Drug Detail:Tegsedi (Inotersen [ in-oh-ter-sen ])
Drug Class: Miscellaneous metabolic agents
Highlights of Prescribing Information
TEGSEDI (inotersen) injection, for subcutaneous use
Initial U.S. Approval: 2018
WARNING: THROMBOCYTOPENIA AND GLOMERULONEPHRITIS
See full prescribing information for complete boxed warning.
Thrombocytopenia
- TEGSEDI causes reductions in platelet count that may result in sudden and unpredictable thrombocytopenia, which can be life-threatening. (5.1)
- Testing prior to treatment and monitoring during treatment is required (2.3, 2.4, 5.1)
Glomerulonephritis
- TEGSEDI can cause glomerulonephritis that may require immunosuppressive treatment and may result in dialysis-dependent renal failure. (5.2)
- Testing prior to treatment and monitoring during treatment is required (2.3, 2.4, 5.2)
TEGSEDI is available only through a restricted distribution program called the TEGSEDI REMS Program (5.3).
Indications and Usage for Tegsedi
TEGSEDI is a transthyretin-directed antisense oligonucleotide indicated for treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults (1).
Tegsedi Dosage and Administration
- The recommended dosage is 284 mg administered by subcutaneous injection once weekly. (2.1)
- Laboratory tests must be measured prior to treatment, continue to be monitored after treatment initiation, and for 8 weeks following discontinuation of treatment, as directed. (2.3, 2.4)
Dosage Forms and Strengths
Injection: 284 mg/ 1.5 mL in a single-dose prefilled syringe (3)
Contraindications
- Platelet count less than 100 x 109/L (4, 5.1)
- History of acute glomerulonephritis caused by TEGSEDI (4, 5.2)
- Patients with a history of a hypersensitivity reaction to TEGSEDI (4, 5.7)
Warnings and Precautions
-
Stroke and Cervicocephalic Arterial Dissection: These adverse events occurred within 2 days of first dose and with symptoms of cytokine release. Educate patients on symptoms of stroke and central nervous system arterial dissection. (5.4)
-
Inflammatory and Immune Effects: Serious neurologic adverse reactions consistent with inflammatory and immune effects occurred. (5.5)
-
Liver Injury: Monitor alanine amino-transferase, aspartate aminotransferase, and total bilirubin every 4 months during treatment and in case of symptoms of hepatic dysfunction. (5.6)
-
Hypersensitivity Reactions: If these occur, discontinue and initiate appropriate therapy. (5.7)
-
Uninterpretable Platelet Counts: Reaction between Antiplatelet Antibodies and ethylenediaminetetra-acetic acid: Platelet clumping can cause uninterpretable platelet measurement; repeat test if this is suspected. (5.8)
-
Reduced Serum Vitamin A Levels and Recommended Supplementation: Supplement with the recommended daily allowance of vitamin A. Refer to an ophthalmologist if ocular symptoms suggestive of vitamin A deficiency occur. (5.9)
Adverse Reactions/Side Effects
The most common adverse reactions (those that occurred in at least 20% of TEGSEDI-treated patients and more frequently than on placebo) were injection site reactions, nausea, headache, fatigue, thrombocytopenia, and fever (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Sobi, Inc. at 1-833-642-5232 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2022
Full Prescribing Information
WARNING: THROMBOCYTOPENIA AND GLOMERULONEPHRITIS
Thrombocytopenia
TEGSEDI causes reductions in platelet count that may result in sudden and unpredictable thrombocytopenia, which can be life-threatening. One clinical trial patient died from intracranial hemorrhage.
TEGSEDI is contraindicated in patients with a platelet count below 100 x 109/L [see Contraindications (4) and Warnings and Precautions (5.2)].
Prior to starting TEGSEDI, obtain a platelet count [see Dosage and Administration (2.3)]. During treatment, monitor platelet counts weekly if values are 75 x 109/L or greater, and more frequently if values are less than 75 x 109/L [see Dosage and Administration (2.4) and Warnings and Precautions (5.1)].
If a patient develops signs or symptoms of thrombocytopenia, obtain a platelet count as soon as possible. The patient should not receive additional TEGSEDI unless a platelet count is determined to be interpretable and acceptable by a medical professional [see Warnings and Precautions (5.1)].
Following discontinuation of treatment for any reason, continue to monitor platelet count for 8 weeks, or longer if platelet counts are less than 100 x 109/L, to verify that platelet counts remain above 75 x 109/L [see Dosage and Administration (2.4)].
Glomerulonephritis
TEGSEDI can cause glomerulonephritis that may require immunosuppressive treatment and may result in dialysis-dependent renal failure. One clinical trial patient who developed glomerulonephritis and did not receive immunosuppressive treatment remained dialysis-dependent. In clinical trials, cases of glomerulonephritis were accompanied by nephrotic syndrome, which can have manifestations of edema, hypercoagulability with venous or arterial thrombosis, and increased susceptibility to infection [see Warnings and Precautions (5.2)].
TEGSEDI should generally not be initiated in patients with urinary protein to creatinine ratio (UPCR) of 1000 mg/g or higher [see Dosage and Administration (2.4) and Warnings and Precautions (5.2)].
Prior to starting TEGSEDI, measure the serum creatinine, estimated glomerular filtration rate (eGFR), urine protein to creatinine ratio (UPCR), and perform a urinalysis [see Dosage and Administration (2.3)]. During treatment, monitor serum creatinine, eGFR urinalysis, and UPCR every two weeks. TEGSEDI should not be given to patients who develop a UPCR of 1000 mg/g or higher, or eGFR below 45 mL/minute/1.73 m2, pending further evaluation of the cause.
If a dose is held, once eGFR increases to ≥45 mL/minute/1.73 m2, UPCR decreases to below 1000 mg/g, or the underlying cause of the decline in renal function is corrected, weekly dosing may be reinitiated. In patients with UPCR of 2000 mg/g or higher, perform further evaluation for acute glomerulonephritis, as clinically indicated. If acute glomerulonephritis is confirmed, TEGSEDI should be permanently discontinued [see Dosage and Administration (2.4) and Warnings and Precautions (5.2)].
TEGSEDI REMS Program
Because of the risks of serious bleeding caused by severe thrombocytopenia and because of glomerulonephritis, both of which require frequent monitoring, TEGSEDI is available only through a restricted distribution program under a Risk Evaluation and Mitigation Strategy (REMS) called the TEGSEDI REMS Program [see Warnings and Precautions (5.3)].
1. Indications and Usage for Tegsedi
TEGSEDI is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.
2. Tegsedi Dosage and Administration
2.1 Dosing Information
The recommended dose of TEGSEDI is 284 mg injected subcutaneously once weekly.
For consistency of dosing, patients should be instructed to give the injection on the same day every week.
If a dose is missed, patients should be instructed to take the missed dose as soon as possible, unless the next scheduled dose is within 2 days. In this situation, the patient should be directed to skip the missed dose and take the next scheduled dose on the scheduled day.
2.2 Administration
- TEGSEDI is intended for subcutaneous use only.
- The first injection administered by the patient or caregiver should be performed under the guidance of an appropriately qualified healthcare professional. Patients and/or caregivers should be trained in the subcutaneous administration of TEGSEDI in accordance with the Instructions for Use.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit [see How Supplied/Storage and Handling (16)].
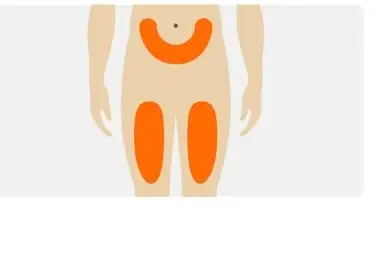
- Sites for injection include the abdomen, upper thigh region, or outer area of the upper arm. It is important to rotate sites for injection.
- If injected in the upper arm, the injection should be administered by a person other than the patient.
- Injection should be avoided at the waistline and other sites where pressure or rubbing from clothing may occur.
- TEGSEDI should not be injected into areas of skin disease or injury.
- Tattoos and scars should also be avoided.
- TEGSEDI prefilled syringe should be allowed to reach room temperature prior to injection.
- Remove from refrigerated storage at least 30 minutes prior to use.
- Other warming methods should not be used.
- Use each prefilled syringe only once.
2.3 Assessment Prior to Initiating TEGSEDI
Measure platelet count, serum creatinine, estimated glomerular filtration rate (eGFR), urine protein to creatinine ratio (UPCR), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin, and perform urinalysis prior to treatment with TEGSEDI and as directed following treatment initiation [see Dosage and Administration (2.4) and Warnings and Precautions (5.1 and 5.2)].
2.4 Laboratory Testing and Monitoring to Assess Safety after Initiating TEGSEDI
Monitor platelet count, serum creatinine, estimated glomerular filtration rate (eGFR), urinalysis, urine protein to creatinine ratio (UPCR), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin during treatment with TEGSEDI, and for 8 weeks following discontinuation of treatment.
Platelet Count
Do not initiate TEGSEDI in patients with a platelet count less than 100 x 109/L. Recommendations for platelet monitoring frequency and TEGSEDI dosing are specified in Table 1. If a patient develops signs or symptoms of thrombocytopenia, obtain a platelet count as soon as possible, and hold dosing until platelet count is confirmed. Recheck the platelet count as soon as possible if a platelet measurement is uninterpretable (e.g., clumped sample) [see Warnings and Precautions (5.8].
| * It is strongly recommended that, unless the patient has a medical contraindication to receiving glucocorticoids, the patient receive glucocorticoid therapy to reverse the platelet decline [see Warnings and Precautions (5.1)]. # Additional risk factors for bleeding include age >60 years, receiving anticoagulant or antiplatelet medicinal products, or prior history of major bleeding events. †Patients who discontinue therapy with TEGSEDI because of platelet counts below 25 x109/L should not reinitiate therapy. |
||
| Platelet count (x109/L) | Monitoring Frequency | Dosing |
| At least 100 | Weekly | Continue to dose weekly. |
| At least 75 to less than 100 | Weekly | Stop treatment. Do not restart unless platelet count is greater than 100. |
| At least 50 to less than 75 | Twice weekly until 3 successive values above 75; then weekly monitoring. | Stop treatment. Do not restart TEGSEDI in patients with thrombocytopenia, unless there have been 3 successive values above 100 and the benefit of TEGSEDI outweighs the risk of thrombocytopenia and potential bleeding. |
| At least 25 to less than 50* |
Twice weekly until 3 successive values above 75; then weekly monitoring. Consider more frequent monitoring if additional risk factors for bleeding are present.# |
Stop treatment. Do not restart TEGSEDI in patients with thrombocytopenia, unless there have been 3 successive values above 100 and the benefit of TEGSEDI outweighs the risk of thrombocytopenia and potential bleeding. Corticosteroids recommended. Consider discontinuation of any antiplatelet agents or anticoagulants. |
| Less than 25*† | Daily until 2 successive values above 25. Then monitor twice weekly until 3 successive values above 75. Then weekly monitoring until stable. |
Stop TEGSEDI. Corticosteroids recommended. Consider discontinuation of any antiplatelet agents or anticoagulants. |
Renal Monitoring
TEGSEDI should generally not be initiated in patients with a urine protein to creatinine ratio (UPCR) of 1000 mg/g or higher. Monitor serum creatinine, estimated glomerular filtration rate (eGFR), urinalysis, and UPCR every 2 weeks during treatment with TEGSEDI. Hold TEGSEDI in patients who develop a UPCR of 1000 mg/g or higher, or estimated glomerular filtration rate (eGFR) below 45 mL/minute/1.73 m2, pending further evaluation of the cause.
If a dose is held, once eGFR increases to ≥45 mL/minute/1.73 m2, UPCR decreases to below 1000 mg/g, or the underlying cause of the decline in renal function is corrected, weekly dosing may be reinitiated. In the case of UPCR of 2000 mg/g or higher, perform further evaluation for acute glomerulonephritis, as clinically indicated. If acute glomerulonephritis is confirmed, TEGSEDI should be permanently discontinued.
Liver Tests
Monitor alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin every four months during treatment with TEGSEDI; monitor monthly for patients who have received a liver transplant [see Warnings and Precautions (5.6)].
3. Dosage Forms and Strengths
Injection: 284 mg/1.5 mL clear, colorless to pale yellow solution in a single-dose prefilled syringe.
4. Contraindications
TEGSEDI is contraindicated in patients with:
- Platelet count below 100 x 109/L [see Warnings and Precautions (5.1)]
- History of acute glomerulonephritis caused by TEGSEDI [see Warnings and Precautions (5.2)]
- History of a hypersensitivity reaction to TEGSEDI [see Warnings and Precautions (5.7)].
5. Warnings and Precautions
5.1 Thrombocytopenia
TEGSEDI causes reductions in platelet count that may result in sudden and unpredictable thrombocytopenia that can be life-threatening. In Study 1 [see Clinical studies (14)] , platelet counts below 100 x 109/L occurred in 25% of TEGSEDI-treated patients, compared with 2% of patients on placebo. Platelet counts below 75 x 109/L occurred in 14% of TEGSEDI-treated patients, compared to no patient on placebo. In Study 1 and its extension study, 39% of TEGSEDI-treated patients with a baseline platelet count below 200 x109/L had a nadir platelet count below 75 x 109/L, compared to 6% of patients with baseline platelet counts 200 x109/L or higher.
Three TEGSEDI-treated patients (3%) had sudden severe thrombocytopenia (platelet count below 25 x 109/L), which can have potentially fatal bleeding complications, including spontaneous intracranial or intrapulmonary hemorrhage. One patient in a clinical trial experienced a fatal intracranial hemorrhage.
In clinical trials, all 3 patients with severe thrombocytopenia had treatment-emergent antiplatelet IgG antibodies detected shortly before or at the time of the severe thrombocytopenia. In 2 patients, platelet clumping caused uninterpretable platelet measurements that delayed the diagnosis and treatment of severe thrombocytopenia. Platelet clumping can be caused by a reaction between antiplatelet antibodies and ethylenediaminetetraacetic acid (EDTA) [see Warnings and Precautions (5.8)].
Monitoring and Dosing
Patients who are not able to adhere to the recommended laboratory monitoring or to the related treatment recommendations must not receive TEGSEDI. Do not initiate TEGSEDI in patients with a platelet count below 100 x 109/L. Follow recommended monitoring and treatment recommendations for platelet count [see Dosage and Administration (2.4)]. If a patient develops signs or symptoms of thrombocytopenia, obtain a platelet count as soon as possible, and hold TEGSEDI dosing unless the platelet count is confirmed to be acceptable. Recheck the platelet count as soon as possible if a platelet measurement is uninterpretable (e.g., clumped sample) [see Warnings and Precautions (5.8)]. Hold TEGSEDI dosing until an acceptable platelet count is confirmed with an interpretable blood sample.
Concomitant Medications with Platelet Effects
When considering use of TEGSEDI concomitantly with antiplatelet drugs or anticoagulants, be aware of the risk of potential bleeding from thrombocytopenia with TEGSEDI, and consider discontinuation of these drugs in patients with a platelet count less than 50 x 109/L [see Drug Interactions (7.1)].
Symptoms of Thrombocytopenia
Symptoms of thrombocytopenia can include unusual or prolonged bleeding (e.g., petechiae, easy bruising, hematoma, subconjunctival bleeding, gingival bleeding, epistaxis, hemoptysis, irregular or heavier than normal menstrual bleeding, hematemesis, hematuria, hematochezia, melena), neck stiffness or atypical severe headache. Patients and caregivers should be instructed to be vigilant for symptoms of thrombocytopenia and seek immediate medical help if they have concerns.
Severe Thrombocytopenia: Treatment with Glucocorticoids
Glucocorticoid therapy is strongly recommended in patients with a platelet count below 50 x 109/L, and in patients with suspected immune-mediated thrombocytopenia. Avoid using TEGSEDI in patients for whom glucocorticoid treatment is not advised.
5.2 Glomerulonephritis and Renal Toxicity
TEGSEDI can cause glomerulonephritis that may result in dialysis-dependent renal failure. In Study 1 [see Clinical studies (14)], glomerulonephritis occurred in three (3%) TEGSEDI-treated patients vs. no patient on placebo. In these patients, stopping TEGSEDI alone was not sufficient to resolve manifestations of glomerulonephritis, and treatment with an immunosuppressive medication was necessary. One patient did not receive immunosuppressive treatment and remained dialysis-dependent. If glomerulonephritis is suspected, pursue prompt diagnosis and initiate immunosuppressive treatment as soon as possible.
Cases of glomerulonephritis were accompanied by nephrotic syndrome. Possible complications of nephrotic syndrome can include edema, hypercoagulability with venous or arterial thrombosis, and increased susceptibility to infection. TEGSEDI-treated patients who develop glomerulonephritis will require monitoring and treatment for nephrotic syndrome and its manifestations.
Accumulation of antisense oligonucleotides in proximal tubule cells of the kidney, sometimes leading to increased tubular proteinuria, has been described in nonclinical studies. Urine protein to creatinine ratio (UPCR) greater than 5 times the upper limit of normal occurred in 15% of TEGSEDI-treated patients, compared to 8% of patients on placebo. Increase from baseline in serum creatinine greater than 0.5 mg/dL occurred in 11% of TEGSEDI-treated patients, compared to 2% of patients on placebo.
Follow recommended monitoring and treatment recommendations for renal parameters [see Dosage and Administration (2.4)] . TEGSEDI should generally not be initiated in patients with a UPCR of 1000 mg/g or greater. If acute glomerulonephritis is confirmed, TEGSEDI should be permanently discontinued [see Contraindications (4)].
Use caution with nephrotoxic drugs and other drugs that may impair renal function. Because immunosuppressive treatment is necessary for the treatment of glomerulonephritis, avoid using TEGSEDI in patients for whom immunosuppressive treatment is not advised.
5.3 TEGSEDI REMS Program
TEGSEDI is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the TEGSEDI REMS Program, because of risks of serious bleeding caused by severe thrombocytopenia and because of glomerulonephritis [see Warnings and Precautions (5.1, 5.2)].
Important requirements of the TEGSEDI Prescribing Program include:
- Prescribers must be certified within the program by enrolling and completing training.
- Patients must enroll in the program and comply with ongoing monitoring requirements [see Warnings and Precautions (5.1) and Dosage and Administration (2.4)]. Pharmacies must be certified with the program and must only dispense to patients who are authorized to receive TEGSEDI.
5.4 Stroke and Cervicocephalic Arterial Dissection
TEGSEDI may cause stroke and cervicocephalic arterial dissection. In clinical studies, 1 of 161 (0.6%) TEGSEDI-treated patients experienced carotid artery dissection and stroke. These events occurred within 2 days of the first TEGSEDI dose, a time when the patient also had symptoms of cytokine release (e.g., nausea, vomiting, muscular pain and weakness) and a high sensitivity C-reactive protein level greater than 100 mg/L.
Educate patients on the symptoms of stroke and central nervous system arterial dissection. Instruct patients to seek help as soon as possible if symptoms of stroke or arterial dissection occur.
5.5 Inflammatory and Immune Effects
Inflammatory and immune changes are an effect of some antisense oligonucleotide drugs, including TEGSEDI. In clinical studies, serious inflammatory and immune adverse reactions occurred in TEGSEDI-treated patients, including immune thrombocytopenia and glomerulonephritis, as well as a single case of antineutrophil cytoplasmic autoantibody (ANCA)-positive systemic vasculitis [see Warnings and Precautions (5.2) and (5.3)].
Neurologic Serious Adverse Reactions
In clinical studies, neurologic serious adverse reactions consistent with inflammatory and immune effects occurred in TEGSEDI-treated patients, in addition to stroke and carotid artery dissection [see Warnings and Precautions (5.5)]. Two months after the first TEGSEDI dose, one patient developed a change in gait that progressed over 6 months to paraparesis, in the absence of radiologic evidence of spinal cord compression. Another patient developed progressive lumbar pain, weight loss, headache, vomiting, and impaired speech 7 months after starting TEGSEDI. Cerebrospinal fluid analysis findings included elevated protein, a lymphocyte-predominant pleocytosis, and testing that was negative for infection. The patient recovered after empiric therapy (high-dose steroids, antibiotics) and resumed TEGSEDI without recurrence of symptoms.
5.6 Liver Injury
The liver is a site of accumulation of antisense oligonucleotides. In clinical studies, 8% of TEGSEDI-treated patients had an increased alanine aminotransferase (ALT) at least 3 times the upper limit of normal (ULN), compared to 3% of patients on placebo; 3% of TEGSEDI-treated patients had an ALT at least 8 times the ULN, compared to no patient on placebo. One clinical study patient experienced an increased ALT more than 30 times the ULN. After a course of corticosteroids and discontinuation of TEGSEDI, the patient’s ALT returned to normal levels. Some patients had resolution of the liver laboratory abnormalities with continued use of TEGSEDI.
In clinical studies, demonstrated or possible cases of immune-mediated biliary disease occurred in TEGSEDI-treated patients. There was a single case of autoimmune hepatitis with primary biliary cirrhosis in a patient with a family history of primary biliary cirrhosis, as well as a single case of biliary obstruction of unclear etiology.
Monitor alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin at baseline and every four months during treatment with TEGSEDI. If a patient develops clinical signs or symptoms suggestive of hepatic dysfunction (e.g., unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine), promptly measure serum transaminases and total bilirubin and interrupt or discontinue treatment with TEGSEDI, as appropriate.
Liver Transplant Rejection
In a clinical study, cases of liver transplant rejection were reported 2-4 months after starting TEGSEDI in patients whose liver allografts had previously been clinically stable (for over 10 years) prior to starting TEGSEDI. In these cases, the patients clinically improved and transaminase levels normalized after glucocorticoid administration and cessation of TEGSEDI.
In patients with a history of liver transplant, monitor ALT, AST, and total bilirubin monthly. Discontinue TEGSEDI in patients who develop signs of liver transplant rejection.
5.7 Hypersensitivity Reactions/Antibody Formation
TEGSEDI can cause hypersensitivity reactions. In clinical studies, 6 of 161 (4%) TEGSEDI-treated patients stopped treatment because of a hypersensitivity reaction. Antibodies to TEGSEDI were present when the reactions occurred. These reactions generally occurred within 2 hours of administration of TEGSEDI and included headache, chest pain, hypertension, chills, flushing, dysphagia, palmar erythema, eosinophilia, involuntary choreaform movements, arthralgia, myalgia, and flu-like symptoms.
If a hypersensitivity reaction occurs, discontinue administration of TEGSEDI, and initiate appropriate therapy. Do not use in patients who have a history of hypersensitivity reaction to TEGSEDI.
5.8 Uninterpretable Platelet Counts: Reaction between Antiplatelet Antibodies and ethylenediaminetetra-acetic acid (EDTA)
In Study 1 [see Clinical Studies (14)], 23% of TEGSEDI-treated patients had at least 1 uninterpretable platelet count caused by platelet clumping, compared to 13% of patients on placebo. In 2 cases of severe thrombocytopenia with platelet count below 25 x 109/L, one of which resulted in death, clumped platelet samples caused a delay in diagnosis and treatment. Both subjects had tested positive for treatment-emergent anti-platelet IgG antibodies detected shortly before, or at the time of the severe reduction in platelet count.
Although platelet clumping can have a variety of causes (e.g., incompletely mixed or inadequately anticoagulated samples), platelet clumping can be caused by a reaction between antiplatelet antibodies and ethylenediaminetetra-acetic acid (EDTA). In Study 1, 7 of the 9 (78%) TEGSEDI-treated patients with treatment-emergent positive antiplatelet antibody testing had at least 1 clumped platelet sample.
If there is suspicion of EDTA-mediated platelet clumping, perform a repeat platelet count using a different anticoagulant (e.g., sodium citrate, heparin) in the blood collection tube. Recheck the platelet count as soon as possible if a platelet measurement is uninterpretable. Hold TEGSEDI dosing until an acceptable platelet count is confirmed with an interpretable blood sample.
5.9 Reduced Serum Vitamin A Levels and Recommended Supplementation
TEGSEDI treatment leads to a decrease in serum vitamin A levels. Supplementation at the recommended daily allowance of vitamin A is advised for patients taking TEGSEDI. Higher doses than the recommended daily allowance of vitamin A should not be given to try to achieve normal serum vitamin A levels during treatment with TEGSEDI, as serum vitamin A levels do not reflect the total vitamin A in the body.
Patients should be referred to an ophthalmologist if they develop ocular symptoms suggestive of vitamin A deficiency (e.g., night blindness).
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed in greater detail in other sections of the labeling:
- Thrombocytopenia [see Warnings and Precautions (5.1)]
- Glomerulonephritis and Renal Toxicity [see Warnings and Precautions (5.2)]
- Stroke and Cervicocephalic Arterial Dissection [see Warnings and Precautions (5.4)]
- Inflammatory and Immune Effects [see Warnings and Precautions (5.5)]
- Liver Injury [see Warnings and Precautions (5.6)]
- Hypersensitivity [see Warnings and Precautions (5.7)]
- Reducted Serum Vitamin A Levels and Recommended Supplementation [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of TEGSEDI cannot be directly compared to rates in clinical trials of other drugs and may not reflect the rates observed in practice.
A total of 112 adult patients with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis (hATTR) received TEGSEDI in Study 1 and 60 patients received placebo. The, mean age of the study patients was 59 years (27 to 78 years of age). Of the TEGSEDI-treated patients, 69% were male and 94% were Caucasian, with a mean exposure of 385 days, and median exposure of 449 days. Baseline disease characteristics were largely similar in TEGSEDI-treated patients and patients in the placebo control group. Sixty-seven percent of patients were in Stage 1 of the disease at baseline, and 33% in Stage 2. Fifty-two percent of patients had Val30Met mutations in the TTR gene, with the remaining 48% comprised of 26 different other point mutations.
Table 2 presents common adverse reactions that occurred in at least 5% of TEGSEDI-treated patients and that occurred at least 5% more frequently or two times more frequently than on placebo.
The most common adverse reactions that occurred in at least 20% of TEGSEDI-treated patients and more frequently than on placebo were injection site reactions, nausea, headache, fatigue, thrombocytopenia, and fever. Serious adverse reactions were more frequent in TEGSEDI-treated patients (32%) than in patients on placebo (21%). The most common adverse reactions leading to discontinuation were thrombocytopenia and cachexia.
| a Includes bruising, erythema, hematoma, hemorrhage, induration, inflammation, mass, edema, pain, pruritus, rash, swelling, and urticaria. b Includes arrhythmia, atrial fibrillation, atrial flutter, bradyarrhythmia, bradycardia, extrasystoles, sinus arrhythmia, sinus bradycardia, supraventricular extrasystoles, tachycardia, and ventricular extrasystoles. c Includes bacteremia, cellulitis staphylococcal, clostridium difficile infection, conjunctivitis bacterial, cystitis Escherichia, Helicobacter gastritis, Helicobacter infection, Staphylococcal infection. |
||
| TEGSEDI (N=112) % | Placebo (N=60) % |
|
| Injection site reactionsa | 49 | 10 |
| Nausea | 31 | 12 |
| Headache | 26 | 12 |
| Fatigue | 25 | 20 |
| Thrombocytopenia | 24 | 2 |
| Fever | 20 | 8 |
| Peripheral edema | 19 | 10 |
| Chills | 18 | 3 |
| Anemia | 17 | 3 |
| Vomiting | 15 | 5 |
| Myalgia | 15 | 10 |
| Decreased renal function | 14 | 5 |
| Arrhythmiab | 13 | 5 |
| Arthralgia | 13 | 8 |
| Pre-syncope or syncope | 13 | 5 |
| Decreased appetite | 10 | 0 |
| Paresthesia | 10 | 3 |
| Dyspnea | 9 | 3 |
| Elevated liver function test | 9 | 3 |
| Orthostasis | 8 | 2 |
| Influenza-like illness | 8 | 3 |
| Contusion | 7 | 2 |
| Bacterial infectionc | 7 | 3 |
| Eosinophilia | 5 | 0 |
| Dry mouth | 5 | 2 |
6.2 Immunogenicity
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. In addition, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to TEGSEDI in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In Study 1, 30% of TEGSEDI-treated patients tested positive for anti-drug antibodies (ADA) following 65 weeks of treatment [see Warnings and Precautions (5.7, 5.8)]. However, the assay measured only IgG isotypes and the existence of other isotypes may be possible. In many cases adverse reactions occurred in patients with ADA, although the available data are too limited to make definitive conclusions about the relationship.
7. Drug Interactions
7.1 Antiplatelet Drugs or Anticoagulant Medications
Because of the risk of thrombocytopenia, caution should be used when using antiplatelet drugs (e.g., adenosine, clopidogrel, prasugrel, ticagrelor, or ticlopidine), including non-prescription products that affect platelets (e.g., aspirin, nonsteroidal anti-inflammatory drugs), or anticoagulants (e.g., heparin, warfarin), concomitantly with TEGSEDI [see Warnings and Precautions (5.1)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to TEGSEDI during pregnancy. Health care providers are encouraged to register patients and pregnant women are encouraged to register themselves by calling: 1-877-465-7510, emailing: [email protected], or visiting online at: www.tegsedipregnancystudy.com.
Risk Summary
There are no data on the developmental risk associated with the use of TEGSEDI in pregnant women. TEGSEDI treatment leads to a decrease in serum vitamin A levels, and vitamin A supplementation is advised for patients taking TEGSEDI. Vitamin A is essential for normal embryofetal development; however, excessive levels of Vitamin A are associated with adverse developmental effects. The effects on the fetus of a reduction in maternal serum TTR caused by TEGSEDI and of vitamin A supplementation are unknown [see Clinical Pharmacology (12.2), Warnings and Precautions (5.9)].
In animal studies, subcutaneous administration of inotersen to pregnant rabbits resulted in premature delivery and reduced fetal body weight at the highest dose tested, which was associated with maternal toxicity. No adverse developmental effects were observed when inotersen or a pharmacologically-active surrogate was administered to pregnant mice.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Subcutaneous administration of inotersen (0, 3, 15, or 25 mg/kg) or a rodent-specific surrogate (15 mg/kg) to male and female mice every other day prior to and during mating and continuing in females throughout the period of organogenesis produced no adverse effects on embryofetal development.
Subcutaneous administration of inotersen (0, 2.5, 5, or 15 mg/kg) to pregnant rabbits every other day throughout the period of organogenesis resulted in premature delivery and reduced fetal body weight at the highest dose tested, which was associated with maternal toxicity (reduced body weight and food consumption).
Subcutaneous administration of inotersen (0, 2.9, 11.4, or 22.9 mg/kg) or a rodent-specific surrogate (11.4 mg/kg) to mice every other day throughout pregnancy and lactation produced no adverse effects on pre- or postnatal development.
8.2 Lactation
Risk Summary
There is no information regarding the presence of TEGSEDI in human milk, the effects on the breast-fed infant, or the effects on milk production. A study in lactating mice has shown excretion of inotersen in milk. The development and health benefits of breastfeeding should be considered along with the mother’s clinical need for TEGSEDI and any potential adverse effects on the breastfed infant from TEGSEDI or from the underlying maternal condition.
8.5 Geriatric Use
Clinical studies of TEGSEDI included 69 patients (45%) aged 65 and over. No differences in pharmacokinetics or effectiveness were observed between these patients and younger patients. Patients 65 years and older may be at increased risk of certain adverse reactions, such as congestive heart failure, chills, myalgia, and extremity pain.
8.6 Renal Impairment
No dose adjustment is necessary in patients with mild to moderate renal impairment (estimated glomerular filtration rate [eGFR] ≥30 to <90 mL/min/1.73 m2) [see Clinical Pharmacology (12.3)]. TEGSEDI has not been studied in patients with severe renal impairment or end-stage renal disease.
11. Tegsedi Description
Inotersen is an antisense oligonucleotide (ASO) inhibitor of human transthyretin (TTR) protein synthesis.
TEGSEDI contains inotersen sodium as the active ingredient. Inotersen sodium is a white to pale yellow solid and it is freely soluble in water and in phosphate buffer (pH 7.5 to 8.5). The chemical name of inotersen sodium is DNA, d(P-thio)([2'-O-(2-methoxyethyl)]m5rU-[2'-O-(2-methoxyethyl)]m5rC-[2'-O-(2-methoxyethyl)]m5rU-[2'-O-(2-methoxyethyl)]m5rU-[2'-O-(2-methoxyethyl)]rG-G-T-T-A-m5C-A-T-G-A-A-[2'-O-(2-methoxyethyl)]rA-[2'-O-(2-methoxyethyl)]m5rU-[2'-O-(2-methoxyethyl)]m5rC-[2'-O-(2-methoxyethyl)]m5rC-[2'-O-(2-methoxyethyl)]m5rC). The molecular formula of inotersen sodium is C230H299N69 Na19O121P19S19 and the molecular weight is 7600.73 Da. It has the following structural formula:
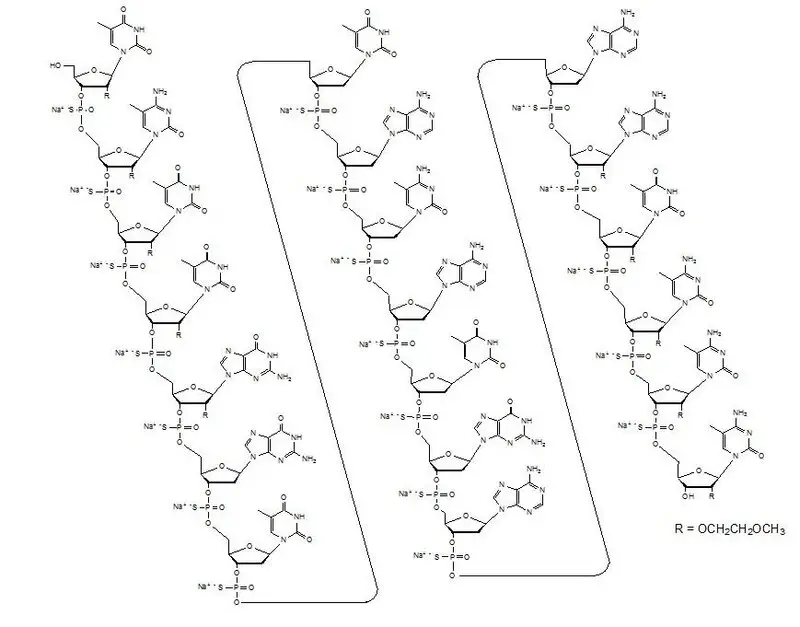
The molecular formula of inotersen free base is C230H318N69O121P19S19 and its molecular weight is 7183.08.
TEGSEDI is a sterile, preservative-free, aqueous solution for subcutaneous injection. It is supplied in a prefilled syringe (PFS). Each PFS contains 1.5 mL of solution containing 284 mg inotersen (equivalent to 300 mg inotersen sodium salt) TEGSEDI is formulated in Water for Injection and may include hydrochloric acid and/or sodium hydroxide for pH adjustment to 7.5-8.5.
12. Tegsedi - Clinical Pharmacology
12.1 Mechanism of Action
Inotersen is an antisense oligonucleotide that causes degradation of mutant and wild-type TTR mRNA through binding to the TTR mRNA, which results in a reduction of serum TTR protein and TTR protein deposits in tissues.
12.2 Pharmacodynamics
The pharmacodynamic effects of TEGSEDI were evaluated in hATTR amyloidosis patients treated with 284 mg TEGSEDI via subcutaneous injection once weekly.
With repeat dosing, the mean percent decreases from baseline in serum TTR from Week 13 to Week 65 of treatment ranged from 68% to 74% (median range: 75% to 79%). Similar TTR reductions were observed regardless of TTR mutation, sex, age, or race.
Serum TTR is a carrier of retinol binding protein, which is involved in the transport of vitamin A in the blood. Mean reductions in serum retinol binding of 71%, and serum vitamin A of 63%, were observed at Week 65 [see Warnings and Precautions (5.6)].
Cardiac Electrophysiology
Formal QTc studies have not been conducted with TEGSEDI. The potential for QTc prolongation with inotersen was evaluated in a randomized, placebo-controlled trial in healthy volunteers. No large changes in the mean QTc interval (>20 ms) were detected in the trial.
In the 66-week controlled efficacy trial, 5.4% of TEGSEDI-treated patients had evidence of QRS prolongation on their electrocardiograms (ECGs) to greater than 160 msec and greater than 25% above baseline, compared to and in 1.7% of patients on placebo.
12.3 Pharmacokinetics
Following subcutaneous administration, systemic exposure to inotersen increased in a dose-proportional manner over the range of 150-400 mg of inotersen sodium salt. At the recommended TEGSEDI dosing regimen of 284 mg every week, steady state is reached after approximately 3 months. The estimated geometric mean (90% confidence interval) steady state peak concentrations (Cmax), trough concentrations (Ctrough), and area under the curve (AUCτ) were 6.39 (5.65, 7.20) µg/mL, 0.034 (0.031, 0.038) µg/mL, and 90 (82.4, 97.4) µg·h/mL, respectively. Plasma Cmax and AUC do not exhibit accumulation at steady state.
Absorption
Following subcutaneous administration, TEGSEDI is absorbed rapidly into systemic circulation in a dose-dependent fashion, with the median time to maximum plasma concentrations (Cmax) of 2 to 4 hours.
Distribution
TEGSEDI is highly bound to human plasma proteins (>94%) and the fraction bound is independent of drug concentration. Based on animal studies (mouse, rat and monkey), TEGSEDI rapidly distributes broadly to tissues, with the highest concentrations observed in the kidney and liver. TEGSEDI does not cross the blood-brain barrier. The apparent volume of distribution of TEGSEDI at steady-state (mean and 90% confidence interval) is 293 (268, 320) L in patients with hATTR.
Elimination
The terminal elimination half-life (mean and 90% confidence interval) for TEGSEDI is 32.3 (29.4, 35.5) days. Inotersen is mainly cleared through metabolism, and the total body clearance (mean and 90% confidence interval) is 3.18 (3.08, 3.29) L/h.
Metabolism
Inotersen is metabolized by nucleases to nucleotides of various lengths.
Excretion
Less than 1% of the administered dose of inotersen is excreted unchanged into urine within 24 hours.
Specific Populations
Age, race, and sex had no impact on the steady state pharmacokinetics of inotersen or TTR reduction. Population pharmacokinetic and pharmacodynamic analyses indicated no impact of mild or moderate renal impairment (eGFR ≥30 to <90 mL/min/1.73 m2) or mild hepatic impairment (bilirubin less than or equal to 1.5 x ULN and/or AST less than 1.9 x ULN) on inotersen exposure or TTR reduction. TEGSEDI has not been studied in patients with severe renal impairment, end-stage renal disease, moderate or severe hepatic impairment, or in patients with prior liver transplant.
Drug Interaction Studies
No formal clinical drug interaction studies have been performed. TEGSEDI is not a substrate or inhibitor/inducer of major CYP enzymes or a substrate or inhibitor of major transporters. In a population pharmacokinetic analysis, concomitant use of diuretics, antithrombotic, and analgesics did not impact the pharmacokinetic parameters of inotersen. TEGSEDI is not expected to cause drug-drug interactions or to be affected by inhibitors or inducers of cytochrome P450 enzymes.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 26-week carcinogenicity study in transgenic (TgRasH2) mice, weekly subcutaneous administration of inotersen (0, 10, 30, or 80 mg/kg) or a rodent-specific (pharmacologically active) surrogate (30 mg/kg) did not result in an increase in tumors.
In a 94-week carcinogenicity study in rats, weekly subcutaneous administration of inotersen (0, 0.5, 2, or 6 mg/kg) resulted in an increase in tumors at or near the injection site in males at all but the lowest dose (0.5 mg/kg) tested. Subcutaneous malignant pleomorphic fibrosarcoma was increased at the mid and high doses and combined subcutaneous malignant pleomorphic fibrosarcoma and monomorphic fibrosarcoma were increased at the high dose. These tumors are considered a response to chronic tissue irritation and inflammation caused by repeated subcutaneous injection.
Mutagenesis
Inotersen was negative for genotoxicity in in vitro (bacterial mutagenicity, chromosomal aberration in Chinese hamster lung) and in vivo (mouse bone marrow micronucleus) assays.
Impairment of Fertility
Subcutaneous administration of inotersen (0, 3, 15, or 25 mg/kg) or a rodent-specific surrogate (15 mg/kg) to male and female mice every other day prior to and during mating and continuing in females throughout the period of organogenesis produced no adverse effects on fertility.
14. Clinical Studies
The efficacy of TEGSEDI was demonstrated in a randomized, double-blind, placebo-controlled, multicenter clinical trial in adult patients with polyneuropathy caused by hATTR amyloidosis (Study 1; NCT 01737398). Patients were randomized in a 2:1 ratio to receive either TEGSEDI (284 mg inotersen) (N=113) or placebo (N=60), respectively, as a subcutaneous injection administered once per week for 65 weeks (3 doses were administered during the first week of treatment). Seventy seven percent of TEGSEDI-treated patients and 87% of patients on placebo completed 66 weeks of the assigned treatment.
The co-primary efficacy endpoints were the change from baseline to Week 66 in the modified Neuropathy Impairment Scale+7 (mNIS+7) composite score and the Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) total score
The mNIS+7 is an objective assessment of neuropathy, and comprises the NIS and Modified +7 composite scores. In the version of the mNIS+7 used in the trial, the NIS objectively measures deficits in cranial nerve function, muscle strength, reflexes, and sensations, and the Modified +7 assesses heart rate response to deep breathing, postural blood pressure, quantitative sensory testing (touch-pressure and heat-pain), and peripheral nerve electrophysiology. The maximum possible score was 346.32 points, with higher scores representing a greater severity of disease.
The clinical meaningfulness of effects on the mNIS+7 was assessed by the change from baseline to Week 66 in Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) total score. The Norfolk QoL-DN scale is a patient-reported assessment that evaluates the subjective experience of neuropathy in the following domains: physical functioning/large fiber neuropathy, activities of daily living, symptoms, small fiber neuropathy, and autonomic neuropathy. The version of the Norfolk QoL-DN that was used in the trial had a maximum possible total score of 136 points, with higher scores representing greater impairment.
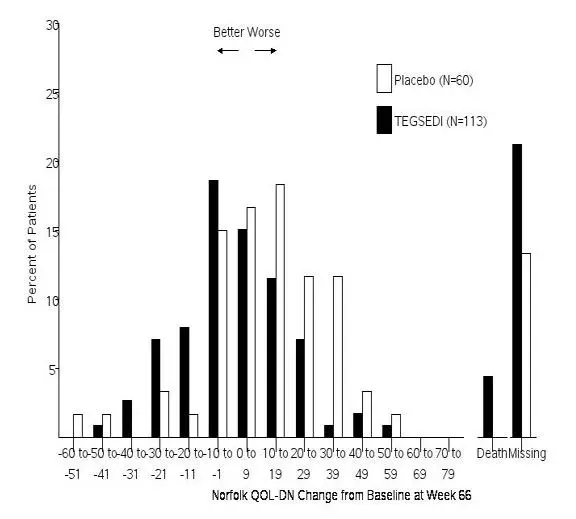
The changes from baseline to Week 66 on both the mNIS+7 and the Norfolk QoL-DN significantly favored TEGSEDI (Table 3, Figures 1 and 3). The distributions of changes in mNIS+7 and Norfolk QoL-DN scores from baseline to Week 66 by percent of patients are shown in Figure 2 and Figure 4, respectively.
| CI, confidence interval; LS, least squares; mNIS, modified Neuropathy Impairment Score; QoL-DN, Quality of Life – Diabetic Neuropathy a All endpoints analyzed using the mixed-effect model repeated measures (MMRM) method. b A lower value indicates less impairment/fewer symptoms. cThe primary analysis population for the mNIS+7 analysis included N=95 TEGSEDI patients and N=56 placebo patients dThe primary analysis population for the Norfolk QOL-DN analysis included N=94 TEGSEDI patients and N=57 placebo patients |
||||||
| Endpoint | Baseline | Change from Baseline to Week 66 (LS Mean) | TEGSEDI – placebo Treatment Difference LS Mean (95% CI) | p- value |
||
| TEGSEDI | Placebo | TEGSEDI | Placebo | |||
| Primarya | ||||||
| mNIS+7b, c | 80.2 | 75.3 | 5.8 | 25.5 | -19.7
[-26.4, -13.0] | <0.001 |
| Norfolk QOL- DNb, d | 48.7 | 48.7 | 1.0 | 12.7 | -11.7
[-18.3, -5.1] | <0.001 |
Figure 1: Change from Baseline in mNIS+7
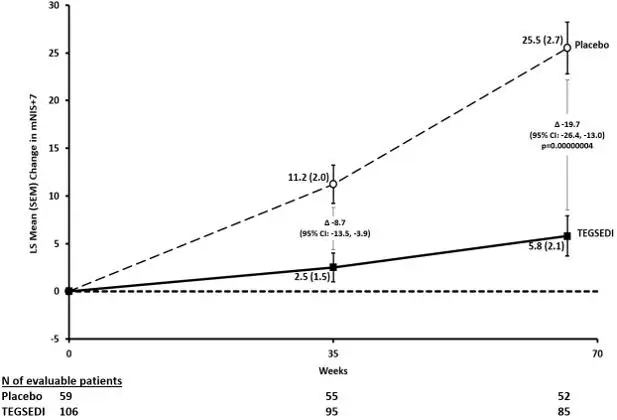
Figure 2: Histogram of mNIS+7 Change from Baseline at Week 66
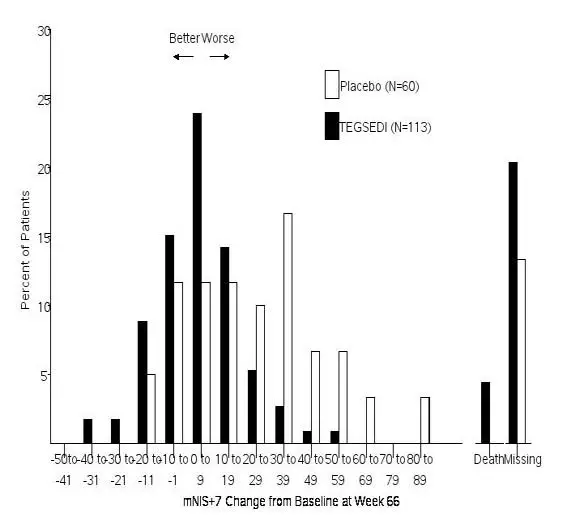
Figure 3: Change from Baseline in Norfolk QoL-DN Score
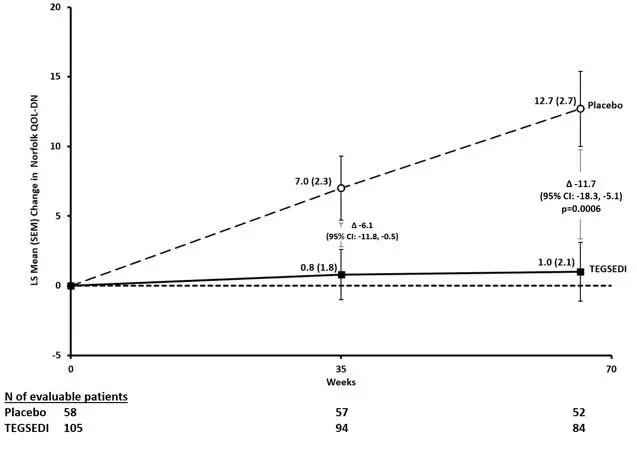
Figure 4: Histogram of Norfolk QoL-DN Change from Baseline at Week 66
Patients receiving TEGSEDI experienced similar improvements relative to placebo in mNIS+7 and Norfolk QoL-DN score across all subgroups including age, sex, race, region, NIS score, Val30Met mutation status, and disease stage.
16. How is Tegsedi supplied
TEGSEDI is a clear, colorless to pale yellow solution supplied in a single-dose, prefilled syringe with a SSD. Each prefilled syringe of TEGSEDI is filled to deliver 1.5 mL of solution containing 284 mg of inotersen (equivalent to 300 mg inotersen sodium salt).
TEGSEDI is available in cartons containing 1 or 4 prefilled syringes supplied in individual trays.
- Pack of 1 prefilled syringe: NDC 72126-007-03
- Pack of 4 prefilled syringes: NDC 72126-007-01
The individual tray of 1 syringe is NDC 72126-007-02.
Pharmacy
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original container and protect from direct light. Do not freeze.
For Patients/Caregivers
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original container. Do not freeze. TEGSEDI can be kept at room temperature (up to 30°C [86°F]) in the original container for up to 6 weeks; if not used within the 6 weeks, discard TEGSEDI.
Remove from refrigerated storage (2°C to 8°C [36°F to 46°F]) at least 30 minutes before use. [TEGSEDI] prefilled syringe should be allowed to reach room temperature prior to injection.
Avoid exposure to temperatures above 30°C (86°F).
17. Patient Counseling Information
Advise the patient and caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Thrombocytopenia
Inform patients that TEGSEDI can cause reductions in platelet count that may result in thrombocytopenia. Instruct patients to notify a healthcare provider immediately if they show symptoms of thrombocytopenia (e.g., unusual or prolonged bleeding, neck stiffness, or atypical severe headache). Advise patients of the importance of monitoring during treatment with TEGSEDI [see Warnings and Precautions (5.1)]. Also instruct patients to notify their healthcare provider of all medications, including over-the-counter, that they are taking [see Drug Interactions (7.1)].
Glomerulonephritis and Renal Toxicity
Inform patients that glomerulonephritis has occurred in patients treated with TEGSEDI. Advise patients of the importance of monitoring of urine protein to creatinine ratio (UPCR during treatment with TEGSEDI) [see Warnings and Precautions (5.2)].
TEGSEDI REMS Program
TEGSEDI is available only through a restricted program called the TEGSEDI REMS Program [see Warnings and Precautions (5.3)]. Inform the patient of the following notable requirements:
- Patients must enroll in the program and comply with ongoing monitoring requirements.
- TEGSEDI is available only from certified pharmacies participating in the program. Therefore, provide patients with the telephone number and website for information on how to obtain the product.
Stroke and Cervicocephalic Arterial Dissection
Educate patient on symptoms of stroke and central nervous system arterial dissection and instruct them to seek help as soon as possible if symptoms of these or other serious neurologic adverse reactions occur [see Warnings and Precautions (5.4)].
Liver Injury
Instruct patients to inform a healthcare professional of symptoms suggestive of hepatic dysfunction that occur after administration of TEGSEDI [see Warnings and Precautions (5.6)].
Hypersensitivity
Instruct patients to inform a healthcare professional of symptoms suggestive of hypersensitivity that occur after administration of TEGSEDI [see Warnings and Precautions (5.7)].
Recommended Vitamin A Supplementation
Inform patients that TEGSEDI treatment leads to a decrease in vitamin A levels measured in the serum. Instruct patients to take the recommended daily allowance of vitamin A. Advise patients to contact their healthcare provider if they experience ocular symptoms suggestive of vitamin A deficiency (e.g., night blindness) and refer them to an ophthalmologist if they develop these symptoms [see Warnings and Precautions (5.9)].
Administration Instructions
Train patients and caregivers on proper subcutaneous administration technique and how to use the single-dose prefilled syringe. Instruct patients and/or caregivers to read and follow the Instructions for Use each time they use TEGSEDI.
Pregnancy
Instruct patients that if they are pregnant or plan to become pregnant while taking TEGSEDI they should inform their healthcare provider. Advise female patients of childbearing potential of the potential risk to the fetus. Encourage patients to enroll in the TEGSEDI Pregnancy Registry if they become pregnant while taking TEGSEDI [see Use in Specific Populations (8.1)].
For more information about TEGSEDI, go to www.TEGSEDIREMS.com or call 1-844-483-4736
Distributed by:
Sobi, Inc.
Waltham, MA 02451
TEGSEDI is a registered trademark of Akcea Therapeutics, Inc.
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Issued: 05/2021 | |
|
MEDICATION GUIDE
TEGSEDI (Teg-SED-ee)
|
||
|
What is the most important information I should know about TEGSEDI? TEGSEDI can cause serious side effects, including:
Your healthcare provider will do laboratory tests to check your platelet count and kidneys before you start TEGSEDI and while you are using it. Your healthcare provider should also do laboratory tests for 8 weeks after you stop TEGSEDI. It is important that you make sure you get these laboratory tests done.
|
||
|
What is TEGSEDI? TEGSEDI is a medicine used to treat the polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults. It is not known if TEGSEDI is safe and effective in children. |
||
|
Do not use TEGSEDI if you have:
|
||
|
Before you start using TEGSEDI, tell your healthcare provider about all your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your healthcare provider if you take:
|
||
|
How should I use TEGSEDI?
|
||
|
What are possible side effects of TEGSEDI? TEGSEDI may cause serious side effects, including:
Get emergency help immediately if you have symptoms of stroke.
|
||
|
|
|
The most common side effects of TEGSEDI include: injection site reactions (such as redness or pain at the injection site), nausea, headache, tiredness, low platelet counts (thrombocytopenia), and fever. These are not all the possible side effects of TEGSEDI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
|
How should I store TEGSEDI?
Keep TEGSEDI and all medicines out of the reach of children. |
||
|
General information about the safe and effective use of TEGSEDI Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TEGSEDI for a condition for which it has not been prescribed. Do not give TEGSEDI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TEGSEDI that was written for health professionals. |
||
|
What are the ingredients in TEGSEDI? Active ingredients: inotersen Inactive ingredients: purified water (water for injection), hydrochloric acid and or sodium hydroxide for pH adjustment |
||
|
Distributed by Sobi, Inc, Waltham, MA
|
||
| This Instructions for Use has been approved by the U.S. Food and Drug Administration | Issued:10/2018 |
|
TEGSEDI (Teg-SED-ee)
|
|
|
Introduction |
|
|
Before using your TEGSEDI prefilled syringe, your healthcare provider should show you or your caregiver how to use it the right way. If you or your caregiver have any questions, ask your healthcare provider. Read this Instructions for Use before you start using your TEGSEDI prefilled syringe and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment. |
|
|
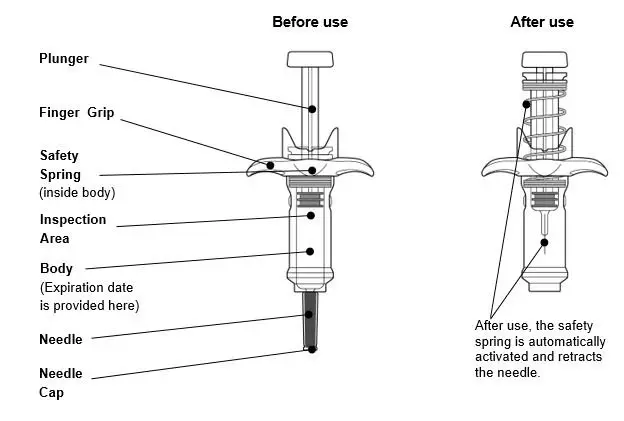
Guide to parts |
|
|
|
|
|
Important information |
|
Each TEGSEDI prefilled syringe contains 1 dose of TEGSEDI and is for 1-time use only. |
|
|
Warnings |
|
If any of the above happens, throw away the prefilled syringe in a puncture-resistant (sharps) container and use a new prefilled syringe. |
|
|
Preparation |
|
|
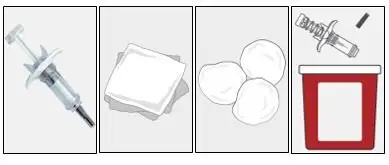
Step 1. Gather supplies |
|
|
|
Do not perform the injection without all the supplies listed. |
|
Step 2. Prepare to use your TEGSEDI prefilled syringe |
|
|
|
|
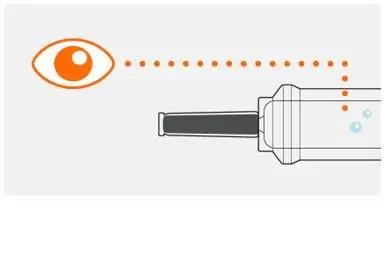
Step 3. Check medicine in the syringe |
|
|
|
|
|
Step 4. Choose the injection site |
|
|
|
|
|
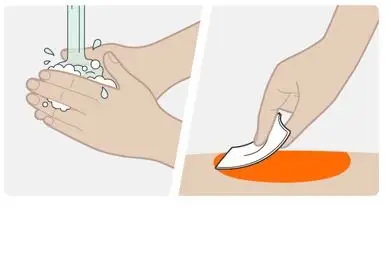
Step 5. Clean the injection site |
|
|
|
|
|
Injection |
|
|
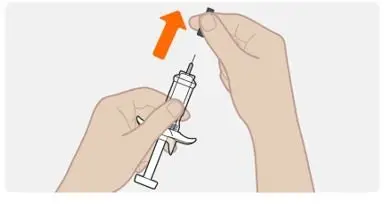
Step 6. Remove the needle cap |
|
|
|
Do not remove the needle cap until right before you inject. Do not pull the cap off while holding the syringe by the plunger. Always hold by the body of the syringe. Do not let the needle touch any surface. Do not remove any air bubbles from the syringe. Do not put the needle cap back onto the syringe. |
|
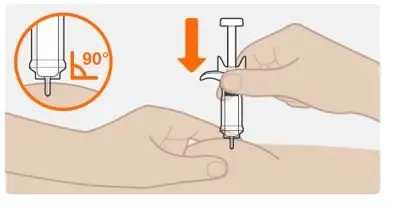
Step 7. Insert the needle |
|
|
|
Do not hold the syringe by the plunger or push against the plunger to insert the needle. |
|
Step 8. Start the injection |
|
|
|
Do not let go of the plunger. |
|
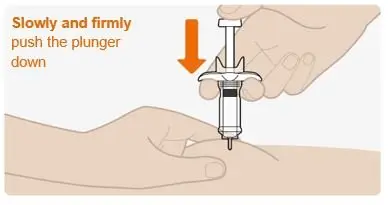
Step 9. Push the plunger down |
|
|
|
|
|
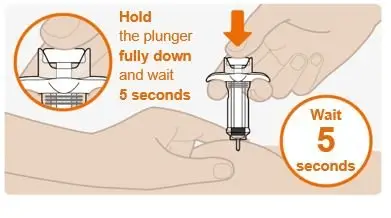
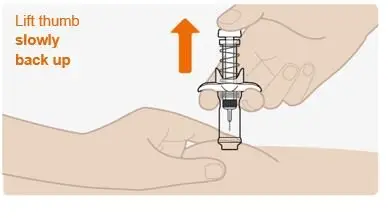
Step 10. Complete the injection |
|
|
|
Do not pull the plunger up by hand. Lift the whole syringe straight up. Do not try to replace the cap on the retracted needle. Do not rub the injection site. |
|
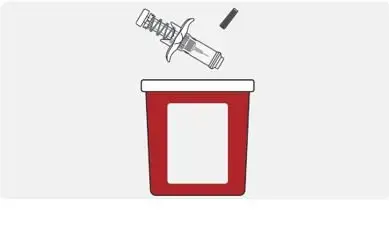
Disposal and care |
|
|
|
http://www.fda.gov/safesharpsdisposal |




| TEGSEDI
inotersen injection, solution |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Akcea Therapeutics, Inc. (079911419) |
