Drug Detail:Arimidex (Anastrozole [ an-as-troe-zole ])
Drug Class: Aromatase inhibitors Hormones / antineoplastics
Highlights of Prescribing Information
ARIMIDEX® (anastrozole) tablet for oral use
Initial U.S. Approval: 1995
Indications and Usage for Arimidex
ARIMIDEX is an aromatase inhibitor indicated for:
- •
- Adjuvant treatment of postmenopausal women with hormone receptor-positive early breast cancer (1.1)
- •
- First-line treatment of postmenopausal women with hormone receptor-positive or hormone receptor unknown locally advanced or metastatic breast cancer (1.2)
- •
- Treatment of advanced breast cancer in postmenopausal women with disease progression following tamoxifen therapy. Patients with ER-negative disease and patients who did not respond to previous tamoxifen therapy rarely responded to ARIMIDEX (1.3)
Arimidex Dosage and Administration
One 1 mg tablet taken once daily (2.1)
Dosage Forms and Strengths
1 mg tablets (3)
Contraindications
- •
- Women of premenopausal endocrine status, including pregnant women (4.1, 8.1)
- •
- Patients with demonstrated hypersensitivity to ARIMIDEX or any excipient (4.2)
Warnings and Precautions
- •
- In women with pre-existing ischemic heart disease, an increased incidence of ischemic cardiovascular events occurred with ARIMIDEX use compared to tamoxifen use. Consider risks and benefits. (5.1, 6.1)
- •
- Decreases in bone mineral density may occur. Consider bone mineral density monitoring. (5.2, 6.1)
- •
- Increases in total cholesterol may occur. Consider cholesterol monitoring. (5.3, 6.1)
Adverse Reactions/Side Effects
In the early breast cancer (ATAC) study, the most common (occurring with an incidence of ≥10%) side effects occurring in women taking ARIMIDEX included: hot flashes, asthenia, arthritis, pain, arthralgia, pharyngitis, hypertension, depression, nausea and vomiting, rash, osteoporosis, fractures, back pain, insomnia, headache, peripheral edema and lymphedema, regardless of causality. (6.1)
In the advanced breast cancer studies, the most common (occurring with an incidence of >10%) side effects occurring in women taking ARIMIDEX included: hot flashes, nausea, asthenia, pain, headache, back pain, bone pain, increased cough, dyspnea, pharyngitis and peripheral edema. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca Pharmaceuticals LP at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Drug Interactions
- •
- Tamoxifen: Do not use in combination with ARIMIDEX. No additional benefit seen over tamoxifen monotherapy. (7.1, 14.1)
- •
- Estrogen-containing products: Combination use may diminish activity of ARIMIDEX. (7.2)
Use In Specific Populations
- •
- Pediatric patients: Efficacy has not been demonstrated for pubertal boys of adolescent age with gynecomastia or girls with McCune-Albright Syndrome and progressive precocious puberty. (8.4)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2014
Full Prescribing Information
1. Indications and Usage for Arimidex
1.1 Adjuvant Treatment
ARIMIDEX is indicated for adjuvant treatment of postmenopausal women with hormone receptor-positive early breast cancer.
2. Arimidex Dosage and Administration
2.1 Recommended Dose
The dose of ARIMIDEX is one 1 mg tablet taken once a day. For patients with advanced breast cancer, ARIMIDEX should be continued until tumor progression. ARIMIDEX can be taken with or without food.
For adjuvant treatment of early breast cancer in postmenopausal women, the optimal duration of therapy is unknown. In the ATAC trial, ARIMIDEX was administered for five years [see Clinical Studies (14.1)].
No dosage adjustment is necessary for patients with renal impairment or for elderly patients [see Use in Specific Populations (8.6)].
3. Dosage Forms and Strengths
The tablets are white, biconvex, film-coated containing 1 mg of anastrozole. The tablets are impressed on one side with a logo consisting of a letter “A” (upper case) with an arrowhead attached to the foot of the extended right leg of the “A” and on the reverse with the tablet strength marking “Adx 1”.
4. Contraindications
4.1 Pregnancy and Premenopausal Women
ARIMIDEX may cause fetal harm when administered to a pregnant woman and offers no clinical benefit to premenopausal women with breast cancer. ARIMIDEX is contraindicated in women who are or may become pregnant. There are no adequate and well-controlled studies in pregnant women using ARIMIDEX. If ARIMIDEX is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus or potential risk for loss of the pregnancy [see Use in Specific Populations (8.1)].
5. Warnings and Precautions
5.1 Ischemic Cardiovascular Events
In women with pre-existing ischemic heart disease, an increased incidence of ischemic cardiovascular events was observed with ARIMIDEX in the ATAC trial (17% of patients on ARIMIDEX and 10% of patients on tamoxifen). Consider risk and benefits of ARIMIDEX therapy in patients with pre-existing ischemic heart disease [see Adverse Reactions (6.1)].
5.2 Bone Effects
Results from the ATAC trial bone substudy at 12 and 24 months demonstrated that patients receiving ARIMIDEX had a mean decrease in both lumbar spine and total hip bone mineral density (BMD) compared to baseline. Patients receiving tamoxifen had a mean increase in both lumbar spine and total hip BMD compared to baseline. Consider bone mineral density monitoring in patients treated with ARIMIDEX [see Adverse Reactions (6.1)].
6. Adverse Reactions/Side Effects
Serious adverse reactions with ARIMIDEX occurring in less than 1 in 10,000 patients, are: 1) skin reactions such as lesions, ulcers, or blisters; 2) allergic reactions with swelling of the face, lips, tongue, and/or throat. This may cause difficulty in swallowing and/or breathing; and 3) changes in blood tests of the liver function, including inflammation of the liver with symptoms that may include a general feeling of not being well, with or without jaundice, liver pain or liver swelling [see Adverse Reactions (6.2)].
Common adverse reactions (occurring with an incidence of ≥10%) in women taking ARIMIDEX included: hot flashes, asthenia, arthritis, pain, arthralgia, hypertension, depression, nausea and vomiting, rash, osteoporosis, fractures, back pain, insomnia, headache, bone pain, peripheral edema, increased cough, dyspnea, pharyngitis and lymphedema.
In the ATAC trial, the most common reported adverse reaction (>0.1%) leading to discontinuation of therapy for both treatment groups was hot flashes, although there were fewer patients who discontinued therapy as a result of hot flashes in the ARIMIDEX group.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience
Adjuvant Therapy
Adverse reaction data for adjuvant therapy are based on the ATAC trial [seeClinical Studies (14.1)]. The median duration of adjuvant treatment for safety evaluation was 59.8 months and 59.6 months for patients receiving ARIMIDEX 1 mg and tamoxifen 20 mg, respectively.
Adverse reactions occurring with an incidence of at least 5% in either treatment group during treatment or within 14 days of the end of treatment are presented in Table 1.
| Body system and adverse reactions by
COSTART†‡ | ARIMIDEX 1 mg
(N§ = 3092) | Tamoxifen 20 mg
(N§ = 3094) |
|---|---|---|
|
||
|
Body as a whole | ||
|
575 (19) |
544 (18) |
|
Pain |
533 (17) |
485 (16) |
|
Back pain |
321 (10) |
309 (10) |
|
Headache |
314 (10) |
249 (8) |
|
Abdominal pain |
271 (9) |
276 (9) |
|
Infection |
285 (9) |
276 (9) |
|
Accidental injury |
311 (10) |
303 (10) |
|
Flu syndrome |
175 (6) |
195 (6) |
|
Chest pain |
200 (7) |
150 (5) |
|
Neoplasm |
162 (5) |
144 (5) |
|
Cyst |
138 (5) |
162 (5) |
|
Cardiovascular | ||
|
Vasodilatation |
1104 (36) |
1264 (41) |
|
Hypertension |
402 (13) |
349 (11) |
|
Digestive | ||
|
Nausea |
343 (11) |
335 (11) |
|
Constipation |
249 (8) |
252 (8) |
|
Diarrhea |
265 (9) |
216 (7) |
|
Dyspepsia |
206 (7) |
169 (6) |
|
Gastrointestinal disorder |
210 (7) |
158 (5) |
|
Hemic and lymphatic | ||
|
Lymphedema |
304 (10) |
341 (11) |
|
Anemia |
113 (4) |
159 (5) |
|
Metabolic and nutritional | ||
|
311 (10) |
343 (11) |
|
Weight gain |
285 (9) |
274 (9) |
|
Hypercholesterolemia |
278 (9) |
108 (3.5) |
|
Musculoskeletal | ||
|
Arthritis |
512 (17) |
445 (14) |
|
Arthralgia |
467 (15) |
344 (11) |
|
Osteoporosis |
325 (11) |
226 (7) |
|
Fracture |
315 (10) |
209 (7) |
|
Bone pain |
201 (7) |
185 (6) |
|
Arthrosis |
207 (7) |
156 (5) |
|
Joint Disorder |
184 (6) |
160 (5) |
|
Myalgia |
179 (6) |
160 (5) |
|
Nervous system | ||
|
Depression |
413 (13) |
382 (12) |
|
Insomnia |
309 (10) |
281 (9) |
|
Dizziness |
236 (8) |
234 (8) |
|
Anxiety |
195 (6) |
180 (6) |
|
Paresthesia |
215 (7) |
145 (5) |
|
Respiratory | ||
|
Pharyngitis |
443 (14) |
422 (14) |
|
Cough increased |
261 (8) |
287 (9) |
|
Dyspnea |
234 (8) |
237 (8) |
|
Sinusitis |
184 (6) |
159 (5) |
|
Bronchitis |
167 (5) |
153 (5) |
|
Skin and appendages | ||
|
Rash |
333 (11) |
387 (13) |
|
Sweating |
145 (5) |
177 (6) |
|
Special Senses | ||
|
Cataract Specified |
182 (6) |
213 (7) |
|
Urogenital | ||
|
Leukorrhea |
86 (3) |
286 (9) |
|
Urinary tract infection |
244 (8) |
313 (10) |
|
Breast pain |
251 (8) |
169 (6) |
|
Breast Neoplasm |
164 (5) |
139 (5) |
|
Vulvovaginitis |
194 (6) |
150 (5) |
|
Vaginal Hemorrhage¶ |
122 (4) |
180 (6) |
|
Vaginitis |
125 (4) |
158 (5) |
Certain adverse reactions and combinations of adverse reactions were prospectively specified for analysis, based on the known pharmacologic properties and side effect profiles of the two drugs (see Table 2).
| ARIMIDEX
N=3092 (%) | Tamoxifen
N=3094 (%) | Odds-ratio | 95% CI | |
|---|---|---|---|---|
|
||||
|
Hot Flashes |
1104 (36) |
1264 (41) |
0.80 |
0.73 − 0.89 |
|
Musculoskeletal Events† |
1100 (36) |
911 (29) |
1.32 |
1.19 − 1.47 |
|
Fatigue/Asthenia |
575 (19) |
544 (18) |
1.07 |
0.94 − 1.22 |
|
Mood Disturbances |
597 (19) |
554 (18) |
1.10 |
0.97 − 1.25 |
|
Nausea and Vomiting |
393 (13) |
384 (12) |
1.03 |
0.88 − 1.19 |
|
All Fractures |
315 (10) |
209 (7) |
1.57 |
1.30 − 1.88 |
|
Fractures of Spine, Hip, or Wrist |
133 (4) |
91 (3) |
1.48 |
1.13 − 1.95 |
|
Wrist/Colles’ fractures |
67 (2) |
50 (2) | ||
|
Spine fractures |
43 (1) |
22 (1) | ||
|
Hip fractures |
28 (1) |
26 (1) | ||
|
Cataracts |
182 (6) |
213 (7) |
0.85 |
0.69 − 1.04 |
|
Vaginal Bleeding |
167 (5) |
317 (10) |
0.50 |
0.41 − 0.61 |
|
Ischemic Cardiovascular Disease |
127 (4) |
104 (3) |
1.23 |
0.95 − 1.60 |
|
Vaginal Discharge |
109 (4) |
408 (13) |
0.24 |
0.19 − 0.30 |
|
Venous Thromboembolic events |
87 (3) |
140 (5) |
0.61 |
0.47 − 0.80 |
|
Deep Venous Thromboembolic Events |
48 (2) |
74 (2) |
0.64 |
0.45 − 0.93 |
|
Ischemic Cerebrovascular Event |
62 (2) |
88 (3) |
0.70 |
0.50 − 0.97 |
|
Endometrial Cancer‡ |
4 (0.2) |
13 (0.6) |
0.31 |
0.10 − 0.94 |
Ischemic Cardiovascular Events
Between treatment arms in the overall population of 6186 patients, there was no statistical difference in ischemic cardiovascular events (4% ARIMIDEX vs. 3% tamoxifen). In the overall population, angina pectoris was reported in 71/3092 (2.3%) patients in the ARIMIDEX arm and 51/3094 (1.6%) patients in the tamoxifen arm; myocardial infarction was reported in 37/3092 (1.2%) patients in the ARIMIDEX arm and 34/3094 (1.1%) patients in the tamoxifen arm.
In women with pre-existing ischemic heart disease 465/6186 (7.5%), the incidence of ischemic cardiovascular events was 17% in patients on ARIMIDEX and 10% in patients on tamoxifen. In this patient population, angina pectoris was reported in 25/216 (11.6%) patients receiving ARIMIDEX and 13/249 (5.2%) patients receiving tamoxifen; myocardial infarction was reported in 2/216 (0.9%) patients receiving ARIMIDEX and 8/249 (3.2%) patients receiving tamoxifen.
Bone Mineral Density Findings
Results from the ATAC trial bone substudy at 12 and 24 months demonstrated that patients receiving ARIMIDEX had a mean decrease in both lumbar spine and total hip bone mineral density (BMD) compared to baseline. Patients receiving tamoxifen had a mean increase in both lumbar spine and total hip BMD compared to baseline.
Because ARIMIDEX lowers circulating estrogen levels it may cause a reduction in bone mineral density.
A post-marketing trial assessed the combined effects of ARIMIDEX and the bisphosphonate risedronate on changes from baseline in BMD and markers of bone resorption and formation in postmenopausal women with hormone receptor-positive early breast cancer. All patients received calcium and vitamin D supplementation. At 12 months, small reductions in lumbar spine bone mineral density were noted in patients not receiving bisphosphonates. Bisphosphonate treatment preserved bone density in most patients at risk of fracture.
Postmenopausal women with early breast cancer scheduled to be treated with ARIMIDEX should have their bone status managed according to treatment guidelines already available for postmenopausal women at similar risk of fragility fracture.
Cholesterol
During the ATAC trial, more patients receiving ARIMIDEX were reported to have an elevated serum cholesterol compared to patients receiving tamoxifen (9% versus 3.5%, respectively).
A post-marketing trial also evaluated any potential effects of ARIMIDEX on lipid profile. In the primary analysis population for lipids (ARIMIDEX alone), there was no clinically significant change in LDL-C from baseline to 12 months and HDL-C from baseline to 12 months.
In secondary population for lipids (ARIMIDEX+risedronate), there also was no clinically significant change in LDL-C and HDL-C from baseline to 12 months.
In both populations for lipids, there was no clinically significant difference in total cholesterol (TC) or serum triglycerides (TG) at 12 months compared with baseline.
In this trial, treatment for 12 months with ARIMIDEX alone had a neutral effect on lipid profile. Combination treatment with ARIMIDEX and risedronate also had a neutral effect on lipid profile.
The trial provides evidence that postmenopausal women with early breast cancer scheduled to be treated with ARIMIDEX should be managed using the current National Cholesterol Education Program guidelines for cardiovascular risk-based management of individual patients with LDL elevations.
Other Adverse Reactions
Patients receiving ARIMIDEX had an increase in joint disorders (including arthritis, arthrosis and arthralgia) compared with patients receiving tamoxifen. Patients receiving ARIMIDEX had an increase in the incidence of all fractures (specifically fractures of spine, hip and wrist) [315 (10%)] compared with patients receiving tamoxifen [209 (7%)].
Patients receiving ARIMIDEX had a higher incidence of carpal tunnel syndrome [78 (2.5%)] compared with patients receiving tamoxifen [22 (0.7%)].
Vaginal bleeding occurred more frequently in the tamoxifen-treated patients versus the ARIMIDEX-treated patients 317 (10%) versus 167 (5%), respectively.
Patients receiving ARIMIDEX had a lower incidence of hot flashes, vaginal bleeding, vaginal discharge, endometrial cancer, venous thromboembolic events and ischemic cerebrovascular events compared with patients receiving tamoxifen.
10-year median follow-up Safety Results from the ATAC Trial
Results are consistent with the previous analyses.
Serious adverse reactions were similar between ARIMIDEX (50%) and tamoxifen (51%).
- •
- Cardiovascular events were consistent with the known safety profiles of ARIMIDEX and tamoxifen.
- •
- The cumulative incidences of all first fractures (both serious and non-serious, occurring either during or after treatment) was higher in the ARIMIDEX group (15%) compared to the tamoxifen group (11%). This increased first fracture rate during treatment did not continue in the post-treatment follow-up period.
- •
- The cumulative incidence of new primary cancers was similar in the ARIMIDEX group (13.7%) compared to the tamoxifen group (13.9%). Consistent with the previous analyses, endometrial cancer was higher in the tamoxifen group (0.8%) compared to the ARIMIDEX group (0.2%).
- •
- The overall number of deaths (during or off-trial treatment) was similar between the treatment groups. There were more deaths related to breast cancer in the tamoxifen than in the ARIMIDEX treatment group.
First-Line Therapy
Adverse reactions occurring with an incidence of at least 5% in either treatment group of trials 0030 and 0027 during or within 2 weeks of the end of treatment are shown in Table 3.
| Body system
Adverse Reaction* | Number (%) of subjects | |
|---|---|---|
| ARIMIDEX
(N=506) | Tamoxifen
(N=511) |
|
|
||
|
Whole body | ||
|
Asthenia |
83 (16) |
81 (16) |
|
Pain |
70 (14) |
73 (14) |
|
Back pain |
60 (12) |
68 (13) |
|
Headache |
47 (9) |
40 (8) |
|
Abdominal pain |
40 (8) |
38 (7) |
|
Chest pain |
37 (7) |
37 (7) |
|
Flu syndrome |
35 (7) |
30 (6) |
|
Pelvic pain |
23 (5) |
30 (6) |
|
Cardiovascular | ||
|
Vasodilation |
128 (25) |
106 (21) |
|
Hypertension |
25 (5) |
36 (7) |
|
Digestive | ||
|
Nausea |
94 (19) |
106 (21) |
|
Constipation |
47 (9) |
66 (13) |
|
Diarrhea |
40 (8) |
33 (6) |
|
Vomiting |
38 (8) |
36 (7) |
|
Anorexia |
26 (5) |
46 (9) |
|
Metabolic and Nutritional | ||
|
Peripheral edema |
51 (10) |
41 (8) |
|
Musculoskeletal | ||
|
Bone pain |
54 (11) |
52 (10) |
|
Nervous | ||
|
Dizziness |
30 (6) |
22 (4) |
|
Insomnia |
30 (6) |
38 (7) |
|
Depression |
23 (5) |
32 (6) |
|
Hypertonia |
16 (3) |
26 (5) |
|
Respiratory | ||
|
Cough increased |
55 (11) |
52 (10) |
|
Dyspnea |
51 (10) |
47 (9) |
|
Pharyngitis |
49 (10) |
68 (13) |
|
Skin and appendages | ||
|
Rash |
38 (8) |
34 (8) |
|
Urogenital | ||
|
Leukorrhea |
9 (2) |
31 (6) |
Less frequent adverse experiences reported in patients receiving ARIMIDEX 1 mg in either Trial 0030 or Trial 0027 were similar to those reported for second-line therapy.
Based on results from second-line therapy and the established safety profile of tamoxifen, the incidences of 9 pre-specified adverse event categories potentially causally related to one or both of the therapies because of their pharmacology were statistically analyzed. No significant differences were seen between treatment groups.
| Number (n) and Percentage of Patients | ||
|---|---|---|
| Adverse Reaction* | ARIMIDEX
1 mg (N=506) n (%) | NOLVADEX
20 mg (N=511) n (%) |
|
||
|
Depression |
23 (5) |
32 (6) |
|
Tumor Flare |
15 (3) |
18 (4) |
|
Thromboembolic Disease† |
18 (4) |
33 (6) |
|
Venous† |
5 |
15 |
|
Coronary and Cerebral‡ |
13 |
19 |
|
Gastrointestinal Disturbance |
170 (34) |
196 (38) |
|
Hot Flushes |
134 (26) |
118 (23) |
|
Vaginal Dryness |
9 (2) |
3 (1) |
|
Lethargy |
6 (1) |
15 (3) |
|
Vaginal Bleeding |
5 (1) |
11 (2) |
|
Weight Gain |
11 (2) |
8 (2) |
Second-Line Therapy
ARIMIDEX was tolerated in two controlled clinical trials (i.e., Trials 0004 and 0005), with less than 3.3% of the ARIMIDEX-treated patients and 4.0% of the megestrol acetate-treated patients withdrawing due to an adverse reaction.
The principal adverse reaction more common with ARIMIDEX than megestrol acetate was diarrhea. Adverse reactions reported in greater than 5% of the patients in any of the treatment groups in these two controlled clinical trials, regardless of causality, are presented below:
| Adverse Reaction* | ARIMIDEX | ARIMIDEX | Megestrol Acetate | |||
|---|---|---|---|---|---|---|
| 1 mg | 10 mg | 160 mg | ||||
| (N=262) | (N=246) | (N=253) | ||||
| n | % | n | % | n | % | |
|
||||||
|
Asthenia |
42 |
(16) |
33 |
(13) |
47 |
(19) |
|
Nausea |
41 |
(16) |
48 |
(20) |
28 |
(11) |
|
Headache |
34 |
(13) |
44 |
(18) |
24 |
(9) |
|
Hot Flashes |
32 |
(12) |
29 |
(11) |
21 |
(8) |
|
Pain |
28 |
(11) |
38 |
(15) |
29 |
(11) |
|
Back Pain |
28 |
(11) |
26 |
(11) |
19 |
(8) |
|
Dyspnea |
24 |
(9) |
27 |
(11) |
53 |
(21) |
|
Vomiting |
24 |
(9) |
26 |
(11) |
16 |
(6) |
|
Cough Increased |
22 |
(8) |
18 |
(7) |
19 |
(8) |
|
Diarrhea |
22 |
(8) |
18 |
(7) |
7 |
(3) |
|
Constipation |
18 |
(7) |
18 |
(7) |
21 |
(8) |
|
Abdominal Pain |
18 |
(7) |
14 |
(6) |
18 |
(7) |
|
Anorexia |
18 |
(7) |
19 |
(8) |
11 |
(4) |
|
Bone Pain |
17 |
(6) |
26 |
(12) |
19 |
(8) |
|
Pharyngitis |
16 |
(6) |
23 |
(9) |
15 |
(6) |
|
Dizziness |
16 |
(6) |
12 |
(5) |
15 |
(6) |
|
Rash |
15 |
(6) |
15 |
(6) |
19 |
(8) |
|
Dry Mouth |
15 |
(6) |
11 |
(4) |
13 |
(5) |
|
Peripheral Edema |
14 |
(5) |
21 |
(9) |
28 |
(11) |
|
Pelvic Pain |
14 |
(5) |
17 |
(7) |
13 |
(5) |
|
Depression |
14 |
(5) |
6 |
(2) |
5 |
(2) |
|
Chest Pain |
13 |
(5) |
18 |
(7) |
13 |
(5) |
|
Paresthesia |
12 |
(5) |
15 |
(6) |
9 |
(4) |
|
Vaginal Hemorrhage |
6 |
(2) |
4 |
(2) |
13 |
(5) |
|
Weight Gain |
4 |
(2) |
9 |
(4) |
30 |
(12) |
|
Sweating |
4 |
(2) |
3 |
(1) |
16 |
(6) |
|
Increased Appetite |
0 |
(0) |
1 |
(0) |
13 |
(5) |
Other less frequent (2% to 5%) adverse reactions reported in patients receiving ARIMIDEX 1 mg in either Trial 0004 or Trial 0005 are listed below. These adverse experiences are listed by body system and are in order of decreasing frequency within each body system regardless of assessed causality.
Body as a Whole: Flu syndrome; fever; neck pain; malaise; accidental injury; infection
Cardiovascular: Hypertension; thrombophlebitis
Hepatic: Gamma GT increased; SGOT increased; SGPT increased
Hematologic: Anemia; leukopenia
Metabolic and Nutritional: Alkaline phosphatase increased; weight loss
Mean serum total cholesterol levels increased by 0.5 mmol/L among patients receiving ARIMIDEX. Increases in LDL cholesterol have been shown to contribute to these changes.
Musculoskeletal: Myalgia; arthralgia; pathological fracture
Nervous: Somnolence; confusion; insomnia; anxiety; nervousness
Respiratory: Sinusitis; bronchitis; rhinitis
Skin and Appendages: Hair thinning (alopecia); pruritus
Urogenital: Urinary tract infection; breast pain
The incidences of the following adverse reaction groups potentially causally related to one or both of the therapies because of their pharmacology, were statistically analyzed: weight gain, edema, thromboembolic disease, gastrointestinal disturbance, hot flushes, and vaginal dryness. These six groups, and the adverse reactions captured in the groups, were prospectively defined. The results are shown in the table below.
| ARIMIDEX | ARIMIDEX | Megestrol Acetate | ||||
|---|---|---|---|---|---|---|
| 1 mg | 10 mg | 160 mg | ||||
| (N=262) | (N=246) | (N=253) | ||||
| Adverse Reaction Group | n | (%) | n | (%) | n | (%) |
|
Gastrointestinal Disturbance |
77 |
(29) |
81 |
(33) |
54 |
(21) |
|
Hot Flushes |
33 |
(13) |
29 |
(12) |
35 |
(14) |
|
Edema |
19 |
(7) |
28 |
(11) |
35 |
(14) |
|
Thromboembolic Disease |
9 |
(3) |
4 |
(2) |
12 |
(5) |
|
Vaginal Dryness |
5 |
(2) |
3 |
(1) |
2 |
(1) |
|
Weight Gain |
4 |
(2) |
10 |
(4) |
30 |
(12) |
6.2 Post-Marketing Experience
These adverse reactions are reported voluntarily from a population of uncertain size. Therefore, it is not always possible to estimate reliably their frequency or establish a causal relationship to drug exposure. The following have been reported in post-approval use of Arimidex:
- •
- Hepatobiliary events including increases in alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, gamma-GT, and bilirubin; hepatitis
- •
- Rash including cases of mucocutaneous disorders such as erythema multiforme and Stevens-Johnson syndrome
- •
- Cases of allergic reactions including angioedema, urticaria and anaphylaxis [see Contraindications (4.2)]
- •
- Myalgia, trigger finger and hypercalcemia (with or without an increase in parathyroid hormone)
7. Drug Interactions
7.1 Tamoxifen
Co-administration of anastrozole and tamoxifen in breast cancer patients reduced anastrozole plasma concentration by 27%. However, the co-administration of anastrozole and tamoxifen did not affect the pharmacokinetics of tamoxifen or N-desmethyltamoxifen. At a median follow-up of 33 months, the combination of ARIMIDEX and tamoxifen did not demonstrate any efficacy benefit when compared with tamoxifen in all patients as well as in the hormone receptor-positive subpopulation. This treatment arm was discontinued from the trial [see Clinical Studies (14.1)]. Based on clinical and pharmacokinetic results from the ATAC trial, tamoxifen should not be administered with anastrozole.
7.2 Estrogen
Estrogen-containing therapies should not be used with ARIMIDEX as they may diminish its pharmacological action.
8. Use In Specific Populations
8.1 Pregnancy
PREGNANCY CATEGORY X [see Contraindications (4.1)]
ARIMIDEX may cause fetal harm when administered to a pregnant woman and offers no clinical benefit to premenopausal women with breast cancer. ARIMIDEX is contraindicated in women who are or may become pregnant. In animal studies, anastrozole caused pregnancy failure, increased pregnancy loss, and signs of delayed fetal development. There are no studies of ARIMIDEX use in pregnant women. If ARIMIDEX is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus and potential risk for pregnancy loss.
In animal reproduction studies, pregnant rats and rabbits received anastrozole during organogenesis at doses equal to or greater than 1 (rats) and 1/3 (rabbits) the recommended human dose on a mg/m2 basis. In both species, anastrozole crossed the placenta, and there was increased pregnancy loss (increased pre- and/or post-implantation loss, increased resorption, and decreased numbers of live fetuses). In rats, these effects were dose related, and placental weights were significantly increased. Fetotoxicity, including delayed fetal development (i.e., incomplete ossification and depressed fetal body weights), occurred in rats at anastrozole doses that produced peak plasma levels 19 times higher than serum levels in humans at the therapeutic dose (AUC0-24hr 9 times higher). In rabbits, anastrozole caused pregnancy failure at doses equal to or greater than 16 times the recommended human dose on a mg/m2 basis [see Animal Toxicology and/or Pharmacology (13.2)].
8.3 Nursing Mothers
It is not known if anastrozole is excreted in human milk. Because many drugs are excreted in human milk and because of the tumorigenicity shown for anastrozole in animal studies, or the potential for serious adverse reactions in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
Clinical studies in pediatric patients included a placebo-controlled trial in pubertal boys of adolescent age with gynecomastia and a single-arm trial in girls with McCune-Albright Syndrome and progressive precocious puberty. The efficacy of ARIMIDEX in the treatment of pubertal gynecomastia in adolescent boys and in the treatment of precocious puberty in girls with McCune-Albright Syndrome has not been demonstrated.
Gynecomastia Study
A randomized, double-blind, placebo-controlled, multi-center study enrolled 80 boys with pubertal gynecomastia aged 11 to 18 years. Patients were randomized to a daily regimen of either ARIMIDEX 1 mg or placebo. After 6 months of treatment there was no statistically significant difference in the percentage of patients who experienced a ≥50% reduction in gynecomastia (primary efficacy analysis). Secondary efficacy analyses (absolute change in breast volume, the percentage of patients who had any reduction in the calculated volume of gynecomastia, breast pain resolution) were consistent with the primary efficacy analysis. Serum estradiol concentrations at Month 6 of treatment were reduced by 15.4% in the ARIMIDEX group and 4.5% in the placebo group.
Adverse reactions that were assessed as treatment-related by the investigators occurred in 16.3% of the ARIMIDEX-treated patients and 8.1% of the placebo-treated patients with the most frequent being acne (7% ARIMIDEX and 2.7% placebo) and headache (7% ARIMIDEX and 0% placebo); all other adverse reactions showed small differences between treatment groups. One patient treated with ARIMIDEX discontinued the trial because of testicular enlargement. The mean baseline-subtracted change in testicular volume after 6 months of treatment was + 6.6 ± 7.9 cm3 in the ARIMIDEX-treated patients and + 5.2 ± 8.0 cm3 in the placebo group.
McCune-Albright Syndrome Study
A multi-center, single-arm, open-label study was conducted in 28 girls with McCune-Albright Syndrome and progressive precocious puberty aged 2 to <10 years. All patients received a 1 mg daily dose of ARIMIDEX. The trial duration was 12 months. Patients were enrolled on the basis of a diagnosis of typical (27/28) or atypical (1/27) McCune-Albright Syndrome, precocious puberty, history of vaginal bleeding, and/or advanced bone age. Patients’ baseline characteristics included the following: a mean chronological age of 5.9 ± 2.0 years, a mean bone age of 8.6 ± 2.6 years, a mean growth rate of 7.9 ± 2.9 cm/year and a mean Tanner stage for breast of 2.7 ± 0.81. Compared to pre-treatment data there were no on-treatment statistically significant reductions in the frequency of vaginal bleeding days, or in the rate of increase of bone age (defined as a ratio between the change in bone age over the change of chronological age). There were no clinically significant changes in Tanner staging, mean ovarian volume, mean uterine volume and mean predicted adult height. A small but statistically significant reduction of growth rate from 7.9 ± 2.9 cm/year to 6.5 ± 2.8 cm/year was observed but the absence of a control group precludes attribution of this effect to treatment or to other confounding factors such as variations in endogenous estrogen levels commonly seen in McCune-Albright Syndrome patients.
Five patients (18%) experienced adverse reactions that were considered possibly related to ARIMIDEX. These were nausea, acne, pain in an extremity, increased alanine transaminase and aspartate transaminase, and allergic dermatitis.
Pharmacokinetics in Pediatric Patients
Following 1 mg once daily multiple administration in pediatric patients, the mean time to reach the maximum anastrozole concentration was 1 hr. The mean (range) disposition parameters of anastrozole in pediatric patients were described by a CL/F of 1.54 L/h (0.77-4.53 L/h) and V/F of 98.4 L (50.7-330.0 L). The terminal elimination half-life was 46.8 h, which was similar to that observed in postmenopausal women treated with anastrozole for breast cancer. Based on a population pharmacokinetic analysis, the pharmacokinetics of anastrozole was similar in boys with pubertal gynecomastia and girls with McCune-Albright Syndrome.
8.5 Geriatric Use
In studies 0030 and 0027, about 50% of patients were 65 or older. Patients ≥ 65 years of age had moderately better tumor response and time to tumor progression than patients < 65 years of age regardless of randomized treatment. In studies 0004 and 0005, 50% of patients were 65 or older. Response rates and time to progression were similar for the over 65 and younger patients.
In the ATAC study, 45% of patients were 65 years of age or older. The efficacy of ARIMIDEX compared to tamoxifen in patients who were 65 years or older (N=1413 for ARIMIDEX and N=1410 for tamoxifen, the hazard ratio for disease-free survival was 0.93 [95% CI: 0.80, 1.08]) was less than efficacy observed in patients who were less than 65 years of age (N=1712 for ARIMIDEX and N=1706 for tamoxifen, the hazard ratio for disease-free survival was 0.79 [95% CI: 0.67, 0.94]).
The pharmacokinetics of anastrozole are not affected by age.
8.6 Renal Impairment
Since only about 10% of anastrozole is excreted unchanged in the urine, the renal impairment does not influence the total body clearance. Dosage adjustment in patients with renal impairment is not necessary [seeDosage and Administration (2.1)andClinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The plasma anastrozole concentrations in the subjects with hepatic cirrhosis were within the range of concentrations seen in normal subjects across all clinical trials. Therefore, dosage adjustment is also not necessary in patients with stable hepatic cirrhosis. ARIMIDEX has not been studied in patients with severe hepatic impairment [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
10. Overdosage
Clinical trials have been conducted with ARIMIDEX, up to 60 mg in a single dose given to healthy male volunteers and up to 10 mg daily given to postmenopausal women with advanced breast cancer; these dosages were tolerated. A single dose of ARIMIDEX that results in life-threatening symptoms has not been established. There is no specific antidote to overdosage and treatment must be symptomatic. In the management of an overdose, consider that multiple agents may have been taken. Vomiting may be induced if the patient is alert. Dialysis may be helpful because ARIMIDEX is not highly protein bound. General supportive care, including frequent monitoring of vital signs and close observation of the patient, is indicated.
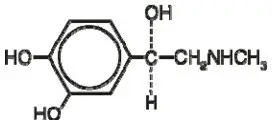
11. Arimidex Description
ARIMIDEX (anastrozole) tablets for oral administration contain 1 mg of anastrozole, a non-steroidal aromatase inhibitor. It is chemically described as 1,3-Benzenediacetonitrile, a, a, a', a'-tetramethyl-5-(1H-1,2,4-triazol-1-ylmethyl). Its molecular formula is C17H19N5 and its structural formula is:
Anastrozole is an off-white powder with a molecular weight of 293.4. Anastrozole has moderate aqueous solubility (0.5 mg/mL at 25°C); solubility is independent of pH in the physiological range. Anastrozole is freely soluble in methanol, acetone, ethanol, and tetrahydrofuran, and very soluble in acetonitrile.
Each tablet contains as inactive ingredients: lactose, magnesium stearate, hydroxypropylmethylcellulose, polyethylene glycol, povidone, sodium starch glycolate, and titanium dioxide.
12. Arimidex - Clinical Pharmacology
12.1 Mechanism of Action
The growth of many cancers of the breast is stimulated or maintained by estrogens.
In postmenopausal women, estrogens are mainly derived from the action of the aromatase enzyme, which converts adrenal androgens (primarily androstenedione and testosterone) to estrone and estradiol. The suppression of estrogen biosynthesis in peripheral tissues and in the cancer tissue itself can therefore be achieved by specifically inhibiting the aromatase enzyme.
Anastrozole is a selective non-steroidal aromatase inhibitor. It significantly lowers serum estradiol concentrations and has no detectable effect on formation of adrenal corticosteroids or aldosterone.
12.2 Pharmacodynamics
Effect on Estradiol
Mean serum concentrations of estradiol were evaluated in multiple daily dosing trials with 0.5, 1, 3, 5, and 10 mg of ARIMIDEX in postmenopausal women with advanced breast cancer. Clinically significant suppression of serum estradiol was seen with all doses. Doses of 1 mg and higher resulted in suppression of mean serum concentrations of estradiol to the lower limit of detection (3.7 pmol/L). The recommended daily dose, ARIMIDEX 1 mg, reduced estradiol by approximately 70% within 24 hours and by approximately 80% after 14 days of daily dosing. Suppression of serum estradiol was maintained for up to 6 days after cessation of daily dosing with ARIMIDEX 1 mg.
The effect of ARIMIDEX in premenopausal women with early or advanced breast cancer has not been studied. Because aromatization of adrenal androgens is not a significant source of estradiol in premenopausal women, ARIMIDEX would not be expected to lower estradiol levels in premenopausal women.
Effect on Corticosteroids
In multiple daily dosing trials with 3, 5, and 10 mg, the selectivity of anastrozole was assessed by examining effects on corticosteroid synthesis. For all doses, anastrozole did not affect cortisol or aldosterone secretion at baseline or in response to ACTH. No glucocorticoid or mineralocorticoid replacement therapy is necessary with anastrozole.
Other Endocrine Effects
In multiple daily dosing trials with 5 and 10 mg, thyroid stimulating hormone (TSH) was measured; there was no increase in TSH during the administration of ARIMIDEX. ARIMIDEX does not possess direct progestogenic, androgenic, or estrogenic activity in animals, but does perturb the circulating levels of progesterone, androgens, and estrogens.
12.3 Pharmacokinetics
Absorption
Inhibition of aromatase activity is primarily due to anastrozole, the parent drug. Absorption of anastrozole is rapid and maximum plasma concentrations typically occur within 2 hours of dosing under fasted conditions. Studies with radiolabeled drug have demonstrated that orally administered anastrozole is well absorbed into the systemic circulation. Food reduces the rate but not the overall extent of anastrozole absorption. The mean Cmax of anastrozole decreased by 16% and the median Tmax was delayed from 2 to 5 hours when anastrozole was administered 30 minutes after food. The pharmacokinetics of anastrozole are linear over the dose range of 1 to 20 mg, and do not change with repeated dosing. The pharmacokinetics of anastrozole were similar in patients and healthy volunteers.
Distribution
Steady-state plasma levels are approximately 3- to 4-fold higher than levels observed after a single dose of ARIMIDEX. Plasma concentrations approach steady-state levels at about 7 days of once daily dosing. Anastrozole is 40% bound to plasma proteins in the therapeutic range.
Metabolism
Metabolism of anastrozole occurs by N-dealkylation, hydroxylation and glucuronidation. Three metabolites of anastrozole (triazole, a glucuronide conjugate of hydroxy-anastrozole, and a glucuronide conjugate of anastrozole itself) have been identified in human plasma and urine. The major circulating metabolite of anastrozole, triazole, lacks pharmacologic activity.
Anastrozole inhibited reactions catalyzed by cytochrome P450 1A2, 2C8/9, and 3A4 in vitro with Ki values which were approximately 30 times higher than the mean steady-state Cmax values observed following a 1 mg daily dose. Anastrozole had no inhibitory effect on reactions catalyzed by cytochrome P450 2A6 or 2D6 in vitro. Administration of a single 30 mg/kg or multiple 10 mg/kg doses of anastrozole to healthy subjects had no effect on the clearance of antipyrine or urinary recovery of antipyrine metabolites.
Excretion
Eighty-five percent of radiolabeled anastrozole was recovered in feces and urine. Hepatic metabolism accounts for approximately 85% of anastrozole elimination. Renal elimination accounts for approximately 10% of total clearance. The mean elimination half-life of anastrozole is 50 hours.
Effect of Gender and Age
Anastrozole pharmacokinetics have been investigated in postmenopausal female volunteers and patients with breast cancer. No age-related effects were seen over the range <50 to >80 years.
Effect of Race
Estradiol and estrone sulfate serum levels were similar between Japanese and Caucasian postmenopausal women who received 1 mg of anastrozole daily for 16 days. Anastrozole mean steady-state minimum plasma concentrations in Caucasian and Japanese postmenopausal women were 25.7 and 30.4 ng/mL, respectively.
Effect of Renal Impairment
Anastrozole pharmacokinetics have been investigated in subjects with renal impairment. Anastrozole renal clearance decreased proportionally with creatinine clearance and was approximately 50% lower in volunteers with severe renal impairment (creatinine clearance < 30 mL/min/1.73m2) compared to controls. Total clearance was only reduced 10%. No dosage adjustment is needed for renal impairment [see Dosage and Administration (2.1) and Use in Specific Populations (8.6)].
Effect of Hepatic Impairment
Anastrozole pharmacokinetics have been investigated in subjects with hepatic cirrhosis related to alcohol abuse. The apparent oral clearance (CL/F) of anastrozole was approximately 30% lower in subjects with stable hepatic cirrhosis than in control subjects with normal liver function. However, these plasma concentrations were still with the range of values observed in normal subjects. The effect of severe hepatic impairment was not studied. No dose adjustment is necessary for stable hepatic cirrhosis [see Dosage and Administration (2.2) and Use in Specific Populations (8.7)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A conventional carcinogenesis study in rats at doses of 1.0 to 25 mg/kg/day (about 10 to 243 times the daily maximum recommended human dose on a mg/m2 basis) administered by oral gavage for up to 2 years revealed an increase in the incidence of hepatocellular adenoma and carcinoma and uterine stromal polyps in females and thyroid adenoma in males at the high dose. A dose-related increase was observed in the incidence of ovarian and uterine hyperplasia in females. At 25 mg/kg/day, plasma AUC0-24 hr levels in rats were 110 to 125 times higher than the level exhibited in postmenopausal volunteers at the recommended dose. A separate carcinogenicity study in mice at oral doses of 5 to 50 mg/kg/day (about 24 to 243 times the daily maximum recommended human dose on a mg/m2 basis) for up to 2 years produced an increase in the incidence of benign ovarian stromal, epithelial and granulosa cell tumors at all dose levels. A dose-related increase in the incidence of ovarian hyperplasia was also observed in female mice. These ovarian changes are considered to be rodent-specific effects of aromatase inhibition and are of questionable significance to humans. The incidence of lymphosarcoma was increased in males and females at the high dose. At 50 mg/kg/day, plasma AUC levels in mice were 35 to 40 times higher than the level exhibited in postmenopausal volunteers at the recommended dose.
ARIMIDEX has not been shown to be mutagenic in in vitro tests (Ames and E. coli bacterial tests, CHO-K1 gene mutation assay) or clastogenic either in vitro (chromosome aberrations in human lymphocytes) or in vivo (micronucleus test in rats).
Oral administration of anastrozole to female rats (from 2 weeks before mating to pregnancy day 7) produced significant incidence of infertility and reduced numbers of viable pregnancies at 1 mg/kg/day (about 10 times the recommended human dose on a mg/m2 basis and 9 times higher than the AUC0-24 hr found in postmenopausal volunteers at the recommended dose). Pre-implantation loss of ova or fetus was increased at doses equal to or greater than 0.02 mg/kg/day (about one-fifth the recommended human dose on a mg/m2 basis). Recovery of fertility was observed following a 5-week non-dosing period which followed 3 weeks of dosing. It is not known whether these effects observed in female rats are indicative of impaired fertility in humans.
Multiple-dose studies in rats administered anastrozole for 6 months at doses equal to or greater than 1 mg/kg/day (which produced plasma anastrozole Cssmax and AUC0-24 hr that were 19 and 9 times higher than the respective values found in postmenopausal volunteers at the recommended dose) resulted in hypertrophy of the ovaries and the presence of follicular cysts. In addition, hyperplastic uteri were observed in 6-month studies in female dogs administered doses equal to or greater than 1 mg/kg/day (which produced plasma anastrozole Cssmax and AUC0-24 hr that were 22 times and 16 times higher than the respective values found in postmenopausal women at the recommended dose). It is not known whether these effects on the reproductive organs of animals are associated with impaired fertility in premenopausal women.
13.2 Animal Toxicology and/or Pharmacology
Reproductive Toxicology
Anastrozole has been found to cross the placenta following oral administration of 0.1 mg/kg in rats and rabbits (about 1 and 1.9 times the recommended human dose, respectively, on a mg/m2 basis). Studies in both rats and rabbits at doses equal to or greater than 0.1 and 0.02 mg/kg/day, respectively (about 1 and 1/3, respectively, the recommended human dose on a mg/m2 basis), administered during the period of organogenesis showed that anastrozole increased pregnancy loss (increased pre- and/or post-implantation loss, increased resorption, and decreased numbers of live fetuses); effects were dose related in rats. Placental weights were significantly increased in rats at doses of 0.1 mg/kg/day or more.
Evidence of fetotoxicity, including delayed fetal development (i.e., incomplete ossification and depressed fetal body weights), was observed in rats administered doses of 1 mg/kg/day (which produced plasma anastrozole Cssmax and AUC0-24 hr that were 19 times and 9 times higher than the respective values found in postmenopausal volunteers at the recommended dose). There was no evidence of teratogenicity in rats administered doses up to 1.0 mg/kg/day. In rabbits, anastrozole caused pregnancy failure at doses equal to or greater than 1.0 mg/kg/day (about 16 times the recommended human dose on a mg/m2 basis); there was no evidence of teratogenicity in rabbits administered 0.2 mg/kg/day (about 3 times the recommended human dose on a mg/m2 basis).
14. Clinical Studies
14.1 Adjuvant Treatment of Breast Cancer in Postmenopausal Women
A multicenter, double-blind trial (ATAC) randomized 9,366 postmenopausal women with operable breast cancer to adjuvant treatment with ARIMIDEX 1 mg daily, tamoxifen 20 mg daily, or a combination of the two treatments for five years or until recurrence of the disease.
The primary endpoint of the trial was disease-free survival (i.e., time to occurrence of a distant or local recurrence, or contralateral breast cancer or death from any cause). Secondary endpoints of the trial included distant disease-free survival, the incidence of contralateral breast cancer and overall survival. At a median follow-up of 33 months, the combination of ARIMIDEX and tamoxifen did not demonstrate any efficacy benefit when compared with tamoxifen in all patients as well as in the hormone receptor positive subpopulation. This treatment arm was discontinued from the trial. Based on clinical and pharmacokinetic results from the ATAC trial, tamoxifen should not be administered with anastrozole [see Drug Interactions (7.1)].
Demographic and other baseline characteristics were similar among the three treatment groups (see Table 7).
| Demographic Characteristic | ARIMIDEX
1 mg (N*=3125) | Tamoxifen
20 mg (N*=3116) | ARIMIDEX 1 mg
plus Tamoxifen† 20 mg (N*=3125) |
|---|---|---|---|
|
|||
|
Mean age (yrs.) |
64.1 |
64.1 |
64.3 |
|
Age Range (yrs.) |
38.1 - 92.8 |
32.8 - 94.9 |
37.0 - 92.2 |
|
Age Distribution (%) | |||
|
<45 yrs. |
0.7 |
0.4 |
0.5 |
|
45-60 yrs. |
34.6 |
35.0 |
34.5 |
|
>60 <70 yrs. |
38.0 |
37.1 |
37.7 |
|
>70 yrs. |
26.7 |
27.4 |
27.3 |
|
Mean Weight (kg) |
70.8 |
71.1 |
71.3 |
|
Receptor Status (%) | |||
|
Positive‡ |
83.5 |
83.1 |
84.0 |
|
Negative§ |
7.4 |
8.0 |
7.0 |
|
Other¶ |
8.8 |
8.6 |
9.0 |
|
Other Treatment (%) prior to Randomization | |||
|
Mastectomy |
47.8 |
47.3 |
48.1 |
|
Breast conservation# |
52.3 |
52.8 |
51.9 |
|
Axillary surgery |
95.5 |
95.7 |
95.2 |
|
Radiotherapy |
63.3 |
62.5 |
61.9 |
|
Chemotherapy |
22.3 |
20.8 |
20.8 |
|
Neoadjuvant Tamoxifen |
1.6 |
1.6 |
1.7 |
|
Primary Tumor Size (%) | |||
|
T1 (≤2 cm) |
63.9 |
62.9 |
64.1 |
|
T2 (>2 cm and ≤5 cm) |
32.6 |
34.2 |
32.9 |
|
T3 (>5 cm) |
2.7 |
2.2 |
2.3 |
|
Nodal Status (%) | |||
|
Node positive |
34.9 |
33.6 |
33.5 |
|
1-3 (# of nodes) |
24.4 |
24.4 |
24.3 |
|
4-9 |
7.5 |
6.4 |
6.8 |
|
>9 |
2.9 |
2.7 |
2.3 |
|
Tumor Grade (%) | |||
|
Well-differentiated |
20.8 |
20.5 |
21.2 |
|
Moderately differentiated |
46.8 |
47.8 |
46.5 |
|
Poorly/undifferentiated |
23.7 |
23.3 |
23.7 |
|
Not assessed/recorded |
8.7 |
8.4 |
8.5 |
Patients in the two monotherapy arms of the ATAC trial were treated for a median of 60 months (5 years) and followed for a median of 68 months. Disease-free survival in the intent-to-treat population was statistically significantly improved [Hazard Ratio (HR) = 0.87, 95% CI: 0.78, 0.97, p=0.0127] in the ARIMIDEX arm compared to the tamoxifen arm. In the hormone receptor-positive subpopulation representing about 84% of the trial patients, disease-free survival was also statistically significantly improved (HR = 0.83, 95% CI: 0.73, 0.94, p=0.0049) in the ARIMIDEX arm compared to the tamoxifen arm.
Figure 1 — Disease-Free Survival Kaplan Meier Survival Curve for all Patients Randomized to ARIMIDEX or Tamoxifen Monotherapy in the ATAC trial (Intent-to-Treat)
Figure 2 — Disease-free Survival for Hormone Receptor-Positive Subpopulation of Patients Randomized to ARIMIDEX or Tamoxifen Monotherapy in the ATAC Trial
The survival data with 68 months follow-up is presented in Table 9.
In the group of patients who had previous adjuvant chemotherapy (N=698 for ARIMIDEX and N=647 for tamoxifen), the hazard ratio for disease-free survival was 0.91 (95% CI: 0.73 to 1.13) in the ARIMIDEX arm compared to the tamoxifen arm.
The frequency of individual events in the intent-to-treat population and the hormone receptor-positive subpopulation are described in Table 8.
| Intent-To-Treat Population† | Hormone Receptor-Positive Subpopulation† | |||
|---|---|---|---|---|
| ARIMIDEX
1 mg (N‡=3125) | Tamoxifen
20 mg (N‡=3116) | ARIMIDEX
1 mg (N‡=2618) | Tamoxifen
20 mg (N‡=2598) |
|
|
||||
|
Median Duration of Therapy (mo) |
60 |
60 |
60 |
60 |
|
Median Efficacy Follow-up (mo) |
68 |
68 |
68 |
68 |
|
Loco-regional recurrence |
119 (3.8) |
149 (4.8) |
76 (2.9) |
101 (3.9) |
|
Contralateral breast cancer |
35 (1.1) |
59 (1.9) |
26 (1.0) |
54 (2.1) |
|
Invasive |
27 (0.9) |
52 (1.7) |
21 (0.8) |
48 (1.8) |
|
Ductal carcinoma in situ |
8 (0.3) |
6 (0.2) |
5 (0.2) |
5 (0.2) |
|
Unknown |
0 |
1 (<0.1) |
0 |
1 (<0.1) |
|
Distant recurrence |
324 (10.4) |
375 (12.0) |
226 (8.6) |
265 (10.2) |
|
Death from Any Cause |
411 (13.2) |
420 (13.5) |
296 (11.3) |
301 (11.6) |
|
Death breast cancer |
218 (7.0) |
248 (8.0) |
138 (5.3) |
160 (6.2) |
|
Death other reason (including unknown) |
193 (6.2) |
172 (5.5) |
158 (6.0) |
141 (5.4) |
A summary of the study efficacy results is provided in Table 9.
|
||||
|
Intent-To-Treat Population |
Hormone Receptor-Positive Subpopulation |
|||
|
ARIMIDEX 1 mg (N=3125) |
Tamoxifen 20 mg (N=3116) |
ARIMIDEX 1 mg (N=2618) |
Tamoxifen 20 mg (N=2598) |
|
|
Number of Events |
Number of Events |
|||
|
Disease-free Survival |
575 |
651 |
424 |
497 |
|
Hazard ratio |
0.87 |
0.83 |
||
|
2-sided 95% CI |
0.78 to 0.97 |
0.73 to 0.94 |
||
|
p-value |
0.0127 |
0.0049 |
||
|
Distant Disease-free Survival |
500 |
530 |
370 |
394 |
|
Hazard ratio |
0.94 |
0.93 |
||
|
2-sided 95% CI |
0.83 to 1.06 |
0.80 to 1.07 |
||
|
Overall Survival |
411 |
420 |
296 |
301 |
|
Hazard ratio |
0.97 |
0.97 |
||
|
2-sided 95% CI |
0.85 to 1.12 |
0.83 to 1.14 |
||
10-year median follow-up Efficacy Results from the ATAC Trial
In a subsequent analysis of the ATAC trial, patients in the two monotherapy arms were followed for a median of 120 months (10 years). Patients received study treatment for a median of 60 months (5 years) (see Table 10).
|
Intent-To-Treat Population |
Hormone Receptor-Positive Subpopulation |
|||
|
ARIMIDEX 1 mg (N=3125) |
Tamoxifen 20 mg (N=3116) |
ARIMIDEX 1 mg (N=2618) |
Tamoxifen 20 mg (N=2598) |
|
|
Number of Events |
||||
|
Disease-free Survival |
953 |
1022 |
735 |
924 |
|
Hazard ratio |
0.91 |
0.86 |
||
|
2-sided 95% CI |
0.83 to 0.99 |
0.78 to 0.95 |
||
|
p-value |
0.0365 |
0.0027 |
||
|
Overall Survival |
734 |
747 |
563 |
586 |
|
Hazard ratio |
0.97 |
0.95 |
||
|
2-sided 95% CI |
0.88 to 1.08 |
0.84 to 1.06 |
||
Figure 3 - Disease-Free Survival Kaplan Meier Survival Curve for all Patients Randomized to ARIMIDEX or Tamoxifen Monotherapy in the ATAC Trial (Intent-to-Treat)(a)
a The proportion of patients with 120 months’ follow-up was 29.4%.
Figure 4 - Disease-Free Survival for Hormone Receptor-Positive Subpopulation of Patients Randomized to ARIMIDEX or Tamoxifen Monotherapy in the ATAC Trial(b)

bThe proportion of patients with 120 months’ follow-up was 29.8%.
14.2 First-Line Therapy in Postmenopausal Women with Advanced Breast Cancer
Two double-blind, controlled clinical studies of similar design (0030, a North American study and 0027, a predominately European study) were conducted to assess the efficacy of ARIMIDEX compared with tamoxifen as first-line therapy for hormone receptor positive or hormone receptor unknown locally advanced or metastatic breast cancer in postmenopausal women. A total of 1021 patients between the ages of 30 and 92 years old were randomized to receive trial treatment. Patients were randomized to receive 1 mg of ARIMIDEX once daily or 20 mg of tamoxifen once daily. The primary endpoints for both trials were time to tumor progression, objective tumor response rate, and safety.
Demographics and other baseline characteristics, including patients who had measurable and no measurable disease, patients who were given previous adjuvant therapy, the site of metastatic disease and ethnic origin were similar for the two treatment groups for both trials. The following table summarizes the hormone receptor status at entry for all randomized patients in trials 0030 and 0027.
| Number (%) of subjects | ||||
|---|---|---|---|---|
| Trial 0030 | Trial 0027 | |||
| Receptor status | ARIMIDEX
1 mg (N=171) | Tamoxifen
20 mg (N=182) | ARIMIDEX
1 mg (N=340) | Tamoxifen
20 mg (N=328) |
|
||||
|
ER* and/or PgR† |
151 (88.3) |
162 (89.0) |
154 (45.3) |
144 (43.9) |
|
ER* unknown, PgR† unknown |
19 (11.1) |
20 (11.0) |
185 (54.4) |
183 (55.8) |
For the primary endpoints, trial 0030 showed that ARIMIDEX had a statistically significant advantage over tamoxifen (p=0.006) for time to tumor progression; objective tumor response rates were similar for ARIMIDEX and tamoxifen. Trial 0027 showed that ARIMIDEX and tamoxifen had similar objective tumor response rates and time to tumor progression (see Table 12 and Figures 5 and 6).
Table 12 below summarizes the results of trial 0030 and trial 0027 for the primary efficacy endpoints.
| Endpoint | Trial 0030 | Trial 0027 | ||
|---|---|---|---|---|
| ARIMIDEX
1 mg (N=171) | Tamoxifen
20 mg (N=182) | ARIMIDEX
1 mg (N=340) | Tamoxifen
20 mg (N=328) |
|
|
||||
| ||||
|
Median TTP (months) |
11.1 |
5.6 |
8.2 |
8.3 |
|
Number (%) of subjects who progressed |
114 (67%) |
138 (76%) |
249 (73%) |
247 (75%) |
|
Hazard ratio (LCL* )† |
1.42 (1.15) |
1.01 (0.87) |
||
|
2-sided 95% CI‡ |
(1.11, 1.82) |
(0.85, 1.20) |
||
|
p-value§ |
0.006 |
0.920 |
||
|
Best objective response rate | ||||
|
Number (%) of subjects with CR¶ + PR# |
36 (21.1%) |
31 (17.0%) |
112 (32.9%) |
107 (32.6%) |
|
Odds Ratio (LCL* )Þ |
1.30 (0.83) |
1.01 (0.77) |
||
Figure 5 - Kaplan-Meier probability of time to disease progression for all randomized patients (intent-to-treat) in Trial 0030

Figure 6 - Kaplan-Meier probability of time to progression for all randomized patients (intent-to-treat) in Trial 0027
Results from the secondary endpoints were supportive of the results of the primary efficacy endpoints. There were too few deaths occurring across treatment groups of both trials to draw conclusions on overall survival differences.
14.3 Second-Line Therapy in Postmenopausal Women with Advanced Breast Cancer who had Disease Progression following Tamoxifen Therapy
Anastrozole was studied in two controlled clinical trials (0004, a North American study; 0005, a predominately European study) in postmenopausal women with advanced breast cancer who had disease progression following tamoxifen therapy for either advanced or early breast cancer. Some of the patients had also received previous cytotoxic treatment. Most patients were ER-positive; a smaller fraction were ER-unknown or ER-negative; the ER-negative patients were eligible only if they had a positive response to tamoxifen. Eligible patients with measurable and non-measurable disease were randomized to receive either a single daily dose of 1 mg or 10 mg of ARIMIDEX or megestrol acetate 40 mg four times a day. The studies were double-blinded with respect to ARIMIDEX. Time to progression and objective response (only patients with measurable disease could be considered partial responders) rates were the primary efficacy variables. Objective response rates were calculated based on the Union Internationale Contre le Cancer (UICC) criteria. The rate of prolonged (more than 24 weeks) stable disease, the rate of progression, and survival were also calculated.
Both trials included over 375 patients; demographics and other baseline characteristics were similar for the three treatment groups in each trial. Patients in the 0005 trial had responded better to prior tamoxifen treatment. Of the patients entered who had prior tamoxifen therapy for advanced disease (58% in Trial 0004; 57% in Trial 0005), 18% of these patients in Trial 0004 and 42% in Trial 0005 were reported by the primary investigator to have responded. In Trial 0004, 81% of patients were ER-positive, 13% were ER-unknown, and 6% were ER-negative. In Trial 0005, 58% of patients were ER-positive, 37% were ER-unknown, and 5% were ER-negative. In Trial 0004, 62% of patients had measurable disease compared to 79% in Trial 0005. The sites of metastatic disease were similar among treatment groups for each trial. On average, 40% of the patients had soft tissue metastases; 60% had bone metastases; and 40% had visceral (15% liver) metastases.
Efficacy results from the two studies were similar as presented in Table 13. In both studies there were no significant differences between treatment arms with respect to any of the efficacy parameters listed in the table below.
| ARIMIDEX
1 mg | ARIMIDEX
10 mg | Megestrol
Acetate 160 mg |
|
|---|---|---|---|
|
|||
|
Trial 0004 | |||
|
(N. America) |
(N=128) |
(N=130) |
(N=128) |
|
Median Follow-up (months)* |
31.3 |
30.9 |
32.9 |
|
Median Time to Death (months) |
29.6 |
25.7 |
26.7 |
|
2 Year Survival Probability (%) |
62.0 |
58.0 |
53.1 |
|
Median Time to Progression (months) |
5.7 |
5.3 |
5.1 |
|
Objective Response (all patients) (%) |
12.5 |
10.0 |
10.2 |
|
Stable Disease for >24 weeks (%) |
35.2 |
29.2 |
32.8 |
|
Progression (%) |
86.7 |
85.4 |
90.6 |
|
Trial 0005 | |||
|
(Europe, Australia, S. Africa) |
(N=135) |
(N=118) |
(N=125) |
|
Median Follow-up (months)* |
31.0 |
30.9 |
31.5 |
|
Median Time to Death (months) |
24.3 |
24.8 |
19.8 |
|
2 Year Survival Probability (%) |
50.5 |
50.9 |
39.1 |
|
Median Time to Progression (months) |
4.4 |
5.3 |
3.9 |
|
Objective Response (all patients) (%) |
12.6 |
15.3 |
14.4 |
|
Stable Disease for >24 weeks (%) |
24.4 |
25.4 |
23.2 |
|
Progression (%) |
91.9 |
89.8 |
92.0 |
When data from the two controlled trials are pooled, the objective response rates and median times to progression and death were similar for patients randomized to ARIMIDEX 1 mg and megestrol acetate. There is, in this data, no indication that ARIMIDEX 10 mg is superior to ARIMIDEX 1 mg.
| Trials 0004 & 0005
(Pooled Data) | ARIMIDEX
1 mg N=263 | ARIMIDEX
10 mg N=248 | Megestrol Acetate
160 mg N=253 |
|---|---|---|---|
|
Median Time to Death (months) |
26.7 |
25.5 |
22.5 |
|
2 Year Survival Probability (%) |
56.1 |
54.6 |
46.3 |
|
Median Time to Progression |
4.8 |
5.3 |
4.6 |
|
Objective Response (all patients) (%) |
12.5 |
12.5 |
12.3 |
16. How is Arimidex supplied
These tablets are supplied in bottles of 30 tablets (NDC 0310-0201-30).
17. Patient Counseling Information
See FDA approved patient labeling (Patient Information).
17.1 Pregnancy
Patients should be advised that ARIMIDEX may cause fetal harm. They should also be advised that ARIMIDEX is not for use in premenopausal women; therefore, if they become pregnant, they should stop taking ARIMIDEX and immediately contact their doctor.
17.2 Allergic (Hypersensitivity) Reactions
Patients should be informed of the possibility of serious allergic reactions with swelling of the face, lips, tongue and/or throat (angioedema) which may cause difficulty in swallowing and/or breathing and to seek medical attention immediately.
17.3 Ischemic Cardiovascular Events
Patients with pre-existing ischemic heart disease should be informed that an increased incidence of cardiovascular events has been observed with ARIMIDEX use compared to tamoxifen use. If patients have new or worsening chest pain or shortness of breath they should seek medical attention immediately.
17.4 Bone Effects
Patients should be informed that ARIMIDEX lowers the level of estrogen. This may lead to a loss of the mineral content of bones, which might decrease bone strength. A possible consequence of decreased mineral content of bones is an increase in the risk of fractures.
17.5 Cholesterol
Patients should be informed that an increased level of cholesterol might be seen while receiving ARIMIDEX.
Patient Information
ARIMIDEX®
A-rim-eh-dex
(anastrozole)
tablets
What is the most important information I should know about ARIMIDEX?
ARIMIDEX may cause serious side effects including:
- •
-
heart disease. Women with early breast cancer, who have a history of blockage in their heart arteries (ischemic heart disease) and who take ARIMIDEX, may have an increase in symptoms of decreased blood flow to their heart compared to similar women who take tamoxifen.
Get medical help right away if you have new or worsening chest pain or shortness of breath during treatment with ARIMIDEX.
What is ARIMIDEX?
ARIMIDEX is a prescription medicine used in women after menopause (“the change of life”) for:
- •
- treatment of early breast cancer
- ∘
- after surgery
- ∘
- in women whose breast cancer is hormone receptor-positive
- •
- the first treatment of breast cancer that has spread to nearby tissue or lymph nodes (locally advanced) or has spread to other parts of the body (metastatic), in women whose breast cancer is hormone receptor-positive or the hormone receptors are not known
- •
- treatment of advanced breast cancer, if the cancer has grown, or the disease has spread after tamoxifen therapy
ARIMIDEX does not work in women with breast cancer who have not gone through menopause (premenopausal women).
Who should not take ARIMIDEX?
Do not take ARIMIDEX if you:
- •
- are pregnant or able to become pregnant. ARIMIDEX may harm your unborn baby. If you become pregnant while taking ARIMIDEX, tell your doctor right away.
- •
- have not gone through menopause (are premenopausal)
- •
- have had a severe allergic reaction to anastrozole or any of the ingredients in ARIMIDEX. See the end of this leaflet for a complete list of ingredients in ARIMIDEX. Symptoms of a severe allergic reaction to ARIMIDEX include: swelling of the face, lips, tongue, or throat, trouble breathing or swallowing, hives and itching.
What should I tell my doctor before taking ARIMIDEX?
Before you take ARIMIDEX, tell your doctor if you:
- •
- have not gone through menopause. Talk to your doctor if you are not sure.
- •
- have or had a heart problem
- •
- have been told you have bone thinning or weakness (osteoporosis)
- •
- have high cholesterol
- •
- have any other medical conditions
- •
- are pregnant or plan to become pregnant. ARIMIDEX may harm your unborn baby. See “Who should not take ARIMIDEX?”
- •
- are breastfeeding or plan to breastfeed. It is not known if ARIMIDEX passes into breast milk. You and your doctor should decide if you will take ARIMIDEX or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your doctor if you take:
- •
- tamoxifen. You should not take ARIMIDEX if you take tamoxifen. Taking ARIMIDEX with tamoxifen may lower the amount of ARIMIDEX in your blood and may cause ARIMIDEX not to work as well.
- •
-
Medicines that contain estrogen. ARIMIDEX may not work if taken with any of these medicines:
- ∘
- hormone replacement therapy
- ∘
- birth control pills
- ∘
- estrogen creams
- ∘
- vaginal rings
- ∘
- vaginal suppositories
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
How should I take ARIMIDEX?
- •
- Take ARIMIDEX exactly as your doctor tells you to take it.
- •
- Continue taking ARIMIDEX until your doctor tells you to stop.
- •
- ARIMIDEX can be taken with or without food.
- •
- If you miss a dose, take it as soon as you remember. If it is almost time for your next dose, skip the missed dose. Take your next regularly scheduled dose. Do not take two doses at the same time.
If you take too much ARIMIDEX, call your doctor or go to the nearest hospital emergency room right away.
What are the possible side effects of ARIMIDEX?
ARIMIDEX may cause serious side effects including:
- •
- See “What is the most important information I should know about ARIMIDEX?”
- •
- bone thinning or weakness (osteoporosis). ARIMIDEX lowers estrogen in your body, which may cause your bones to become thinner and weaker. This may increase your risk of fractures, especially of your spine, hip and wrist. Your doctor may order a bone mineral density test before you start and during treatment with ARIMIDEX to check you for bone changes.
- •
- increased blood cholesterol (fat in the blood). Your doctor may do blood tests to check your cholesterol while you are taking ARIMIDEX.
- •
- skin reactions. Stop taking ARIMIDEX and call your doctor right away if you get any skin lesions, ulcers, or blisters.
- •
-
severe allergic reactions. Get medical help right away if you get:
- ∘
- swelling of your face, lips, tongue, or throat
- ∘
- trouble swallowing or breathing
- •
- liver problems. ARIMIDEX can cause inflammation of your liver and changes in liver function blood tests. Your doctor may check you for this.
-
Stop taking ARIMIDEX and call your doctor right away if you have any of these signs or symptoms of a liver problem:
- ∘
- a general feeling of not being well
- ∘
- yellowing of your skin or whites of your eyes
- ∘
- pain on the right side of your stomach-area (abdomen)
Common side effects in women taking ARIMIDEX include:
- •
- hot flashes
- •
- weakness
- •
- joint aches
- •
- joint pain, stiffness or swelling (arthritis)
- •
- pain
- •
- sore throat
- •
- high blood pressure
- •
- depression
- •
- nausea and vomiting
- •
- rash
- •
- back pain
- •
- sleep problems
- •
- bone pain
- •
- headache
- •
- swelling of your legs, ankles, or feet
- •
- increased cough
- •
- shortness of breath
- •
- build up of lymph fluid in the tissues of your affected arm (lymphedema)
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of ARIMIDEX. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ARIMIDEX?
- •
- Store ARIMIDEX at room temperature between 68°F to 77°F (20°C to 25°C).
Keep ARIMIDEX and all medicines out of the reach of children.
General information about the safe and effective use of ARIMIDEX.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not take ARIMIDEX for a condition for which it was not prescribed. Do not give ARIMIDEX to other people, even if they have the same symptoms that you have. It may harm them.
If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about ARIMIDEX that is written for health professionals. For more information call 1-866-992-9276 or go to www.ARIMIDEX.com.
What are the ingredients in ARIMIDEX?
Active ingredient: anastrozole
Inactive ingredients: lactose, magnesium stearate, hydroxypropylmethylcellulose, polyethylene glycol, povidone, sodium starch glycolate, and titanium dioxide.
This Patient Information has been approved by the U.S. Food and Drug Administration.
May/2014
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
ARIMIDEX is a registered trademark of the AstraZeneca group of companies. ©2009 AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850. All rights reserved.

| ARIMIDEX
anastrozole tablet |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - AstraZeneca Pharmaceuticals LP (054743190) |
| Registrant - AstraZeneca PLC (230790719) |
