 mL (70 mg/mL) solution in a single-dose prefilled cartridge for use only with supplied single-use on-body injector (3).
Contraindications
ULTOMIRIS is contraindicated in:
- Patients with unresolved Neisseria meningitidis infection (4).
- Patients who are not currently vaccinated against Neisseria meningitidis, unless the risks of delaying ULTOMIRIS treatment outweigh the risks of developing a meningococcal infection (4, 5.1).
Warnings and Precautions
- Other Infections: Use caution when administering ULTOMIRIS to patients with any other systemic infection (5.3).
- Infusion-Related Reactions: Monitor during infusion, interrupt for reactions, and institute appropriate supportive measures (5.6).
- Allergies to Acrylic Adhesives: The on-body injector of ULTOMIRIS uses acrylic adhesive. For patients with a known allergy to acrylic adhesive, use of this product may result in an allergic reaction (5.7).
Adverse Reactions/Side Effects
Most common adverse reactions in patients with PNH (incidence ≥ 10%) were upper respiratory tract infection and headache. Injection site reactions and diarrhea occurred in patients (incidence ≥ 10%) receiving ULTOMIRIS subcutaneously (6.1).
Most common adverse reactions in patients with aHUS (incidence ≥ 20%) were upper respiratory tract infection, diarrhea, nausea, vomiting, headache, hypertension, and pyrexia (6.1).
Most common adverse reactions in adult patients with gMG (incidence ≥ 10%) were diarrhea and upper respiratory tract infection (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Plasma Exchange, Plasmapheresis, or Intravenous Immunoglobulins: concomitant use requires supplemental dose of ULTOMIRIS (7.1).
- Neonatal Fc Receptor Blockers: Closely monitor for reduced effectiveness of ULTOMIRIS (7.2).
Use In Specific Populations
Pediatric Use: Subcutaneous dosing of ULTOMIRIS is not approved for use in pediatric patients (8.4).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2022
Full Prescribing Information
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
Life-threatening meningococcal infections/sepsis have occurred in patients treated with ULTOMIRIS. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early [see Warnings and Precautions (5.1)].
- Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for meningococcal vaccination in patients with complement deficiencies.
- Immunize patients with meningococcal vaccines at least 2 weeks prior to administering the first dose of ULTOMIRIS, unless the risks of delaying ULTOMIRIS therapy outweigh the risk of developing a meningococcal infection. See Warnings and Precautions (5.1) for additional guidance on the management of the risk of meningococcal infection.
- Vaccination reduces, but does not eliminate, the risk of meningococcal infections. Monitor patients for early signs of meningococcal infections and evaluate immediately if infection is suspected.
Because of the risk of serious meningococcal infections, ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS REMS [see Warnings and Precautions (5.2)].
1. Indications and Usage for Ultomiris
1.1 Paroxysmal Nocturnal Hemoglobinuria
ULTOMIRIS is indicated for the treatment of adult and pediatric patients one month of age and older with paroxysmal nocturnal hemoglobinuria (PNH).
2. Ultomiris Dosage and Administration
2.1 Important Dosage Information
ULTOMIRIS may be administered as an intravenous infusion (ULTOMIRIS vial for intravenous administration) in adult or pediatric patients one month of age and older or as a subcutaneous injection for maintenance (ULTOMIRIS on-body delivery system for subcutaneous administration) in adult patients.
Vials are intended for intravenous use only, and on-body delivery systems (prefilled cartridge and on-body injector) are intended for subcutaneous maintenance use only.
2.2 Recommended Vaccination and Prophylaxis
Vaccinate patients for meningococcal disease according to current ACIP guidelines to reduce the risk of serious infection [see Warnings and Precautions (5.1, 5.3)].
Provide 2 weeks of antibacterial drug prophylaxis to patients if ULTOMIRIS must be initiated immediately and vaccines are administered less than 2 weeks before starting ULTOMIRIS therapy.
Healthcare professionals who prescribe ULTOMIRIS must enroll in the ULTOMIRIS REMS [see Warnings and Precautions (5.1)].
2.3 Intravenous Administration in Adult and Pediatric Patients with PNH, aHUS, or gMG
The recommended intravenous ULTOMIRIS loading and maintenance dosing in adult and pediatric patients, one month of age or older weighing 5 kg or greater, with PNH or aHUS, or in adult patients with gMG weighing 40 kg or greater, is based on the patient's body weight, as shown in Table 1, with maintenance doses administered every 4 or 8 weeks, starting 2 weeks after loading dose.
The intravenous (IV) dosing schedule is allowed to occasionally vary within 7 days of the scheduled infusion day (except for the first maintenance dose of ULTOMIRIS); but subsequent doses should be administered according to the original schedule.
Following a missed intravenous ULTOMIRIS dose, the patient should contact their health care provider immediately.
| Indications | Body Weight Range (kg) | Loading Dose (mg)† | Maintenance Dose (mg) and Dosing Interval | |
|---|---|---|---|---|
|
||||
| PNH and aHUS | 5 to less than 10 | 600 | 300 | Every 4 weeks |
| 10 to less than 20 | 600 | 600 | ||
| 20 to less than 30 | 900 | 2,100 | Every 8 weeks |
|
| 30 to less than 40 | 1,200 | 2,700 | ||
| PNH, aHUS, and gMG | 40 to less than 60 | 2,400 | 3,000 | Every 8 weeks |
| 60 to less than 100 | 2,700 | 3,300 | ||
| 100 or greater | 3,000 | 3,600 | ||
Refer to Table 2 for treatment initiation instructions in patients who are complement inhibitor treatment-naïve, or switching treatment from subcutaneous (SUBQ) administration of ULTOMIRIS or eculizumab.
| Population | Weight-based ULTOMIRIS IV Loading Dose | Time of First ULTOMIRIS IV Weight-based Maintenance Dose |
|---|---|---|
|
||
| Not currently on ULTOMIRIS or eculizumab treatment | At treatment start | 2 weeks after ULTOMIRIS IV loading dose |
| Currently treated with eculizumab | At time of next scheduled eculizumab dose | 2 weeks after ULTOMIRIS IV loading dose |
| Currently treated with ULTOMIRIS on-body delivery system for subcutaneous administration (SUBQ)* | Not applicable | 1 week after last ULTOMIRIS SUBQ maintenance dose |
2.4 Subcutaneous Administration in Adult Patients with PNH or aHUS
Subcutaneous (SUBQ) dosing of ULTOMIRIS is not approved for use in pediatric patients.
The recommended subcutaneous ULTOMIRIS maintenance dose is 490 mg once weekly in adult patients greater than or equal to 40 kg body weight with PNH or aHUS. The 490 mg dose of ULTOMIRIS is delivered using 2 on-body delivery systems. Each on-body delivery system consists of 1 on-body injector and 1 prefilled cartridge containing 245 mg of ravulizumab.
The subcutaneous dosing schedule is allowed to occasionally vary by ± 1 day of the scheduled dose day, but the subsequent dose should be administered according to the original schedule.
Following a missed or partial subcutaneous ULTOMIRIS dose, the patient should contact their health care provider immediately.
Refer to Table 3 for treatment initiation instructions in patients who are complement inhibitor treatment-naïve, or switching treatment from intravenous administration of ULTOMIRIS or eculizumab.
| Population | Weight-based ULTOMIRIS IV Loading Dose* | Time of First ULTOMIRIS 490 mg SUBQ Maintenance Dose |
|---|---|---|
|
||
| Not currently on ULTOMIRIS or eculizumab treatment | At treatment start | 2 weeks after ULTOMIRIS IV loading dose |
| Currently treated with eculizumab | At time of next scheduled eculizumab dose | 2 weeks after ULTOMIRIS IV loading dose |
| Currently treated with ULTOMIRIS intravenous (IV) administration | Not applicable | 8 weeks after last ULTOMIRIS IV maintenance dose |
2.5 Dosing Considerations
Supplemental Dose of ULTOMIRIS
Plasma exchange (PE), plasmapheresis (PP), and intravenous immunoglobulin (IVIg) have been shown to reduce ULTOMIRIS serum levels. A supplemental dose of ULTOMIRIS is required in the setting of PE, PP, or IVIg (Table 4).
| Body Weight Range (kg) | Most Recent ULTOMIRIS Dose (mg) | Supplemental Dose (mg) following each PE or PP Intervention | Supplemental Dose (mg) following Completion of an IVIg Cycle |
|---|---|---|---|
| Abbreviations: IVIg = intravenous immunoglobulin; PE = plasma exchange; PP = plasmapheresis | |||
|
|||
| 40 to less than 60 | 2,400 | 1,200 | 600 |
| 3,000 | 1,500 | ||
| 60 to less than 100 | 2,700 | 1,500 | 600 |
| 3,300 | 1,800 | ||
| 100 or greater | 3,000 | 1,500 | 600 |
| 3,600 | 1,800 | ||
| Timing of ULTOMIRIS Supplemental Dose | Within 4 hours following each PE or PP intervention | Within 4 hours following completion of an IVIg cycle | |
2.6 Preparation and Administration
Intravenous Administration of ULTOMIRIS (Healthcare Providers)
Only administer as an intravenous infusion through a 0.2 or 0.22 micron filter.
Dilute ULTOMIRIS to a final concentration of:
- 50 mg/mL for the 3 mL and 11 mL vial sizes or
- 5 mg/mL for the 30 mL vial size.
Prior to administration, allow the admixture to adjust to room temperature (18°C - 25°C, 64°F - 77°F). Do not heat the admixture in a microwave or with any heat source other than ambient air temperature.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
| Body Weight Range (kg)* | Loading Dose (mg) | ULTOMIRIS Volume (mL) | Volume of NaCl Diluent† (mL) | Total Volume (mL) | Minimum Infusion Time (hr) | Maximum Infusion Rate (mL/hr) |
|---|---|---|---|---|---|---|
|
||||||
| 5 to less than 10‡ | 600 | 6 | 6 | 12 | 1.4 | 9 |
| 10 to less than 20‡ | 600 | 6 | 6 | 12 | 0.8 | 15 |
| 20 to less than 30‡ | 900 | 9 | 9 | 18 | 0.6 | 30 |
| 30 to less than 40‡ | 1,200 | 12 | 12 | 24 | 0.5 | 48 |
| 40 to less than 60 | 2,400 | 24 | 24 | 48 | 0.8 | 60 |
| 60 to less than 100 | 2,700 | 27 | 27 | 54 | 0.6 | 90 |
| 100 or greater | 3,000 | 30 | 30 | 60 | 0.4 | 150 |
| Body Weight Range (kg)* | Maintenance Dose (mg) | ULTOMIRIS Volume (mL) | Volume of NaCl Diluent† (mL) | Total Volume (mL) | Minimum Infusion Time (hr) | Maximum Infusion Rate (mL/hr) |
|---|---|---|---|---|---|---|
|
||||||
| 5 to less than 10‡ | 300 | 3 | 3 | 6 | 0.8 | 8 |
| 10 to less than 20‡ | 600 | 6 | 6 | 12 | 0.8 | 15 |
| 20 to less than 30‡ | 2,100 | 21 | 21 | 42 | 1.3 | 33 |
| 30 to less than 40‡ | 2,700 | 27 | 27 | 54 | 1.1 | 50 |
| 40 to less than 60 | 3,000 | 30 | 30 | 60 | 0.9 | 67 |
| 60 to less than 100 | 3,300 | 33 | 33 | 66 | 0.7 | 95 |
| 100 or greater | 3,600 | 36 | 36 | 72 | 0.5 | 144 |
| Body Weight Range (kg)* | Supplemental Dose (mg) | ULTOMIRIS Volume (mL) | Volume of NaCl Diluent† (mL) | Total Volume (mL) | Minimum Infusion Time (hr) | Maximum Infusion Rate (mL/hr) |
|---|---|---|---|---|---|---|
| Note: Refer to Table 4 for selection of ravulizumab supplemental dose | ||||||
|
||||||
| 40 to less than 60 | 600 | 6 | 6 | 12 | 0.25 | 48 |
| 1,200 | 12 | 12 | 24 | 0.42 | 57 | |
| 1,500 | 15 | 15 | 30 | 0.5 | 60 | |
| 60 to less than 100 | 600 | 6 | 6 | 12 | 0.20 | 60 |
| 1,500 | 15 | 15 | 30 | 0.36 | 83 | |
| 1,800 | 18 | 18 | 36 | 0.42 | 86 | |
| 100 or greater | 600 | 6 | 6 | 12 | 0.17 | 71 |
| 1,500 | 15 | 15 | 30 | 0.25 | 120 | |
| 1,800 | 18 | 18 | 36 | 0.28 | 129 | |
| Body Weight Range (kg)* | Loading Dose (mg) | ULTOMIRIS Volume (mL) | Volume of NaCl Diluent† (mL) | Total Volume (mL) | Minimum Infusion Time (hr) | Maximum Infusion Rate (mL/hr) |
|---|---|---|---|---|---|---|
|
||||||
| 5 to less than 10‡ | 600 | 60 | 60 | 120 | 3.8 | 32 |
| 10 to less than 20‡ | 600 | 60 | 60 | 120 | 1.9 | 64 |
| 20 to less than 30‡ | 900 | 90 | 90 | 180 | 1.5 | 120 |
| 30 to less than 40‡ | 1,200 | 120 | 120 | 240 | 1.3 | 185 |
| 40 to less than 60 | 2,400 | 240 | 240 | 480 | 1.9 | 253 |
| 60 to less than 100 | 2,700 | 270 | 270 | 540 | 1.7 | 318 |
| 100 or greater | 3,000 | 300 | 300 | 600 | 1.8 | 334 |
| Body Weight Range (kg)* | Maintenance Dose (mg) | ULTOMIRIS Volume (mL) | Volume of NaCl Diluent† (mL) | Total Volume (mL) | Minimum Infusion Time (hr) | Maximum Infusion Rate (mL/hr) |
|---|---|---|---|---|---|---|
|
||||||
| 5 to less than 10‡ | 300 | 30 | 30 | 60 | 1.9 | 32 |
| 10 to less than 20‡ | 600 | 60 | 60 | 120 | 1.9 | 64 |
| 20 to less than 30‡ | 2,100 | 210 | 210 | 420 | 3.3 | 128 |
| 30 to less than 40‡ | 2,700 | 270 | 270 | 540 | 2.8 | 193 |
| 40 to less than 60 | 3,000 | 300 | 300 | 600 | 2.3 | 261 |
| 60 to less than 100 | 3,300 | 330 | 330 | 660 | 2 | 330 |
| 100 or greater | 3,600 | 360 | 360 | 720 | 2.2 | 328 |
| Body Weight Range (kg)* | Supplemental Dose (mg) | ULTOMIRIS Volume (mL) | Volume of NaCl Diluent† (mL) | Total Volume (mL) | Minimum Infusion Time (hr) | Maximum Infusion Rate (mL/hr) |
|---|---|---|---|---|---|---|
| Note: Refer to Table 4 for selection of ravulizumab supplemental dose | ||||||
|
||||||
| 40 to less than 60 | 600 | 60 | 60 | 120 | 0.5 | 240 |
| 1,200 | 120 | 120 | 240 | 1.0 | 240 | |
| 1,500 | 150 | 150 | 300 | 1.2 | 250 | |
| 60 to less than 100 | 600 | 60 | 60 | 120 | 0.4 | 300 |
| 1,500 | 150 | 150 | 300 | 1.0 | 300 | |
| 1,800 | 180 | 180 | 360 | 1.1 | 327 | |
| 100 or greater | 600 | 60 | 60 | 120 | 0.4 | 300 |
| 1,500 | 150 | 150 | 300 | 1.0 | 300 | |
| 1,800 | 180 | 180 | 360 | 1.1 | 327 | |
If an adverse reaction occurs during the intravenous administration of ULTOMIRIS, the infusion may be slowed or stopped at the discretion of the physician. Monitor the patient for at least 1 hour following completion of the infusion for signs or symptoms of an infusion-related reaction.
4. Contraindications
ULTOMIRIS is contraindicated in:
- Patients with unresolved Neisseria meningitidis infection [see Warnings and Precautions (5.1)].
- Patients who are not currently vaccinated against Neisseria meningitidis, unless the risks of delaying ULTOMIRIS treatment outweigh the risks of developing a meningococcal infection [see Warnings and Precautions (5.1)].
5. Warnings and Precautions
5.1 Serious Meningococcal Infections
Life-threatening meningococcal infections have occurred in patients treated with ULTOMIRIS. The use of ULTOMIRIS increases a patient's susceptibility to serious meningococcal infections (septicemia and/or meningitis). Meningococcal disease due to any serogroup may occur.
Vaccinate for meningococcal disease according to the most current Advisory Committee on Immunization Practices (ACIP) recommendations for patients with complement deficiencies. Revaccinate patients in accordance with ACIP recommendations considering the duration of ULTOMIRIS therapy.
Immunize patients without a history of meningococcal vaccination at least 2 weeks prior to receiving the first dose of ULTOMIRIS. Patients who initiate ULTOMIRIS treatment less than 2 weeks after receiving meningococcal vaccine(s) must receive appropriate prophylactic antibiotics until 2 weeks after vaccination.
In clinical studies, 59 adult patients with PNH and 2 adult patients with gMG were treated with ULTOMIRIS less than 2 weeks after meningococcal vaccination. All of these patients received antibiotics for prophylaxis of meningococcal infection until at least 2 weeks after meningococcal vaccination. The benefits and risks of antibiotic prophylaxis for prevention of meningococcal infections in patients receiving ULTOMIRIS have not been established.
Vaccination reduces, but does not eliminate, the risk of meningococcal infections. In clinical studies with ULTOMIRIS, < 1% of patients developed serious meningococcal infections/sepsis while receiving treatment with ULTOMIRIS. All were adult patients with PNH who had been vaccinated. These patients recovered while continuing treatment with ULTOMIRIS.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and steps to be taken to seek immediate medical care. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider discontinuation of ULTOMIRIS in patients who are undergoing treatment for serious meningococcal infection.
ULTOMIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions (5.2)].
5.2 ULTOMIRIS REMS
ULTOMIRIS is available only through a restricted program under a REMS called ULTOMIRIS REMS, due to the risk of meningococcal infections [see Warnings and Precautions (5.2)].
Notable requirements of the ULTOMIRIS REMS include the following:
- Prescribers must enroll in the program
- Prescribers must counsel patients about the risk of meningococcal infection/sepsis
- Prescribers must provide the patients with the REMS educational materials
- Prescribers must ensure patients are vaccinated with meningococcal vaccines or receive 2 weeks of antibacterial drug prophylaxis if treatment must be initiated immediately and vaccines are administered less than 2 weeks before starting ULTOMIRIS
- Patients must receive counseling from the prescriber, take antibiotics as directed by the prescriber after vaccination (if they have to start ULTOMIRIS right away), receive meningococcal vaccines as directed by the prescriber, and carry the Patient Safety Card with them at all times during and for 8 months following treatment with ULTOMIRIS.
Further information is available at www.ultomirisrems.com or 1-888-765-4747.
5.3 Other Infections
ULTOMIRIS blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria, such as infections caused by Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Children treated with ULTOMIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP guidelines. If ULTOMIRIS therapy is administered to patients with active systemic infections, monitor closely for signs and symptoms of worsening infection.
5.5 Thromboembolic Event Management
The effect of withdrawal of anticoagulant therapy during ULTOMIRIS treatment has not been established. Therefore, treatment with ULTOMIRIS should not alter anticoagulant management.
5.6 Infusion-Related Reactions
Intravenous or subcutaneous administration of ULTOMIRIS may result in systemic infusion-related reactions, including anaphylaxis [see Adverse Reactions (6.3)] and hypersensitivity reactions. In clinical trials, infusion-related reactions occurred in approximately 1% of patients treated with ULTOMIRIS. These events included lower back pain, drop in blood pressure, elevation in blood pressure, limb discomfort, drug hypersensitivity (allergic reaction), dysgeusia (bad taste), and drowsiness. These reactions did not require discontinuation of ULTOMIRIS. If signs of cardiovascular instability or respiratory compromise occur, interrupt ULTOMIRIS infusion and institute appropriate supportive measures.
5.7 Allergies to Acrylic Adhesives
The on-body injector of ULTOMIRIS uses acrylic adhesive. For patients with a known allergy to acrylic adhesive, use of this product may result in an allergic reaction. Premedication can be considered, and supportive measures should be instituted if signs of allergy appear.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Serious Meningococcal Infections [see Warnings and Precautions (5.1)]
- Other Infections [see Warnings and Precautions (5.3)]
- Infusion-Related Reactions [see Warnings and Precautions (5.6)]
- Allergies to Acrylic Adhesives [see Warnings and Precautions (5.7)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Paroxysmal Nocturnal Hemoglobinuria (PNH)
Adult Population with PNH Treated with Intravenous ULTOMIRIS
The data described below reflect exposure of 441 adult patients with PNH in Phase 3 studies who received intravenously administered ULTOMIRIS (n = 222) or eculizumab (n = 219) at the recommended dosing regimens with median treatment duration of 6 months for ULTOMIRIS and 6 months for eculizumab. The most frequent adverse reactions (≥ 10%) with ULTOMIRIS were upper respiratory tract infection and headache. Table 11 describes adverse reactions that occurred at a rate of 5% or more among patients treated with ULTOMIRIS in PNH studies.
Serious adverse reactions were reported in 15 (6.8%) patients with PNH receiving ULTOMIRIS. The serious adverse reactions in patients treated with ULTOMIRIS included hyperthermia and pyrexia. No serious adverse reaction was reported in more than 1 patient treated with ULTOMIRIS.
One fatal case of sepsis was identified in a patient treated with ULTOMIRIS.
| Body System Adverse Reaction | Number of Patients | |
|---|---|---|
| ULTOMIRIS (IV) (N=222) n (%) | Eculizumab (N=219) n (%) |
|
|
||
| Gastrointestinal disorders | ||
| Diarrhea | 19 (9) | 12 (5) |
| Nausea | 19 (9) | 19 (9) |
| Abdominal pain | 13 (6) | 16 (7) |
| General disorders and administration site conditions | ||
| Pyrexia | 15 (7) | 18 (8) |
| Infections and infestations | ||
| Upper respiratory tract infection* | 86 (39) | 86 (39) |
| Musculoskeletal and connective tissue disorders | ||
| Pain in extremity | 14 (6) | 11 (5) |
| Arthralgia | 11 (5) | 12 (5) |
| Nervous system disorders | ||
| Headache | 71 (32) | 57 (26) |
| Dizziness | 12 (5) | 14 (6) |
Clinically relevant adverse reactions in 1% of patients include infusion-related reactions.
Pediatric Population with PNH Treated with Intravenous ULTOMIRIS
In pediatric patients with PNH (aged 9 to 17 years old) included in the pediatric PNH Phase 3 study, the safety profile appeared similar to that observed in adult patients with PNH and in pediatric and adult patients with aHUS. The most common adverse reactions (> 20%) were upper respiratory tract infection, anemia, abdominal pain, and headache. Table 12 describes the adverse reactions that occurred at a rate of 10% or more among pediatric patients treated with ULTOMIRIS administered intravenously in Study ALXN1210-PNH-304.
| Body System Adverse Reaction | Treatment Naïve (N=5) | Eculizumab Experienced (N=8) | Total (N=13) |
|---|---|---|---|
| n (%) | n (%) | n (%) | |
|
|||
| Blood and lymphatic system disorders | |||
| Anemia* | 1 (20) | 2 (25) | 3 (23) |
| Gastrointestinal disorders | |||
| Abdominal pain | 0 (0) | 3 (38) | 3 (23) |
| Constipation | 0 (0) | 2 (25) | 2 (15) |
| General disorders and administration site conditions | |||
| Pyrexia | 1 (20) | 1 (13) | 2 (15) |
| Infections and infestations | |||
| Upper Respiratory tract infection† | 1 (20) | 6 (75) | 7 (54) |
| Musculoskeletal and connective tissue disorders | |||
| Pain in extremity | 0 (0) | 2 (25) | 2 (15) |
| Nervous system disorders | |||
| Headache | 1 (20) | 2 (25) | 3 (23) |
Subcutaneous Administration of ULTOMIRIS
In the Phase 3 study ALXN1210-PNH-303 with subcutaneous administration of ULTOMIRIS via the on-body injector for adult patients with PNH, during the 10-week randomization period the most common adverse reactions (≥ 10%) with ULTOMIRIS subcutaneous administration were local injection site reactions, diarrhea, and headache. Table 13 describes adverse reactions that occurred at a rate of 10% or more among adult patients treated with ULTOMIRIS in Study ALXN1210-PNH-303 during the 10-week randomized period.
| Number of Patients | ||
|---|---|---|
| Body System Adverse Reaction | ULTOMIRIS (SUBQ) (N = 84) n (%) | ULTOMIRIS (IV) (N = 45) n (%) |
|
||
| Gastrointestinal disorders | ||
| Diarrhea | 11 (13) | 2 (4) |
| General disorders and administration site conditions | ||
| Injection site reaction* | 23 (27) | 0 (0) |
| Infections and infestations | ||
| Upper respiratory tract infection† | 7 (8) | 6 (13) |
| Nervous system disorders | ||
| Headache | 11 (13) | 4 (9) |
Atypical Hemolytic Uremic Syndrome (aHUS)
The data described below reflect exposure of 58 adult and 16 pediatric patients with aHUS in single-arm trials who received ULTOMIRIS administered intravenously at the recommended dose and schedule. The most frequent adverse reactions reported in ≥ 20% of patients treated with ULTOMIRIS were upper respiratory tract infection, diarrhea, nausea, vomiting, headache, hypertension, and pyrexia. Table 14, Table 15 and Table 16 describe adverse reactions that occurred at a rate of 10% or more among patients treated with ULTOMIRIS in aHUS studies. Serious adverse reactions were reported in 42 (57%) patients with aHUS receiving ULTOMIRIS. The most frequent serious adverse reactions reported in more than 2 patients (2.7%) treated with ULTOMIRIS were hypertension, pneumonia, and abdominal pain. Four patients died during the ALXN1210-aHUS-311 study. The cause of death was sepsis in 2 patients and intracranial hemorrhage in 1 patient. The fourth patient, who was excluded from the trial after a diagnosis of STEC-HUS, died due to pretreatment cerebral arterial thrombosis.
| Body System Adverse Reaction | ALXN1210-aHUS-311 (N=58) |
|
|---|---|---|
| All Grades*
(n=53) n (%) | ≥ Grade 3 (n=14) n (%) |
|
|
||
| Blood and lymphatic system disorders | ||
| Anemia | 8 (14) | 0 (0) |
| Gastrointestinal disorders | ||
| Diarrhea | 18 (31) | 2 (3) |
| Nausea | 15 (26) | 2 (3) |
| Vomiting | 15 (26) | 2 (3) |
| Constipation | 8 (14) | 1 (2) |
| Abdominal pain | 7 (12) | 1 (2) |
| General disorders and administration site conditions | ||
| Pyrexia | 11 (19) | 1 (2) |
| Edema peripheral | 10 (17) | 0 (0) |
| Fatigue | 8 (14) | 0 (0) |
| Infections and infestations | ||
| Upper respiratory tract infection† | 15 (26) | 0 (0) |
| Urinary tract infection | 10 (17) | 5 (9) |
| Gastrointestinal infection‡ | 8 (14) | 2 (3) |
| Metabolism and nutrition disorders | ||
| Hypokalemia | 6 (10) | 1 (2) |
| Musculoskeletal and connective tissue disorders | ||
| Arthralgia | 13 (22) | 0 (0) |
| Back pain | 7 (12) | 1 (2) |
| Muscle spasms | 6 (10) | 0 (0) |
| Pain in extremity | 6 (10) | 0 (0) |
| Nervous system disorders | ||
| Headache | 23 (40) | 1 (2) |
| Psychiatric disorders | ||
| Anxiety | 8 (14) | 1 (2) |
| Respiratory, thoracic and mediastinal disorders | ||
| Cough | 10 (17) | 0 (0) |
| Dyspnea | 10 (17) | 1 (2) |
| Skin and subcutaneous tissue disorders | ||
| Alopecia | 6 (10) | 0 (0) |
| Dry skin | 6 (10) | 0 (0) |
| Vascular disorders | ||
| Hypertension | 14 (24) | 7 (12) |
Clinically relevant adverse reactions include viral tonsilitis (in < 10% of patients) and infusion-related reactions (in 3% of patients).
| Body System Adverse Reaction | ALXN1210-aHUS-312 (N=16) |
|
|---|---|---|
| All Grades*
(n=16) n (%) | ≥ Grade 3 (n=6) n (%) |
|
|
||
| Blood and lymphatic system disorders | ||
| Anemia | 2 (13) | 1 (6) |
| Lymphadenopathy | 2 (13) | 0 (0) |
| Gastrointestinal disorders | ||
| Diarrhea | 6 (38) | 0 (0) |
| Constipation | 4 (25) | 0 (0) |
| Vomiting | 4 (25) | 1 (6) |
| Abdominal pain | 3 (19) | 0 (0) |
| Nausea | 2 (13) | 0 (0) |
| General disorders and administration site conditions | ||
| Pyrexia | 8 (50) | 0 (0) |
| Infections and infestations | ||
| Upper respiratory tract infection† | 7 (44) | 1 (6) |
| Gastroenteritis viral | 2 (13) | 2 (13) |
| Pneumonia | 2 (13) | 1 (6) |
| Tonsillitis | 2 (13) | 0 (0) |
| Injury, poisoning and procedural complications | ||
| Contusion | 3 (19) | 0 (0) |
| Investigations | ||
| Vitamin D decreased | 3 (19) | 0 (0) |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 2 (13) | 0 (0) |
| Iron deficiency | 2 (13) | 0 (0) |
| Musculoskeletal and connective tissue disorders | ||
| Myalgia | 3 (19) | 0 (0) |
| Pain in extremity | 2 (13) | 0 (0) |
| Nervous system disorders | ||
| Headache | 5 (31) | 0 (0) |
| Respiratory, thoracic and mediastinal disorders | ||
| Cough | 3 (19) | 0 (0) |
| Dyspnea | 2 (13) | 0 (0) |
| Skin and subcutaneous tissue disorders | ||
| Rash | 3 (19) | 0 (0) |
| Vascular disorders | ||
| Hypertension | 4 (25) | 1 (6) |
| Hypotension | 2 (13) | 0 (0) |
Clinically relevant adverse reactions in < 10% of patients include viral infection.
| Body System Adverse Reaction | ALXN1210-aHUS-312 | |||
|---|---|---|---|---|
| Age 0 to < 2 (N=2) | Age 2 to < 12 (N=12) | Age 12 to 16 (N=1) | Total (N=15) |
|
| n (%) | n (%) | n (%) | n (%) | |
|
||||
| Blood and lymphatic system disorders | ||||
| Lymphadenopathy | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Gastrointestinal disorders | ||||
| Diarrhea | 1 (50) | 3 (25) | 1 (100) | 5 (33) |
| Constipation | 0 (0) | 4 (33) | 0 (0) | 4 (27) |
| Vomiting | 0 (0) | 3 (25) | 0 (0) | 3 (20) |
| Abdominal pain | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| General disorders and administration site conditions | ||||
| Pyrexia | 1 (50) | 5 (42) | 1 (100) | 7 (47) |
| Infections and infestations | ||||
| Upper respiratory tract infection* | 1 (50) | 6 (50) | 0 (0) | 7 (47) |
| Gastroenteritis viral | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Tonsillitis | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Injury, poisoning and procedural complications | ||||
| Contusion | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Investigations | ||||
| Vitamin D decreased | 0 (0) | 2 (17) | 1 (100) | 3 (20) |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Iron deficiency | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Musculoskeletal and connective tissue disorders | ||||
| Myalgia | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Pain in extremity | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Nervous system disorders | ||||
| Headache | 0 (0) | 4 (33) | 0 (0) | 4 (27) |
| Respiratory, thoracic and mediastinal disorders | ||||
| Cough | 0 (0) | 3 (25) | 0 (0) | 3 (20) |
| Dyspnea | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Skin and subcutaneous tissue disorders | ||||
| Rash | 1 (50) | 2 (17) | 0 (0) | 3 (20) |
| Vascular disorders | ||||
| Hypertension | 1 (50) | 3 (25) | 0 (0) | 4 (27) |
| Hypotension | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
Clinically relevant adverse reactions in < 10% of patients include viral infection.
Generalized Myasthenia Gravis (gMG)
Adult Population with gMG Treated with Intravenous ULTOMIRIS
The safety of ULTOMIRIS has been evaluated in 175 adult patients with gMG, including 169 patients who received at least one dose of intravenously administered ULTOMIRIS, 142 patients who were exposed for at least 6 months, and 95 who were exposed for at least 12 months [see Clinical Studies (14.3)]. In a randomized, double-blind, placebo-controlled trial (ALXN1210-MG-306), the most frequent adverse reactions (≥ 10%) with ULTOMIRIS were diarrhea and upper respiratory tract infection. Table 17 describes adverse reactions that occurred at a rate of 5% or more and at greater frequency than placebo. Serious adverse reactions were reported in 20 (23%) patients with gMG receiving ULTOMIRIS and in 14 (16%) patients receiving placebo. The most frequent serious adverse reactions were infections reported in at least 8 (9%) patients treated with ULTOMIRIS and in 4 (4%) patients treated with placebo [see Warnings and Precautions (5.3)]. Of these infections, one fatal case of COVID-19 pneumonia was identified in a patient treated with ULTOMIRIS and one case of infection led to discontinuation of ULTOMIRIS.
| Body System Adverse Reaction | Number of Patients | |
|---|---|---|
| ULTOMIRIS (IV) (N=86) n (%) | Placebo (N=89) n (%) |
|
| Gastrointestinal Disorders | ||
| Diarrhea | 13 (15) | 11 (12) |
| Abdominal pain | 5 (6) | 0 |
| Infections and Infestations | ||
| Upper respiratory tract infection | 12 (14) | 7 (8) |
| Urinary tract infection | 5 (6) | 4 (4) |
| Musculoskeletal and Connective Tissue Disorders | ||
| Back Pain | 7 (8) | 5 (6) |
| Nervous System Disorders | ||
| Dizziness | 8 (9) | 3 (3) |
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibodies) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other ravulizumab-cwvz products may be misleading.
The immunogenicity of ravulizumab-cwvz has been evaluated using an enzyme-linked immunosorbent assay (ELISA) for the detection of binding anti-ravulizumab-cwvz antibodies. For patients whose sera tested positive in the screening immunoassay, an in vitro biological assay was performed to detect neutralizing antibodies.
In clinical studies with intravenous administration of ULTOMIRIS, treatment-emergent antibodies to ravulizumab-cwvz were detected in 1 of 219 (0.5%) patients with PNH and 1 of 71 (1.4%) patients with aHUS. In these 2 patient populations, no apparent correlation of antibody development to altered pharmacokinetic profile, clinical response, or adverse events was observed. No treatment-emergent antibodies to ravulizumab-cwvz were detected in patients with gMG treated with ULTOMIRIS.
In patients receiving subcutaneous administration of ULTOMIRIS (N = 128), no treatment-emergent anti-drug antibodies were observed.
However, the assay used to measure anti-drug antibodies (ADA) is subject to interference by serum ravulizumab-cwvz, possibly resulting in an underestimation of the incidence of antibody formation. Due to the limitation of the assay conditions, the potential clinical impact of antibodies to ravulizumab-cwvz is not known.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ULTOMIRIS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to ULTOMIRIS exposure.
Serious Adverse Reaction: Anaphylaxis [see Warnings and Precautions (5.6)]
7. Drug Interactions
7.1 Plasma Exchange, Plasmapheresis, and Intravenous Immunoglobulins
Concomitant use of ULTOMIRIS with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg) treatment can reduce serum ravulizumab concentrations and requires a supplemental dose of ULTOMIRIS [see Dosage and Administration (2.5)].
8. Use In Specific Populations
8.4 Pediatric Use
The safety and effectiveness of ULTOMIRIS administered intravenously for the treatment of PNH have been established in pediatric patients aged one month and older. Use of ULTOMIRIS for this indication is supported by evidence from adequate and well-controlled trials in adults with additional pharmacokinetic, efficacy and safety data in pediatric patients aged 9 to 17 years [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)]. The safety and efficacy for the treatment of pediatric and adult patients with PNH appear similar. Use of ULTOMIRIS administered intravenously in pediatric patients with PNH aged less than 9 years and body weight < 30 kg is based on extrapolation of pharmacokinetic / pharmacodynamic (PK/PD), and efficacy and safety data from aHUS and PNH clinical studies [see Clinical Pharmacology (12.3) and Clinical Studies (14)].
The safety and effectiveness of ULTOMIRIS administered intravenously for the treatment of aHUS have been established in pediatric patients aged one month and older. Use of ULTOMIRIS for this indication is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic, safety, and efficacy data in pediatric patients aged 10 months to < 17 years. The safety and efficacy of ULTOMIRIS administered intravenously for the treatment of aHUS appear similar in pediatric and adult patients [see Adverse Reactions (6.1), and Clinical Studies (14.2)].
The safety and effectiveness of ULTOMIRIS for the treatment of gMG in pediatric patients have not been established.
Subcutaneous administration of ULTOMIRIS has not been evaluated and is not approved for use in pediatric patients.
8.5 Geriatric Use
Clinical studies of ULTOMIRIS did not include sufficient numbers of subjects aged 65 and over (58 patients with PNH, 9 with aHUS, and 54 with gMG) to determine whether they respond differently from younger subjects.
Other reported clinical experience has not identified differences in responses between elderly and younger patients.
11. Ultomiris Description
Ravulizumab-cwvz, a complement inhibitor, is a humanized monoclonal antibody (mAb) produced in Chinese hamster ovary (CHO) cells. Ravulizumab-cwvz consists of 2 identical 448 amino acid heavy chains and 2 identical 214 amino acid light chains and has a molecular weight of approximately 148 kDa. The constant regions of ravulizumab-cwvz include the human kappa light chain constant region, and the protein engineered "IgG2/4" heavy chain constant region.
The heavy chain CH1 domain, hinge region, and the first 5 amino acids of the CH2 domain match the human IgG2 amino acid sequence, residues 6 to 36 in the CH2 region (common to both human IgG2 and IgG4 amino acid sequences), while the remainder of the CH2 domain and the CH3 domain match the human IgG4 amino acid sequence. The heavy and light chain variable regions that form the human C5 binding site consist of human framework regions grafted to murine complementarity-determining regions.
12. Ultomiris - Clinical Pharmacology
12.1 Mechanism of Action
Ravulizumab-cwvz is a terminal complement inhibitor that specifically binds to the complement protein C5 with high affinity, thereby inhibiting its cleavage to C5a (the proinflammatory anaphylatoxin) and C5b (the initiating subunit of the membrane attack complex [MAC or C5b-9]) thus preventing MAC formation. ULTOMIRIS inhibits terminal complement-mediated intravascular hemolysis in patients with PNH and complement-mediated thrombotic microangiopathy (TMA) in patients with aHUS.
The precise mechanism by which ravulizumab-cwvz exerts its therapeutic effect in gMG patients is unknown, but is presumed to involve reduction of terminal complement complex C5b-9 deposition at the neuromuscular junction.
12.2 Pharmacodynamics
Complete inhibition of serum free C5 (concentration of less than 0.5 mcg/mL) was observed by the end of the first ULTOMIRIS intravenous infusion and sustained throughout the entire 26-week treatment period in both adult and pediatric patients with PNH, in the majority (93%) of adult and pediatric patients with aHUS, and all adult patients with gMG.
Complete inhibition of serum free C5 (concentration of less than 0.5 mcg/mL) was observed by the end of the first ULTOMIRIS subcutaneous administration and was sustained throughout the entire treatment period.
The extent and duration of the pharmacodynamic response in patients with PNH, aHUS, and gMG were exposure-dependent for ULTOMIRIS. Free C5 levels of < 0.5 mcg/mL were correlated with maximal intravascular hemolysis control and complete terminal complement inhibition in patients with PNH.
Complete terminal complement inhibition following initiation of ULTOMIRIS intravenous treatment led to normalization of serum LDH by week 4 in complement-inhibitor naïve patients with PNH, and maintained LDH normalization in patients previously treated with eculizumab with PNH [see Clinical Studies (14)].
The PD results following ULTOMIRIS subcutaneous treatment are consistent with results in adult patients with PNH and aHUS treated with intravenous ULTOMIRIS.
12.3 Pharmacokinetics
Following ULTOMIRIS intravenous (IV) treatment, ravulizumab-cwvz pharmacokinetics increase proportionally over a dose range of 200 to 5400 mg. Ravulizumab-cwvz Cmax and Ctrough parameters are presented in Table 18, Table 19, and Table 20.
| Pediatric Patients | Adult Patients | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ALXN1210-PNH-304 | ALXN1210-PNH-301 | ALXN1210-PNH-302 | |||||||
| N | Complement Inhibitor-Naïve | N | Previously Treated with Eculizumab | N | Complement Inhibitor-Naïve | N | Previously Treated with Eculizumab | ||
| Abbreviations: LD = Loading Dose; MD = Maintenance Dose | |||||||||
| Cmax
(mcg/mL) | LD | 4 | 733 (14.5) | 8 | 885 (19.3) | 125 | 771 (21.5) | 95 | 843 (24.1) |
| MD | 4 | 1490 (26.7) | 8 | 1705 (9.7) | 124 | 1,379 (20.0) | 95 | 1,386 (19.4) | |
| Ctrough
(mcg/mL) | LD | 4 | 368 (14.7) | 8 | 452 (15.1) | 125 | 391 (35.0) | 96 | 405 (29.9) |
| MD | 4 | 495 (21.3) | 8 | 566 (12.2) | 124 | 473 (33.4) | 95 | 501 (28.6) | |
| Pediatric Patients (ALXN1210-aHUS-312) | Adult Patients (ALXN1210-aHUS-311) |
||||||
|---|---|---|---|---|---|---|---|
| N | < 20 kg MD Q4W | N | ≥ 20 to < 40 kg MD Q8W | N | ≥ 40 kg MD Q8W |
||
| Abbreviations: LD = Loading Dose; MD = Maintenance Dose; Q4W = Every 4 Weeks; Q8W = Every 8 Weeks | |||||||
| Cmax
(mcg/mL) | LD | 8 | 656 (38.1) | 4 | 600 (17.3) | 52 | 754 (35.2) |
| MD | 7 | 1,467 (37.8) | 6 | 1,863 (15.3) | 46 | 1,458 (17.6) | |
| Ctrough
(mcg/mL) | LD | 9 | 241 (52.1) | 5 | 186 (16.5) | 55 | 313 (33.9) |
| MD | 7 | 683 (46.1) | 6 | 549 (34.1) | 46 | 507 (42.5) | |
| N | Adult Patients (ALXN1210-MG-306) | ||
|---|---|---|---|
| Abbreviations: LD = Loading Dose; MD=Maintenance Dose | |||
| Cmax
(mcg/mL) | LD | 86 | 874 (21.1) |
| MD | 76 | 1548 (23.2) | |
| Ctrough
(mcg/mL) | LD | 85 | 418 (27.6) |
| MD | 70 | 587 (29.6) | |
Following ULTOMIRIS subcutaneous (SUBQ) treatment, the mean (%CV) ravulizumab-cwvz trough concentration at steady state is 737.7 (28.3%) mcg/mL.
The estimated bioavailability of ULTOMIRIS subcutaneous treatment is approximately 79%.
Elimination
The mean (standard deviation [SD]) terminal elimination half-life and clearance of ravulizumab-cwvz are shown in Table 21.
| Adult and Pediatric Patients with PNH (IV) | Adult and Pediatric Patients with aHUS (IV) | Adult Patients with gMG (IV) | Adult Patients with PNH (SUBQ) |
|
|---|---|---|---|---|
|
||||
| Distribution | ||||
| Volume of distribution at steady state (liters) Mean (SD) | 5.30 (0.95) | 5.22 (1.85) | 5.74 (1.16) | 5.30 (0.95)* |
| Biotransformation and Elimination | ||||
| Terminal elimination half-life (days) Mean (SD) | 49.6 (9.08) | 51.8 (16.2) | 56.6 (8.36) | 52.4 (9.72) |
| Clearance (liters/day) Mean (SD) | 0.08 (0.02) | 0.08 (0.04) | 0.08 (0.02) | 0.07 (0.02)* |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal carcinogenicity studies of ravulizumab-cwvz have not been conducted.
Genotoxicity studies have not been conducted with ravulizumab-cwvz.
Effects of ravulizumab-cwvz upon fertility have not been studied in animals. Intravenous injections of male and female mice with a murine anti-C5 antibody at up to 0.8-2.2 times the equivalent of the clinical dose of ULTOMIRIS had no adverse effects on mating or fertility.
14. Clinical Studies
14.1 Paroxysmal Nocturnal Hemoglobinuria (PNH)
The safety and efficacy of ULTOMIRIS administered intravenously in adult patients with PNH was assessed in 2 open-label, randomized, active-controlled, non-inferiority Phase 3 studies: PNH Study 301 and PNH Study 302. Study 301 enrolled patients with PNH who were complement inhibitor naïve and had active hemolysis. Study 302 enrolled patients with PNH who were clinically stable after having been treated with eculizumab for at least the past 6 months. The safety and efficacy of ULTOMIRIS administered intravenously in pediatric patients with PNH was assessed in PNH Study 304, an open-label, Phase 3 study conducted in eculizumab-experienced and complement inhibitor treatment naïve pediatric patients with PNH.
In Study 301 and Study 302, adults with PNH were dosed with ULTOMIRIS administered intravenously in accordance with the weight-based dosing described in Section 2.2 (4 infusions of ULTOMIRIS over 26 weeks) above. Eculizumab was administered on Days 1, 8, 15, and 22, followed by maintenance treatment with 900 mg of eculizumab on Day 29 and every 2 weeks (q2w) thereafter for a total of 26 weeks of treatment, according to the approved dosing regimen of eculizumab which was the standard-of-care for PNH at the time of the studies.
The safety and efficacy of ULTOMIRIS administered subcutaneously in adult patients with PNH was assessed in a controlled Phase 3 trial: PNH Study 303. Study 303 was a PK non-inferiority study in adult patients with PNH who were clinically stable and previously treated with eculizumab for at least 3 months prior to study entry. This study was also designed to assess the performance of the ULTOMIRIS on-body delivery system.
Patients were vaccinated against meningococcal infection prior to or at the time of initiating treatment with ULTOMIRIS or eculizumab, or received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination. Prophylactic treatment with appropriate antibiotics beyond 2 weeks after vaccination was at the discretion of the provider.
Study in Complement-Inhibitor Naïve Adult Patients with PNH – Intravenous Administration
The Complement-Inhibitor Naïve Study [ALXN1210-PNH-301; NCT02946463] was a 26-week, multicenter, open-label, randomized, active-controlled, non-inferiority Phase 3 study conducted in 246 patients naïve to complement inhibitor treatment prior to study entry.
Patients with PNH with flow cytometric confirmation of at least 5% PNH cells were randomized 1:1 to either intravenously administered ULTOMIRIS or eculizumab. The mean total PNH granulocyte clone size was 85%, the mean total PNH monocyte clone size was 88%, and the mean total PNH RBC clone size was 39%. Ninety-eight percent of patients had a documented PNH-associated condition diagnosed prior to enrollment on the trial: anemia (85%), hemoglobinuria (63%), history of aplastic anemia (32%), history of renal failure (12%), myelodysplastic syndrome (5%), pregnancy complications (3%), and other (16%). Major baseline characteristics were balanced between treatment groups. Table 22 provides the baseline characteristics for the patients enrolled in the complement-inhibitor naïve study.
| Parameter | Statistics | ULTOMIRIS (IV) (N=125) | Eculizumab (N=121) |
|---|---|---|---|
| Abbreviations: LDH = lactate dehydrogenase; max = maximum; min = minimum; MAVE = major adverse vascular event; pRBC = packed red blood cell; SD = standard deviation | |||
| Age (years) at first infusion in study | Mean (SD) Min, max | 44.8 (15.2) 18, 83 | 46.2 (16.2) 18, 86 |
| Sex | |||
| Male | n (%) | 65 (52.0) | 69 (57.0) |
| Race | |||
| Asian | n (%) | 72 (57.6) | 57 (47.1) |
| White | 43 (34.4) | 51 (42.1) | |
| Black or African American | 2 (1.6) | 4 (3.3) | |
| American Indian or Alaska Native | 1 (0.8) | 1 (0.8) | |
| Other | 4 (3.2) | 4 (3.3) | |
| Not reported | 3 (2.4) | 4 (3.3) | |
| Pre-treatment LDH levels (U/L) | Median Min, max | 1513.5 (378.0, 3759.5) | 1445.0 (423.5, 3139.5) |
| Units of pRBC/whole blood transfused within 12 months prior to first dose | Median Min, max | 6.0 (1, 44) | 6.0 (1, 32) |
| Antithrombotic agents used within 28 days prior to first dose | n (%) | 22 (17.6) | 22 (18.2) |
| Patients with a history of MAVE | n (%) | 17 (13.6) | 25 (20.7) |
| Patients with a history of thrombosis | n (%) | 17 (13.6) | 20 (16.5) |
| Patients with concomitant anticoagulant treatment | n (%) | 23 (18.4) | 28 (23.1) |
Efficacy was established based upon transfusion avoidance and hemolysis as directly measured by normalization of LDH levels. Transfusion avoidance was defined as patients who did not receive a transfusion and did not meet the protocol specified guidelines for transfusion from baseline up to Day 183. Supportive efficacy data included the percent change from baseline in LDH levels, the proportion of patients with breakthrough hemolysis defined as at least one new or worsening symptom or sign of intravascular hemolysis in the presence of elevated LDH ≥ 2 × ULN, after prior LDH reduction to < 1.5 × ULN on therapy and the proportion of patients with stabilized hemoglobin.
Non-inferiority of ULTOMIRIS to eculizumab was demonstrated across endpoints in the complement-inhibitor naïve treatment population described in Table 23 below.
| ULTOMIRIS (IV) (N=125) | Eculizumab (N=121) | Statistic for Comparison | Treatment Effect (95% CI) |
|
|---|---|---|---|---|
| For the transfusion avoidance endpoint, treatment differences (95% CIs) are based on estimated differences in percent with 95% CI. For the lactate dehydrogenase normalization endpoint, the adjusted prevalence within each treatment is displayed. Abbreviations: LDH = lactate dehydrogenase; CI = confidence interval |
||||
| Transfusion avoidance rate | 73.6% | 66.1% | Difference in rate | 6.8 (-4.66, 18.14) |
| LDH normalization | 53.6% | 49.4% | Odds ratio | 1.19 (0.80, 1.77) |
| LDH percent change | -76.84% | -76.02% | Difference in % change from baseline | -0.83 (-5.21, 3.56) |
| Breakthrough hemolysis | 4.0% | 10.7% | Difference in rate | -6.7 (-14.21, 0.18) |
| Hemoglobin stabilization | 68.0% | 64.5% | Difference in rate | 2.9 (-8.80, 14.64) |
There was no observable difference in fatigue between ULTOMIRIS and eculizumab after 26 weeks of treatment compared to baseline as measured by the FACIT-fatigue instrument. Patient-reported fatigue may be an under- or over-estimation because patients were not blinded to treatment assignment.
Study in Eculizumab-Experienced Adult Patients with PNH – Intravenous Administration
The study in eculizumab-experienced patients [ALXN1210-PNH-302; NCT03056040] was a 26-week, multicenter, open-label, randomized, active-controlled, non-inferiority Phase 3 study conducted in 195 patients with PNH who were clinically stable after having been treated with eculizumab for at least the past 6 months.
Patients who demonstrated clinically stable disease after being treated with eculizumab for at least the prior 6 months were randomized 1:1 to either continue eculizumab or to switch to ULTOMIRIS administered intravenously. The mean total PNH granulocyte clone size was 83%, the mean total PNH monocyte clone size was 86%, and the mean total PNH RBC clone size was 60%. Ninety-five percent of patients had a documented PNH-associated condition diagnosed prior to enrollment in the trial: anemia (67%), hematuria or hemoglobinuria (49%), history of aplastic anemia (37%), history of renal failure (9%), myelodysplastic syndrome (5%), pregnancy complication (7%), and other (14%). Major baseline characteristics were balanced between the 2 treatment groups. Table 24 provides the baseline characteristics for the patients enrolled in the eculizumab-experienced study.
| Parameter | Statistics | ULTOMIRIS (IV) (N=97) | Eculizumab (N=98) |
|---|---|---|---|
| Abbreviations: LDH = lactate dehydrogenase; max = maximum; min = minimum; MAVE = major adverse vascular event; pRBC = packed red blood cell; SD = standard deviation | |||
| Age (years) at first infusion in study | Mean (SD) Min, max | 46.6 (14.41) 18, 79 | 48.8 (13.97) 23, 77 |
| Race | n (%) | ||
| White | 50 (51.5) | 61 (62.2) | |
| Asian | 23 (23.7) | 19 (19.4) | |
| Black or African American | 5 (5.2) | 3 (3.1) | |
| Other | 2 (2.1) | 1 (1.0) | |
| Not reported | 13 (13.4) | 13 (13.3) | |
| Unknown | 3 (3.1) | 1 (1.0) | |
| Multiple | 1 (1.0) | 0 | |
| Sex | n (%) | ||
| Male | 50 (51.5) | 48 (49.0) | |
| Pre-treatment LDH levels (U/L) | Median Min, max | 224.0 135.0, 383.5 | 234.0 100.0, 365.5 |
| Units of pRBC/whole blood transfused within 12 months prior to first dose | Median Min, max | 4.0 (1, 32) | 2.5 (2, 15) |
| Antithrombotic agents used within 28 days prior to first dose | n (%) | 20 (20.6) | 13 (13.3) |
| Patients with a history of MAVE | n (%) | 28 (28.9) | 22 (22.4) |
| Patients with a history of thrombosis | n (%) | 27 (27.8) | 21 (21.4) |
| Patients with concomitant anticoagulant treatment | n (%) | 22 (22.7) | 16 (16.3) |
Efficacy was established based on hemolysis as measured by LDH percent change from baseline to Day 183 and supportive efficacy data was transfusion avoidance, proportion of patients with stabilized hemoglobin, and the proportion of patients with breakthrough hemolysis through Day 183.
Non-inferiority of ULTOMIRIS to eculizumab was demonstrated across endpoints in the patients with PNH previously treated with eculizumab described in Table 25 below.
| ULTOMIRIS (IV) N = 97 | Eculizumab N = 98 | Statistic for Comparison | Treatment Effect (95% CI) |
|
|---|---|---|---|---|
| Abbreviations: CI = confidence interval; LDH = lactate dehydrogenase | ||||
| LDH percent change | -0.82% | 8.4% | Difference in % change from baseline | 9.2 (-0.42, 18.8) |
| Breakthrough hemolysis | 0% | 5.1% | Difference in rate | 5.1 (-8.9, 19.0) |
| Transfusion avoidance | 87.6 % | 82.7% | Difference in rate | 5.5 (-4.3, 15.7) |
| Hemoglobin stabilization | 76.3% | 75.5% | Difference in rate | 1.4 (-10.4, 13.3) |
There was no observable difference in fatigue between ULTOMIRIS and eculizumab after 26 weeks of treatment compared to baseline as measured by the FACIT-fatigue instrument. Patient-reported fatigue may be an under-or over-estimation because patients were not blinded to treatment assignment.
Study in Eculizumab-Experienced and Complement-Inhibitor Naïve Pediatric Patients with PNH – Intravenous Administration
The pediatric study, ALXN1210-PNH-304 (NCT03406507), was a multi-center, open-label Phase 3 study conducted in eculizumab-experienced and complement inhibitor treatment-naïve pediatric patients with PNH. A total of 13 pediatric patients with PNH completed intravenously administered ULTOMIRIS treatment during the Primary Evaluation Period (26 weeks). Five of the 13 patients had never been treated with complement inhibitors and 8 patients were treated with eculizumab. Eleven of the thirteen patients were between 12 and 17 years of age at first infusion, with 2 patients under 12 years old (11 and 9 years old). Table 26 presents the baseline characteristics of the pediatric patients enrolled in Study ALXN1210-PNH-304.
| Variable | Complement Inhibitor Treatment-naïve Patients (N = 5) | Eculizumab-Experienced Patients (N = 8) | All Patients (N = 13) |
|---|---|---|---|
| Note: Percentages were based on the total number of patients in each cohort, or overall. Abbreviations: LDH = lactate dehydrogenase; kg = kilogram; max = maximum; min = minimum; pRBC = packed red blood cells; SD = standard deviation |
|||
| Sex, n (%) | |||
| Male | 4 (80.0) | 1 (12.5) | 5 (38.5) |
| Female | 1 (20.0) | 7 (87.5) | 8 (61.5) |
| Age at first infusion (years) | |||
| Mean (SD) | 14.4 (2.2) | 14.4 (3.1) | 14.4 (2.7) |
| Median (min, max) | 15.0 (11, 17) | 15.0 (9, 17) | 15.0 (9, 17) |
| Age at first infusion (years) category, n (%) | |||
| < 12 years | 1 (20.0) | 1 (12.5) | 2 (15.4) |
| ≥ 12 years | 4 (80.0) | 7 (87.5) | 11 (84.6) |
| Baseline weight (kg) | |||
| Mean (SD) | 56.3 (11.6) | 56.3 (12.2) | 56.3 (11.5) |
| Median (min, max) | 55.6 (39.5, 72.0) | 55.5 (36.7, 69.0) | 55.6 (36.7, 72.0) |
| Baseline weight (kg) category, n (%) | |||
| ≥ 30 to < 40 kg | 1 (20.0) | 1 (12.5) | 2 (15.4) |
| ≥ 40 to < 60 kg | 3 (60.0) | 4 (50.0) | 7 (53.8) |
| ≥ 60 to < 100 kg | 1 (20.0) | 3 (37.5) | 4 (30.8) |
| Units of pRBC/whole blood transfused within 12 months prior to first dose | |||
| Median (min, max) | 7.0 (3, 11) | 2.0 (2, 2) | - |
| Pre-treatment LDH levels (U/L) | |||
| Median (min, max) | 588.5 (444, 2269.7) | 251.5 (140.5, 487) | - |
Based on body weight, patients received a loading dose of ULTOMIRIS on Day 1, followed by maintenance treatment on Day 15 and once every 8 weeks (q8w) thereafter for patients weighing ≥ 20 kg, or once every 4 weeks (q4w) for patients weighing < 20 kg. For patients who entered the study on eculizumab therapy, Day 1 of study treatment was planned to occur 2 weeks from the patient's last dose of eculizumab.
The weight-based dose regimen of ravulizumab-cwvz provided inhibition of terminal complement in all patients throughout the entire 26-week treatment period regardless of prior experience with eculizumab. Following initiation of ravulizumab-cwvz treatment, steady-state therapeutic serum concentrations of ravulizumab-cwvz were achieved after the first dose and maintained throughout the primary evaluation period in both cohorts. Three of 5 complement inhibitor treatment-naïve patients and 6 out of 8 eculizumab-experienced patients achieved hemoglobin stabilization by Week 26, respectively. Transfusion avoidance was reached for 11 out of 13 of patients during the 26-week Primary Evaluation Period. One patient experienced breakthrough hemolysis during the extension period. Table 27 presents secondary efficacy outcomes for the primary evaluation period.
| Endpoint | Treatment Naïve (N = 5) | Eculizumab Experienced (N = 8) |
|---|---|---|
| Abbreviations: FACIT = Functional Assessment of Chronic Illness Therapy; LDH = lactate dehydrogenase | ||
|
||
| LDH percent change from baseline (%)* | -47.9 (-113.4, 17.5) | 4.7 (-36.7, 46.0) |
| Transfusion avoidance (%)† | 60.0 (14.7, 94.7) | 100.0 (63.1, 100.0) |
| Change in FACIT-Fatigue* | 3.4 (-4.2, 11.0) | 1.3 (-3.1, 5.7) |
| Hemoglobin stabilization (%)† | 60.0 (14.7, 94.7) | 75.0 (34.9, 96.8) |
| Breakthrough hemolysis (%) | 0 | 0‡ |
A clinically relevant improvement from baseline in fatigue as assessed by Pediatric FACIT-Fatigue (i.e., mean improvement of > 3 units for Pediatric FACIT Fatigue scores) was sustained throughout the primary evaluation period in the 5-complement inhibitor treatment naïve patients. A slight improvement was also observed in eculizumab-experienced patients. However, patient-reported fatigue may be an under- or over-estimation because patients were not blinded to treatment assignment.
The efficacy of ULTOMIRIS administered intravenously in pediatric patients with PNH is similar to that observed in adult patients with PNH enrolled in pivotal studies.
Subcutaneous Administration in Adult Patients with PNH
Subcutaneous administration of ULTOMIRIS was assessed in a multi-center, randomized, open-label, Phase 3 study (ALXN1210-PNH-303 [NCT03748823]) conducted in adult patients with PNH who were clinically stable after having been treated with eculizumab for at least 3 months prior to study entry. The study enrolled 136 patients, of which 129 patients were included in the efficacy and safety analyses. The main outcome measure of Study ALXN1210-PNH-303 was the non-inferiority of Ctrough of ULTOMIRIS when administered subcutaneously via an on-body injector compared to ULTOMIRIS administered intravenously. The study was designed to bridge efficacy and safety data from ULTOMIRIS administered intravenously (IV) to ULTOMIRIS administered subcutaneously (SUBQ).
Patients who completed the 10-week Randomized Treatment Period are followed for up to 172 weeks in the long-term Extension Period. During the Randomized Treatment Period, patients were stratified by weight group (≥ 40 kg to < 60 kg and ≥ 60 kg to < 100 kg) and randomized 2:1 to receive ULTOMIRIS subcutaneously or ULTOMIRIS intravenously. On Day 1, all patients received a weight-based ULTOMIRIS intravenous loading dose. Starting on Day 15, patients randomized to the ULTOMIRIS subcutaneous group received once weekly subcutaneous maintenance doses (490 mg), while patients randomized to the ULTOMIRIS intravenous group received the approved weight-based intravenous maintenance dose. Following the Randomized Treatment Period (on Day 71), patients who were randomized to the ULTOMIRIS intravenous group switched to receive 490 mg of ULTOMIRIS subcutaneously weekly through the end of the Extension Period.
The mean total PNH RBC clone size was 48.35%, with a mean total PNH granulocyte clone size of 77.22% and a mean total PNH monocyte clone size of 80.18%. Ninety-two percent of the patients had documented PNH-associated conditions that were diagnosed prior to informed consent. Overall, the disease history and the baseline characteristics were well balanced between the 2 treatment groups. Table 28 presents the baseline characteristics of patients with PNH enrolled in Study ALXN1210-PNH-303.
| Variable | ULTOMIRIS (SUBQ) (N = 84) | ULTOMIRIS (IV) (N = 45) | Total (N = 129) |
|---|---|---|---|
| Note: Percentages are based on the total number of patients. Patients can be counted in more than one category for race. Abbreviations: LDH = lactate dehydrogenase; max = maximum; min = minimum; SD = standard deviation |
|||
| Sex, n (%) | |||
| Male | 40 (47.6) | 20 (44.4) | 60 (46.5) |
| Female | 44 (52.4) | 25 (55.6) | 69 (53.5) |
| Race, n (%) | |||
| White | 63 (75.0) | 29 (64.4) | 92 (71.3) |
| Not reported | 13 (15.5) | 6 (13.3) | 19 (14.7) |
| Black or African American | 3 (3.6) | 4 (8.9) | 7 (5.4) |
| Asian | 0 | 2 (4.4) | 2 (1.6) |
| Unknown or other | 5 (6.0) | 3 (6.7) | 8 (6.2) |
| American Indian or Alaska Native | 0 | 1 (2.2) | 1 (0.8) |
| Age (years) at informed consent | |||
| Mean (SD) | 45.3 (14.47) | 46.4 (13.22) | 45.7 (14.00) |
| Median | 42.5 | 44.0 | 44.0 |
| Min, Max | 18, 79 | 24, 77 | 18, 79 |
| Age (years) at informed consent category, n (%) | |||
| 18 to 65 years | 75 (89.3) | 41 (91.1) | 116 (89.9) |
| > 65 years | 9 (10.7) | 4 (8.9) | 13 (10.1) |
| Baseline weight (kg) | |||
| Mean (SD) | 72.52 (12.61) | 73.68 (12.66) | 72.92 (12.59) |
| Median | 72.15 | 73.00 | 72.30 |
| Min, Max | 43.5, 98.0 | 52.0, 98.4 | 43.5, 98.4 |
| Baseline weight (kg) category, n (%) | |||
| ≥ 40 kg to < 60 kg | 13 (15.5) | 8 (17.8) | 21 (16.3) |
| ≥ 60 kg to < 100 kg | 71 (84.5) | 37 (82.2) | 108 (83.7) |
In Study ALXN1210-PNH-303, treatment with ULTOMIRIS on-body delivery system for subcutaneous administration achieved PK non-inferiority with a margin 0.80, compared with intravenously administered ULTOMIRIS treatment for the serum ravulizumab-cwvz Ctrough at Day 71, with a geometric least squares mean ratio of 1.257 (90% CI: 1.160, 1.361). Serum free C5 concentrations were maintained below the target threshold (< 0.5 mcg/mL) in all patients throughout the one year of subcutaneous treatment.
In patients with PNH randomized to subcutaneous or intravenous ULTOMIRIS, change in LDH, breakthrough hemolysis, transfusion avoidance, hemoglobin stabilization, and FACIT-Fatigue score were evaluated. No noticeable difference between the routes of administration was observed in these endpoints at Day 71.
14.2 Atypical Hemolytic Uremic Syndrome (aHUS)
The efficacy of ULTOMIRIS administered intravenously in patients with aHUS was assessed in 2 open-label, single-arm studies. Study ALXN1210-aHUS-311 enrolled adult patients who displayed signs of TMA. In order to qualify for enrollment, patients were required to have a platelet count ≤ 150 × 109/L, evidence of hemolysis such as an elevation in serum LDH, and serum creatinine above the upper limits of normal or required dialysis.
Study ALXN1210-aHUS-312 enrolled pediatric patients who displayed signs of TMA. In order to qualify for enrollment, patients were required to have a platelet count ≤ 150 × 109/L, evidence of hemolysis such as an elevation in serum LDH, and serum creatinine level ≥ 97.5% percentile at screening or required dialysis. In both studies, enrollment criteria excluded patients presenting with TMA due to a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) deficiency, Shiga toxin Escherichia coli related hemolytic uremic syndrome (STEC-HUS) and genetic defect in cobalamin C metabolism. Patients with confirmed diagnosis of STEC-HUS after enrollment were excluded from the efficacy evaluation.
Study in Adult Patients with aHUS – Intravenous Administration
The adult study [ALXN1210-aHUS-311; NCT02949128] was conducted in patients who were naïve to complement inhibitor treatment prior to study entry. The study consisted of a 26-week Initial Evaluation Period and patients were allowed to enter an extension period for up to 4.5 years.
A total of 56 patients with aHUS were evaluated for efficacy. Ninety-three percent of patients had extra-renal signs (cardiovascular, pulmonary, central nervous system, gastrointestinal, skin, skeletal muscle) or symptoms of aHUS at baseline. At baseline, 71.4% (n = 40) of patients had Stage 5 chronic kidney disease (CKD). Fourteen percent had a medical history of kidney transplant and 51.8% were on dialysis at study entry. Eight patients entered the study with evidence of TMA for > 3 days after childbirth (i.e., postpartum).
Table 29 presents the demographics and baseline characteristics of the 56 adult patients enrolled in Study ALXN1210-aHUS-311 that constituted the Full Analysis Set.
| Parameter | Statistics | ULTOMIRIS (IV) (N=56) |
|---|---|---|
| Note: Percentages are based on the total number of patients. Abbreviations: eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; max = maximum; min = minimum; SD = standard deviation |
||
|
||
| Age at time of first infusion (years) | Mean (SD) Min, max | 42.2 (14.98) 19.5, 76.6 |
| Sex | ||
| Female | n (%) | 37 (66.1) |
| Race * | n (%) | |
| White | 29 (51.8) | |
| Asian | 15 (26.8) | |
| Unknown | 8 (14.3) | |
| Other | 4 (7.1) | |
| Platelets (109/L) blood [normal range 130 to 400 × 109/L] | n Median (min,max) | 56 95.25 (18, 473) |
| Hemoglobin (g/L) blood [normal range 115 to 160 g/L (female), 130 to 175 g/L (male)] | n Median (min,max) | 56 85.00 (60.5, 140) |
| LDH (U/L) serum [normal range 120 to 246 U/L] | n Median (min,max) | 56 508.00 (229.5, 3249) |
| eGFR (mL/min/1.73 m2) [normal range ≥ 60 mL/min/1.73 m2] | n (%) Mean (SD) Median (min,max) | 55 15.86 (14.815) 10.00 (4, 80) |
The efficacy evaluation was based on Complete TMA Response during the 26-week Initial Evaluation Period, as evidenced by normalization of hematological parameters (platelet count and LDH) and ≥ 25% improvement in serum creatinine from baseline. Patients had to meet each Complete TMA Response criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart, and any measurement in between.
Complete TMA Response was observed in 30 of the 56 patients (54%) during the 26-week Initial Evaluation Period as shown in Table 30.
| Total | Responder | ||
|---|---|---|---|
| n | Proportion (95% CI)* | ||
| Abbreviations: CI = confidence interval; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy | |||
|
|||
| Complete TMA Response | 56 | 30 | 0.54 (0.40, 0.67) |
| Components of Complete TMA Response | |||
| Platelet count normalization | 56 | 47 | 0.84 (0.72, 0.92) |
| LDH normalization | 56 | 43 | 0.77 (0.64, 0.87) |
| ≥ 25% improvement in serum creatinine from baseline | 56 | 33 | 0.59 (0.45, 0.72) |
| Hematologic normalization | 56 | 41 | 0.73 (0.60, 0.84) |
One additional patient had a Complete TMA Response that was confirmed after the 26-week Initial Evaluation Period. Complete TMA Response was achieved at a median time of 86 days (range: 7 to 169 days). The median duration of Complete TMA Response was 7.97 months (range: 2.52 to 16.69 months). All responses were maintained through all available follow-up.
Other endpoints included platelet count change from baseline, dialysis requirement, and renal function as evaluated by estimated glomerular filtration rate (eGFR).
An increase in mean platelet count was observed after commencement of ULTOMIRIS intravenous treatment, increasing from 118.52 × 109/L at baseline to 240.34 ×109/L at Day 8 and remaining above 227 × 109/L at all subsequent visits in the Initial Evaluation Period (26 weeks).
Renal function, as measured by eGFR, was improved or maintained during ULTOMIRIS intravenous therapy. The mean eGFR (+/- SD) increased from 15.86 (14.82) at baseline to 51.83 (39.16) by 26 weeks. In patients with Complete TMA Response, renal function continued to improve after the Complete TMA Response was achieved.
Seventeen of the 29 patients (59%) who required dialysis at study entry discontinued dialysis by the end of the available follow-up and 6 of 27 (22%) patients were off dialysis at baseline were on dialysis at last available follow-up.
Study in Pediatric Patients with aHUS – Intravenous Administration
The Pediatric Study [ALXN1210-aHUS-312; NCT03131219] is a 26-week ongoing, multicenter, single-arm study conducted in 16 pediatric patients.
A total of 14 eculizumab-naïve patients with documented diagnosis of aHUS were enrolled and included in this interim analysis. The median age at the time of first infusion was 5.2 years (range 0.9, 17.3 years). The overall mean weight at Baseline was 19.8 kg; half of the patients were in the baseline weight category ≥ 10 to < 20 kg. The majority of patients (71%) had pretreatment extra-renal signs (cardiovascular, pulmonary, central nervous system, gastrointestinal, skin, skeletal muscle) or symptoms of aHUS at baseline. At baseline, 35.7% (n = 5) of patients had a CKD Stage 5. Seven percent had history of prior kidney transplant and 35.7% were on dialysis at study entry.
Table 31 presents the baseline characteristics of the pediatric patients enrolled in Study ALXN1210-aHUS-312.
| Parameter | Statistics | ULTOMIRIS (IV) (N = 14) |
|---|---|---|
| Note: Percentages are based on the total number of patients. Abbreviations: eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; max = maximum; min = minimum |
||
|
||
| Age at time of first infusion (years) category | n (%) | |
| Birth to < 2 years | 2 (14.3) | |
| 2 to < 6 years | 7 (50.0) | |
| 6 to < 12 years | 4 (28.6) | |
| 12 to < 18 years | 1 (7.1) | |
| Sex | n (%) | |
| Female | 9 (64.3) | |
| Race* | n (%) | |
| White | 7 (50.0) | |
| Asian | 4 (28.6) | |
| Black or African American | 2 (14.3) | |
| American Indian or Alaskan Native | 1 (7.1) | |
| Unknown | 1 (7.1) | |
| Platelets (109/L) blood [normal range 229 to 533 × 109/L] | Median (min, max) | 64.00 (14, 125) |
| Hemoglobin (g/L) blood [normal range 107 to 131 g/L] | Median (min, max) | 74.25 (32, 106) |
| LDH (U/L) serum [normal range 165 to 395 U/L] | Median (min, max) | 2077.00 (772, 4985) |
| eGFR (mL/min/1.73 m2) [normal range ≥ 60 mL/min/1.73 m2] | Mean (SD) Median (min, max) | 28.4 (23.11) 22.0 (10, 84) |
Efficacy evaluation was based upon Complete TMA Response during the 26-week Initial Evaluation Period, as evidenced by normalization of hematological parameters (platelet count and LDH) and ≥ 25% improvement in serum creatinine from baseline. Patients had to meet all Complete TMA Response criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart, and any measurement in between.
Complete TMA Response was observed in 10 of the 14 patients (71%) during the 26-week Initial Evaluation Period as shown in Table 32.
| Total | Responder | ||
|---|---|---|---|
| n | Proportion (95% CI)* | ||
| Note: One patient withdrew from study after receiving 2 doses of ravulizumab-cwvz. Abbreviations: CI = confidence interval; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy |
|||
|
|||
| Complete TMA Response | 14 | 10 | 0.71 (0.42, 0.92) |
| Components of Complete TMA Response | |||
| Platelet count normalization | 14 | 13 | 0.93 (0.66, 0.99) |
| LDH normalization | 14 | 12 | 0.86 (0.57, 0.98) |
| ≥ 25% improvement in serum creatinine from baseline | 14 | 11 | 0.79 (0.49, 0.95) |
| Hematologic normalization | 14 | 12 | 0.86 (0.57, 0.98) |
Complete TMA Response during the Initial Evaluation Period was achieved at a median time of 30 days (range:15 to 88 days). The median duration of Complete TMA Response was 5.08 months (range: 3.08 to 5.54 months). All responses were maintained through all available follow-up.
Other endpoints included platelet count change from baseline, dialysis requirement, and renal function as evaluated by eGFR.
An increase in mean platelet count was observed after commencement of ULTOMIRIS intravenous treatment, increasing from 60.50 × 109/L at baseline to 296.67 × 109/L at Day 8 and remained above 296 × 109/L at all subsequent visits in the Initial Evaluation Period (26 weeks). The mean eGFR (+/- SD) increased from 28.4 (23.11) at baseline to 108.0 (63.21) by 26 weeks.
Four of the 5 patients who required dialysis at study entry were able to discontinue dialysis after the first month in study and for the duration of ULTOMIRIS treatment. No patient started dialysis during the study.
14.3 Generalized Myasthenia Gravis (gMG)
The efficacy of ULTOMIRIS for the treatment of gMG was demonstrated in a randomized, double-blind, placebo-controlled, multicenter study (ALXN1210-MG-306; NCT03920293). Patients were randomized 1:1 to either receive ULTOMIRIS (n=86) or placebo (n=89) for 26 weeks. ULTOMIRIS was administered intravenously according to the weight-based recommended dosage [see Dosage and Administration (2.3)].
Patients with gMG with a positive serologic test for anti-AChR antibodies, Myasthenia Gravis Foundation of America (MGFA) clinical classification class II to IV, and Myasthenia Gravis-Activities of Daily Living (MG-ADL) total score ≥ 6 were enrolled. Baseline and disease characteristics were similar between treatment groups (including age at first dose [mean of 58 years for ULTOMIRIS versus 53 years for placebo], gender [51% female for ULTOMIRIS versus 51% female for placebo], race as White, Asian, and Black or African American [78%, 17%, and 2% for ULTOMIRIS versus 69%, 18%, and 5% for placebo, respectively], and duration of MG since diagnosis [mean of 10 years, ranging from 0.5 to 39.5 years, for ULTOMIRIS versus mean of 10.0 years, ranging from 0.5 to 36.1 years, for placebo].
Over 80% of patients were receiving acetylcholinesterase inhibitors, 70% were receiving corticosteroids, and 68% were receiving non-steroidal immunosuppressants (ISTs) at study entry. Patients on concomitant medications to treat gMG were permitted to continue on therapy throughout the course of the study.
The primary efficacy endpoint was a comparison of the change from baseline between treatment groups in the MG-ADL total score at Week 26. The MG-ADL is a categorical scale that assesses the impact on daily function of 8 signs or symptoms that are typically affected in gMG. Each item is assessed on a 4-point scale where a score of 0 represents normal function and a score of 3 represents loss of ability to perform that function. The total score ranges from 0 to 24, with the higher scores indicating more impairment.
The secondary endpoints, also assessed from baseline to Week 26, included the change in the Quantitative MG total score (QMG). The QMG is a 13-item categorical scale assessing muscle weakness. Each item is assessed on a 4-point scale where a score of 0 represents no weakness and a score of 3 represents severe weakness. A total score ranges from 0 to 39, where higher scores indicate more severe impairment.
Other secondary endpoints included the proportion of patients with improvements of at least 5 and 3 points in the QMG and MG-ADL total scores, respectively.
Treatment with ULTOMIRIS demonstrated a statistically significant change in the MG-ADL and QMG total scores from baseline at Week 26 as compared to placebo (Table 33).
| Efficacy Endpoints: Change from Baseline at Week 26 | Placebo (n = 89) LS Mean | ULTOMIRIS (IV) (n = 86) LS Mean | Treatment Effect (95% CI) | p-value* |
|---|---|---|---|---|
| Abbreviations: CI = confidence interval, LS = least squares; MG-ADL = Myasthenia Gravis Activities of Daily Living profile; QMG = Quantitative Myasthenia Gravis score for disease severity | ||||
|
||||
| Primary Endpoint | ||||
| MG-ADL | -1.4 | -3.1 | -1.6 (-2.6, -0.7) | < 0.001 |
| Secondary Endpoint | ||||
| QMG | -0.8 | -2.8 | -2.0 (-3.2, -0.8) | < 0.001 |
The proportion of QMG responders with at least a 5-point improvement at week 26 was greater for ULTOMIRIS (30.0%) compared to placebo (11.3%) p = 0.005. The proportion of MG-ADL responders with at least a 3-point improvement at week 26 was also greater for ULTOMIRIS (56.7%) compared to placebo (34.1%). The proportion of clinical responders at higher response thresholds (≥ 4-, 5-, 6-, 7-, or 8-point improvement on MG-ADL, and ≥ 6-, 7-, 8-, 9-, or 10-point improvement on QMG) was consistently greater for ULTOMIRIS compared to placebo.
Figure 1: Change from Baseline in MG-ADL Total Score (A) and QMG Total Score (B) Through Week 26 of the Randomized Controlled Period of ALXN1210-MG-306 (Mean and 95% CI)
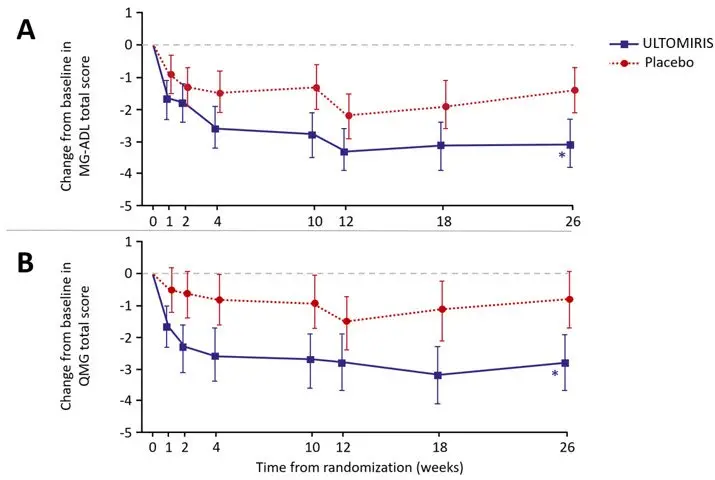
Note: *p<0.001 versus placebo
17. Patient Counseling Information
Advise the patients and/or caregivers to read FDA-approved patient labeling (Medication Guide and Instructions for Use).
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 07/2022 | ||
| MEDICATION GUIDE
ULTOMIRIS® (ul-toe-meer-is) (ravulizumab-cwvz) injection, for intravenous or subcutaneous use |
|||
|
|||
|
|||
|
|
||
| Your healthcare provider will give you a Patient Safety Card about the risk of meningococcal infection. Carry it with you at all times during treatment and for 8 months after your last ULTOMIRIS dose. Your risk of meningococcal infection may continue for several months after your last dose of ULTOMIRIS. It is important to show this card to any healthcare provider or nurse who treats you. This will help them diagnose and treat you quickly. | |||
| ULTOMIRIS is only available through a program called the ULTOMIRIS REMS. Before you can receive ULTOMIRIS, your healthcare provider must: | |||
|
|||
| ULTOMIRIS may also increase the risk of other types of serious infections. | |||
|
|||
| Call your healthcare provider right away if you have any new signs or symptoms of infection. | |||
| What is ULTOMIRIS? | |||
ULTOMIRIS is a prescription medicine called a monoclonal antibody. ULTOMIRIS is used to treat:
|
|||
| It is not known if ULTOMIRIS is safe and effective in children younger than 1 month of age. It is not known if ULTOMIRIS is safe and effective for the treatment of gMG in children. Subcutaneous administration of ULTOMIRIS has not been evaluated and is not approved for use in children. |
|||
| Who should not receive ULTOMIRIS?
Do not receive ULTOMIRIS if you: |
|||
|
|||
| Before you receive ULTOMIRIS, tell your healthcare provider about all of your medical conditions, including if you: | |||
|
|||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. ULTOMIRIS and other medicines can affect each other causing side effects. Know the medicines you take and the vaccines you receive. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|||
| How should I receive ULTOMIRIS? | |||
|
|||
| Adults with PNH, aHUS, or gMG when administered intravenously (by vein) | |||
|
|||
| Children 1 month of age and older with PNH or aHUS when administered intravenously (by vein) | |||
| Your child will be given intravenous ULTOMIRIS infusion by a healthcare provider through a needle placed in a vein | |||
|
|||
| Adults with PNH or aHUS when administered subcutaneously (under your skin) | |||
|
|||
| If you are changing treatment between ULTOMIRIS administered intravenously, ULTOMIRIS administered subcutaneously, or SOLIRIS: | |||
|
|||
|
|
||
|
|||
|
|
||
| If you miss an ULTOMIRIS intravenous or subcutaneous dose, or administer a partial subcutaneous dose of ULTOMIRIS, call your healthcare provider right away. | |||
| ULTOMIRIS can cause serious side effects including: See "What is the most important information I should know about ULTOMIRIS?" | |||
|
|||
|
|
||
|
|||
| The most common side effects of ULTOMIRIS in people treated for PNH are: | |||
|
|
||
| The most common side effects of ULTOMIRIS in people treated for aHUS are: | |||
|
|
||
| The most common side effects of ULTOMIRIS in people with gMG are: | |||
|
|
||
| Tell your healthcare provider about any side effect that bothers you or that does not go away. These are not all of the possible side effects of ULTOMIRIS. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
| General information about the safe and effective use of ULTOMIRIS.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about ULTOMIRIS that is written for health professionals. |
|||
| What are the ingredients in ULTOMIRIS? | |||
| Active ingredient: ravulizumab-cwvz. | |||
| Inactive ingredients: | |||
| Intravenous: | |||
| ULTOMIRIS 100 mg/mL: L-arginine, polysorbate 80 (vegetable origin), sodium phosphate dibasic, sodium phosphate monobasic, sucrose and Water for Injection. | |||
| ULTOMIRIS 10 mg/mL: polysorbate 80 (vegetable origin), sodium chloride, sodium phosphate dibasic, sodium phosphate monobasic and Water for Injection. | |||
| Subcutaneous: | |||
| ULTOMIRIS 70 mg/mL: L-arginine, polysorbate 80 (vegetable origin), sodium phosphate dibasic, sodium phosphate monobasic, sucrose, and Water for Injection. | |||
| Manufactured by Alexion Pharmaceuticals, Inc., 121 Seaport Boulevard, Boston, MA 02210 USA. U.S. License Number 1743 ULTOMIRIS is a trademark of Alexion Pharmaceuticals, Inc. © 2022 Alexion Pharmaceuticals, Inc. For more information, go to www.ULTOMIRIS.com or call: 1-888-765-4747. |
|||
| This Instructions for Use has been approved by the U.S. Food and Drug Administration. | Issued: 07/2022 | ||||||
| INSTRUCTIONS FOR USE On-Body Delivery System for ULTOMIRIS® (uhl-toh-mee-ris) (ravulizumab-cwvz) injection for subcutaneous use |
|||||||
| Single-Use, On-Body Injector and 245 mg/3.5 mL Prefilled Cartridge | |||||||
| This Instructions for Use contains information on how to inject ULTOMIRIS subcutaneously using an on-body delivery system. | |||||||
| Read this Instructions for Use prior to each use of the ULTOMIRIS on-body injector and prefilled cartridge as there may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment. See the Medication Guide that comes with ULTOMIRIS for important information. | |||||||
| Important Information You Need to Know Before Injecting ULTOMIRIS Subcutaneously | |||||||
|
|||||||
 | 1 Full Dose of ULTOMIRIS = 2 ULTOMIRIS On-Body Injectors and 2 Prefilled Cartridges each week |  |
|||||
| Guide to Parts | |||||||
| Prefilled cartridge (Also referred to as "Cartridge") | |||||||
 |
|||||||
| On-Body Injector (Also referred to as "Injector") | |||||||
 |
|||||||
| Important: Needle is inside. | |||||||
| Important | |||||||
| Storing your ULTOMIRIS on-body injector and prefilled cartridge: | |||||||
|
|||||||
| Using your ULTOMIRIS on-body injector and prefilled cartridge: | |||||||
|
|||||||
| Step 1: Prepare | |||||||
| 1A | Remove 2 ULTOMIRIS on-body delivery system cartons from the refrigerator. You will need to use 2 injectors and 2 cartridges for a full dose. (See Figure A) | ||||||
 |
|||||||
| 1B | Wait at least 45 minutes. | ||||||
| Important: Wait at least 45 minutes for the injectors and cartridges in the cartons to naturally reach room temperature. (See Figure B) | |||||||
|
|||||||
| See the Frequently Asked Questions section for additional information on returning the on-body injector and prefilled cartridge to the refrigerator. | |||||||
 |
|||||||
| 1C | Open the cartons and peel away the white paper covers (See Figure C). Remove the plastic covers from the clear trays using the finger hole (See Figure D). | ||||||
 | 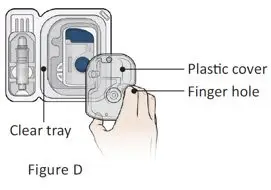 | ||||||
| Leave the injectors and cartridges in the trays until ready to use. | |||||||
|
|||||||
| 1D | Gather all supplies needed for your injection and then wash your hands with soap and water. You can also wear gloves. | ||||||
| On a clean, well-lit work surface, place the: | |||||||
|
|||||||
 |
|||||||
| Additional supplies (See Figure F): | |||||||
|
|||||||
 |
|||||||
| 1E | Prepare and clean 2 different injection sites. Wipe both injection sites with a new alcohol wipe and let your skin air dry. (See Figure G) | ||||||
| The sites you may choose are: | |||||||
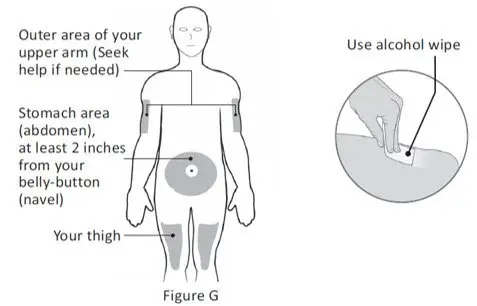 |
|||||||
|
|||||||
| REPEAT Steps 2A - 3C for each injection | |||||||
| Step 2: Get Ready | |||||||
| Swing open the blue cartridge door (See Figure H). Leave the door open. Do not close the cartridge door before the cartridge is loaded. | |||||||
| The door should not be closed when removed from the package. If the door is closed, press firmly on the ribbed side of the door and swing the door to open. (See Figure I) | |||||||
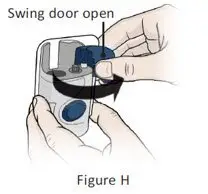 | 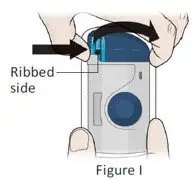 | ||||||
| 2B | Inspect the cartridge. (See Figure J) | ||||||
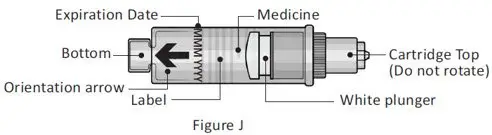 |
|||||||
| Check the expiration date. Do not use the cartridge if the expiration date has passed. | |||||||
| Check the medicine in the cartridge. The medicine should be clear and colorless to slightly yellow in color. | |||||||
|
|||||||
| In any of the above cases, use a new on-body injector and prefilled cartridge and contact 1-888-765-4747. | |||||||
| 2C | Clean the cartridge bottom with a new alcohol wipe. (See Figure K) | ||||||
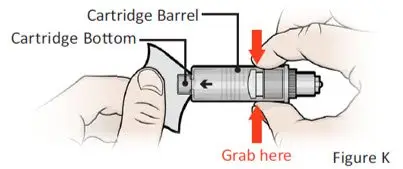 |
|||||||
| With 1 hand, hold the prefilled cartridge barrel in the middle. | |||||||
|
|||||||
| 2D | Load the cleaned cartridge into the injector and gently press on the top until it is secured in place. (See Figure L) | ||||||
| Insert the cartridge into the injector with the arrow pointing down (bottom first). | |||||||
|
|||||||
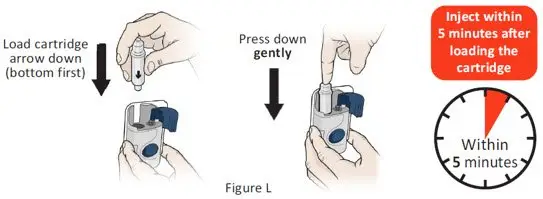 |
|||||||
| Important: Do not insert the cartridge more than 5 minutes before your injection. This can dry out the medicine. See the Frequently Asked Questions section for more information. | |||||||
| 2E | Close the cartridge door. Squeeze it firmly until it snaps shut and there are no gaps between the injector and the cartridge door. (See Figure M) | ||||||
| Do not re-open door or remove the cartridge. | |||||||
| Do not touch the blue start button until you have placed the injector onto your skin or the injector will not work. | |||||||
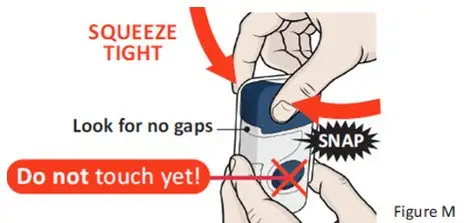 |
|||||||
| Important: Go to the next step right away after loading the cartridge and closing the door. | |||||||
| Step 3: Inject | |||||||
| 3A | Peel away both green pull tabs to show adhesive. (See Figure N) | ||||||
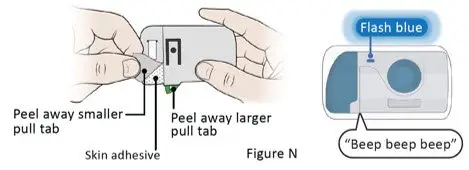 |
|||||||
| The Injector will turn on when the smaller pull tab is removed (along with battery strip). | |||||||
| You will hear 3 "beeps" repeating every 30 seconds until the blue button is pressed. | |||||||
| The status light will begin to flash blue. | |||||||
|
|||||||
| 3B | Stick the injector onto the clean, dry, chosen injection site(s). (See Figure O) | ||||||
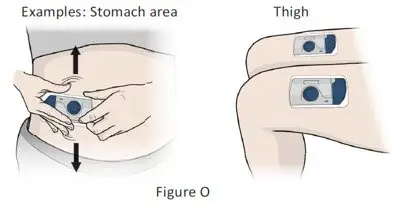 |
|||||||
| Place the injector so that: | |||||||
|
|||||||
| Firmly press the injector onto the skin. | |||||||
| Run your finger around the adhesive edges to make sure the adhesive is secure. | |||||||
|
|||||||
| Important: If placing on stomach, stretch any loose skin as shown with 1 hand. Standing up may make this easier. | |||||||
| 3C | Firmly press and release the blue start button on the injector to start the injection. (See Figure P) | ||||||
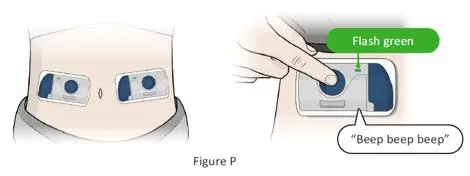 |
|||||||
|
|||||||
| Important: If you have a serious allergic reaction (such as chest pain, trouble breathing, facial swelling, and/or feeling faint), remove the injector(s) to stop the injection and get medical help right away. | |||||||
| 3D | REPEAT Steps 2A - 3C with the second injector. | ||||||
| You must use 2 injectors to receive 1 full dose. | |||||||
| When the second injector is activated, continue to Step 4. | |||||||
 |
|||||||
| Step 4: Monitor | |||||||
| 4A | Monitor both injectors until they are complete. Each injection takes about 10 minutes to finish. (See Figure Q) | ||||||
| The Injection is complete when each injector: | |||||||
|
|||||||
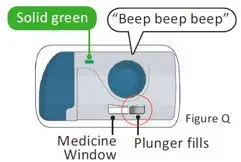 |  | ||||||
| Step 5: Removal and Disposal | |||||||
| 5A | Remove the injectors after both injections are complete. Grip the adhesive and peel the injectors off your skin. (See Figure R) | ||||||
| The green status light will turn off when the injector is removed from your skin. The injector will "beep" 3 times and then stop making noise. |
|||||||
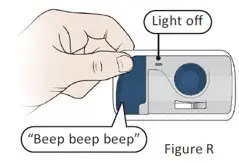 |
|||||||
| 5B | Dispose of (throw away) the used ULTOMIRIS on-body injectors and prefilled cartridges. (See Figure S) | ||||||
|
|||||||
 |
|||||||
| 5C | Check the injection sites. | ||||||
|
|||||||
| Troubleshooting | |||||||
| What do I do if the loaded on-body injector status light flashes red and I hear "beeps"? | |||||||
| Important: If the status light turns red, injector leaks, or either injector does not complete injection, remove and call 1-888-765-4747 immediately (See Figure T). | |||||||
| A red status light indicates that the injector will not work. | |||||||
 |
|||||||
| How many on-body injectors do I need for a full dose? | |||||||
|
|||||||
| What happens if the status light flashes red and I hear beeps? | |||||||
|
|||||||
| I cannot open the blue cartridge door. How do I open it? | |||||||
|
|||||||
| What if the on-body injector does not beep and the blue status light does not blink when I remove the pull tabs? | |||||||
|
|||||||
| What if I push the blue start button before I stick the on-body injector on the skin? | |||||||
|
|||||||
| What if the on-body injector is already on the skin but does not start after pressing the blue start button? | |||||||
|
|||||||
| What if a part of the packaging is missing or damaged (for example: carton, paper cover, plastic cover, etc.)? | |||||||
|
|||||||
| What happens if I do not inject within 5 minutes after loading the prefilled cartridge? | |||||||
|
|||||||
| If I am not ready to inject after waiting at least 45 minutes, can I return the on-body injector and prefilled cartridge to the refrigerator? | |||||||
|
|||||||
| For any additional questions or support, contact 1-888-765-4747. | |||||||
| Additional Environmental Conditions | |||||||
|
|||||||
| Symbol Table | |||||||
 Refer to Instructions for Use Refer to Instructions for Use |  Do not use if packaging is damaged Do not use if packaging is damaged |  Keep Dry Keep Dry |  Single Use Single Use |  Type BF Applied Part Type BF Applied Part |  Sterilized using ethylene oxide Sterilized using ethylene oxide | ||
 Caution Caution |  Use-By Date Use-By Date |  Batch Code Batch Code |  Catalogue Number Catalogue Number |  ULTOMIRIS On-Body Delivery System ULTOMIRIS On-Body Delivery System |  Open Here Open Here | ||
 Serial Number Serial Number |  MR Unsafe MR Unsafe | ||||||
| Manufactured by: Alexion Pharmaceuticals, Inc. Boston, MA 02210 USA U.S. License No. 1743 ULTOMIRIS is a trademark of Alexion Pharmaceuticals, Inc. © 2022 Alexion Pharmaceuticals, Inc. |
|||||||



| ULTOMIRIS
ravulizumab solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ULTOMIRIS
ravulizumab solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ULTOMIRIS
ravulizumab solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ULTOMIRIS
ravulizumab kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Alexion Pharmaceuticals Inc. (789359510) |
