Drug Detail:Azedra (Iobenguane i-131 [ eye-oh-ben-gwane ])
Drug Class: Therapeutic radiopharmaceuticals
Highlights of Prescribing Information
AZEDRA® (iobenguane I 131) injection, for intravenous use
Initial U.S. Approval: 2018
Indications and Usage for Azedra Injection
AZEDRA is a radioactive therapeutic agent indicated for the treatment of adult and pediatric patients 12 years and older with iobenguane scan positive, unresectable, locally advanced or metastatic pheochromocytoma or paraganglioma who require systemic anticancer therapy. (1)
Azedra Injection Dosage and Administration
- Verify pregnancy status in females of reproductive potential prior to administering AZEDRA. (2.1)
- Block thyroid prior to administering AZEDRA. (2.2)
- Do not administer if platelet count is less than 80,000/mcL or absolute neutrophil count is less than 1,200/mcL. (2.4)
- Administer AZEDRA intravenously as a dosimetric dose followed by two therapeutic doses administered 90 days apart. (2.2)
- The recommended dosimetric dose is:
- Patients greater than 50 kg: 185 to 222 MBq (5 to 6 mCi)
- Patients 50 kg or less: 3.7 MBq/kg (0.1 mCi/kg)
- The recommended therapeutic dose for each of the 2 doses is:
- Patients greater than 62.5 kg: 18,500 MBq (500 mCi)
- Patients 62.5 kg or less: 296 MBq/kg (8 mCi/kg)
- Adjust AZEDRA therapeutic doses based on radiation dose estimates results from dosimetry, if needed. (2.2)
Dosage Forms and Strengths
Injection: 555 MBq/mL (15 mCi/ml) at TOC as a clear solution in a single-dose vial. (3)
Contraindications
None. (4)
Warnings and Precautions
- Risk from Radiation Exposure: Minimize radiation exposure consistent with institutional radiation safety practices and patient management procedures. (2.1), (5.1)
- Myelosuppression: Monitor blood cell counts. Withhold and dose reduce AZEDRA as recommended based on severity of cytopenia. (5.2)
- Secondary Myelodysplastic Syndrome, Leukemia and Other Malignancies: The time to development of MDS or acute leukemia ranged from 12 months to 7 years. (5.3)
- Hypothyroidism: Initiate thyroid-blocking medication prior to administration and continue after each dose. Monitor for hypothyroidism and thyroid-stimulating hormone levels before starting AZEDRA and annually thereafter. (2.3, 5.4)
- Elevations in blood pressure: Monitor blood pressure frequently during the first 24 hours after each therapeutic dose. (5.5)
- Renal Toxicity: Monitor renal function during and after treatment. (5.6)
- Pneumonitis: Monitor patients for signs and symptoms of pneumonitis and treat appropriately. (5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females and males of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.8)
- Risk of Infertility: May cause infertility (5.9)
Adverse Reactions/Side Effects
The most common Grade 3-4 adverse reactions (≥ 10%) were lymphopenia, neutropenia, thrombocytopenia, fatigue, anemia, increased international normalized ratio, nausea, dizziness, hypertension, and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Progenics Pharmaceuticals, Inc at 1-800-362-2668 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Drugs that Reduce Catecholamine Uptake or Deplete Stores: Discontinue these drugs prior to and following AZEDRA administration. (2.3), (7.1)
Use In Specific Populations
- Lactation: Advise women not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2023
Full Prescribing Information
1. Indications and Usage for Azedra Injection
AZEDRA is indicated for the treatment of adult and pediatric patients 12 years and older with iobenguane scan positive, unresectable, locally advanced or metastatic pheochromocytoma or paraganglioma who require systemic anticancer therapy.
2. Azedra Injection Dosage and Administration
2.1 Important Safety Information
AZEDRA is a radiopharmaceutical. Handle with appropriate safety measures to minimize radiation exposure [see Warnings and Precautions (5.1)]. Use waterproof gloves and effective radiation shielding when handling AZEDRA. Radiopharmaceuticals, including AZEDRA, should be used by or under the control of physicians who are qualified by specific training and experience in the safe use and handling of radiopharmaceuticals, and whose experience and training have been approved by the appropriate governmental agency authorized to license the use of radiopharmaceuticals.
Verify pregnancy status in females of reproductive potential prior to initiating AZEDRA [see Use in Specific Populations (8.1), (8.3)].
2.2 Recommended Dosage
Administer thyroid blockade and other pre- and concomitant medications as recommended [see Dosage and Administration (2.3)].
Dosimetric Dose
The recommended AZEDRA dosimetric dose administered as an intravenous injection is:
- Patients weighing greater than 50 kg: 185 to 222 MBq (5 or 6 mCi)
- Patients weighing 50 kg or less: 3.7 MBq/kg (0.1 mCi/kg)
Dosimetry and Biodistribution Assessment
Following the AZEDRA dosimetric dose:
- Acquire anterior/posterior whole body gamma camera images within 1 hour of the AZEDRA dosimetric dose and prior to patient voiding (Day 0; Scan 1).
- Acquire additional images on Day 1 or 2 following patient voiding (Scan 2).
- Acquire additional images between Days 2-5 following patient voiding (Scan 3).
For each individual patient, calculate the radiation dose estimates to normal organs and tissues per unit activity [D (organ)] of administered dose using data extracted from these 3 images. Calculate in accordance with the Medical Internal Radiation Dose (MIRD) schema or related methodology. Whenever possible, use patient-specific organ masses (e.g., estimated from imaging).
Therapeutic Dosage
The recommended AZEDRA therapeutic dose is based on body weight and reduced, if necessary, based on the dosimetry data. Administer a total of 2 therapeutic doses intravenously a minimum of 90 days apart.
Weight Based Dose per Therapeutic Cycle
- Patients weighing greater than 62.5 kg: 18,500 MBq (500 mCi)
- Patients weighing 62.5 kg or less: 296 MBq/kg (8 mCi/kg)
Determine if Dose Reduction Needed Based on Critical Organ Limits
- Calculate the estimated critical organ absorbed-dose by multiplying the dosimetry-derived radiation absorbed-dose per unit activity [D (organ)] by weight based therapeutic total activity (Aw).
- If resulting estimated critical organ absorbed-dose is less than threshold absorbed-dose (T) shown in Table 1, no dose adjustment is necessary.
- If resulting estimated critical organ absorbed-dose exceeds threshold absorbed-dose (T) shown in Table 1, calculate the reduced therapeutic total activity (i.e., the cumulative activity that would be administered in 2 therapeutic cycles) using the following equation:
Reduced Therapeutic Total Activity= Aw ×[T ÷ {Aw × D (organ)}] - Example: A 75 kg patient qualifies for a therapeutic total activity of 1000 mCi (Aw). For the kidneys, the dosimetry yields an estimated critical organ absorbed dose per unit activity of 0.027 Gy/mCi [D (kidney)]. Thus, the estimated critical organ absorbed-dose to the kidney is 27 Gy [Aw x D (organ)], which exceeds the threshold absorbed-dose for the kidneys (T) of 18 Gy (Table 1). Using the equation above the reduced therapeutic total activity to be administered to this patient is 666.7 mCi.
1000 mCi × [18 Gy ÷ {1000 mCi × 0.027 Gy/mCi}]
|
*Threshold of ~0.5 Gy for both heart and carotid artery, derived from experience with external-beam radiotherapy and associated with fractionated exposure, has also been proposed to support an ~1% mortality rate of cardiovascular and cerebrovascular deaths in >10-15 years; however, uncertainty is associated with the value ~ 0.5 Gy cited for vascular disease (ICRP publication 118, p.300, Table 4.5). Consider benefits/risks to patients. |
|||
| Organ | ~1%-rate: mortality or organ failure associated with disease | Time to death or organ failure | Threshold* absorbed-dose for ~1%-rate mortality or organ failure (Gy) |
| Red marrow | H-ARS mortality | 1-2 months | 12 |
| Lungs | Pneumonitis mortality | 1-7 months | 16.5 |
| Kidneys | Renal failure | >1 year | 18 |
| Liver | Hepatomegaly, ascites: possible organ failure | 0.5-3 months | 31 |
| Small intestine | GI-ARS mortality | 6-9 days | 40 |
2.3 Thyroid Blockade and Other Pre- and Concomitant Medications
Thyroid Blockade
Administer inorganic iodine starting at least 24 hours before and continuing for 10 days after each AZEDRA dose [see Warnings and Precautions (5.4)].
Hydration
Instruct patients to increase fluid intake to at least two liters a day starting at least 1 day before and continuing for 1 week after each AZEDRA dose to minimize irradiation to the bladder [see Warnings and Precautions (5.1)].
Drugs that Reduce Catecholamine Uptake or Deplete Stores
Discontinue drugs that reduce catecholamine uptake or deplete catecholamine stores for at least 5 half-lives before administration of either the dosimetry dose or a therapeutic dose of AZEDRA. Do not administer these drugs until at least 7 days after each AZEDRA dose [see Drug Interactions (7.1)].
Antiemetic
Administer antiemetics 30 minutes prior to administering each AZEDRA dose.
2.4 Dose Modifications for Adverse Reactions
Recommended dose modifications of AZEDRA for adverse reactions are provided in Table 2 and the recommended dose or dose reduction for the second therapeutic dose of AZEDRA for myelosuppression are provided in Table 3.
| Adverse Reaction | Dose Modification |
| Myelosuppression [see Warnings and Precautions (5.2)] |
Do not administer the first therapeutic dose for platelet counts less than 80,000/mcL or absolute neutrophil counts (ANC) less than 1,200/mcL. Do not administer the second therapeutic dose until platelets and neutrophils return to baseline or to the normal range. Reduce the second therapeutic dose for the following:
|
| Pneumonitis [see Warnings and Precautions (5.7)] | Do not administer the second therapeutic dose if pneumonitis is diagnosed after the first therapeutic dose. |
| Patient Population | If first therapeutic dose was weight based, | If first therapeutic dose was reduced based on critical organ limits, |
| Patients weighing greater than 62.5 kg | Reduce the second therapeutic dose to 425 mCi | Reduce second therapeutic dose to 85% of the first dose |
| Patients weighing 62.5 kg or less | Reduce the second therapeutic dose to 7 mCi/kg | Reduce second therapeutic dose to 85% of the first dose |
2.5 Preparation and Administration
- Refer to the Package Handling Instructions supplied with the frozen vial. Discard if the temperature recording device displays an alarm icon indicating that the temperature exceeded -70ºC during shipment.
- Use aseptic technique and radiation shielding when administering the AZEDRA solution. Use tongs when handling vial to minimize radiation exposure.
- Confirm the amount of radioactivity of AZEDRA in the radiopharmaceutical vial with an appropriate dose calibrator prior to and after AZEDRA administration.
- Inspect visually for particulate matter and discoloration prior to administration whenever solution and container permit. The AZEDRA solution should be a clear, colorless to pale yellow solution without any particulate matter. Discard if particulate matter or discoloration is observed.
Dosimetric Dose Preparation
- Thaw the vial to room temperature in lead pot. Do not heat or refreeze. Confirm complete thawing and gently swirl to ensure homogeneity.
- Insert a venting unit (consisting of a needle, 0.2-micron sterile filter, and a charcoal filter) to avoid pressurizing the contents of the vial during dilution. Swirl gently to ensure homogeneity.
- Add sufficient volume of 0.9% Sodium Chloride Solution, USP to the vial to yield a concentration of 1 mCi/mL (37 MBq/mL). Swirl gently to ensure homogeneity.
- Draw the dosimetric dose into a 10 mL shielded syringe and place in the dose calibrator to ensure that the activity is within ± 10% of dose. Discard unused medicinal product or waste material in accordance with local and federal laws.
- Maintain at room temperature and administer within 8 hours of retrieval from frozen storage.
Dosimetric Dose Administration
- Administer the dosimetric dose over 60 seconds.
Therapeutic Dose Preparation
- Thaw the appropriate number of vials (2 or 3) to room temperature in lead pots. Do not heat or refreeze.
- Swirl each AZEDRA vial to ensure homogeneity.
- Insert a venting unit into each AZEDRA vial to avoid pressurizing the contents of the vial during dilution.
- Insert a venting unit into a sterile 50-mL glass vial. Transfer the entire contents of the two therapeutic vials into a 50-mL glass vial. Measure the radioactivity.
- If radioactivity in the 50-mL glass vial exceeds the therapeutic dose, withdraw and discard the appropriate volume using a shielded syringe. Add 0.9% Sodium Chloride Solution, USP to a total volume of 50 mL.
- If radioactivity in the 50-mL glass vial is less than the therapeutic dose, use a shielded syringe to withdraw the appropriate volume from a third AZEDRA vial and add to the 50-mL glass vial. Add 0.9% Sodium Chloride Solution, USP to a total volume of 50 mL.
- Swirl gently to ensure homogeneity.
- Remove the venting unit and place the 50-mL glass vial into a dose calibrator to ensure that the activity is within ± 10% of therapeutic dose.
- Maintain at room temperature and administer within 8 hours of retrieval from frozen storage.
- Discard unused medicinal product or waste material in accordance with local and federal laws.
Therapeutic Dose Administration
- Verify line patency by infusing 250 mL of 0.9% Sodium Chloride Solution, USP (primary intravenous line) at recommended rate of 200 mL/hour.
- Insert a venting unit into the 50-mL glass vial containing the AZEDRA therapeutic dose.
- Assemble a second intravenous line using a 19 Gauge x 5-inch aspirating needle, 24-inch M-M arterial pressure tubing and a primary set specific connector.
- Clamp the second intravenous line and connect it to the primary intravenous line using the primary set specific connector. Flush the second intravenous line by releasing the clamp and then re-clamp the second intravenous line.
- Insert the needle of the second intravenous line into the 50-mL glass vial containing the AZEDRA therapeutic dose. Ensure the needle reaches the bottom of the glass vial without touching the sides of the vial.
- Clamp the primary intravenous line just above the second intravenous line and remove the clamp from the secondary intravenous line.
- Administer the AZEDRA therapeutic dose over 30 minutes at a recommended rate of 100 mL/hour for adults; for pediatric patients 12 years and older administer over 60 minutes at a recommended rate of 50 mL/hr. Clamp the secondary intravenous line when the first air bubbles form.
- Remove the clamp from the primary intravenous line to flush any residual AZEDRA therapeutic dose within this intravenous line with at least 50 mL of 0.9% Sodium Chloride Solution, USP.
- Remove the clamp from the secondary intravenous line to flush any residual drug in the secondary intravenous line into the 50-mL glass vial.
2.6 Radiation Dosimetry
The mean of the estimated radiation absorbed doses for AZEDRA are shown in Table 4.
|
* Table 1 tends to yield underestimates of absorbed dose for patients weighing less than 65 kg, and tends to yield overestimates for patients weighing more than 65 kg. 1-LLI Wall- Lower Large Intestine Wall. 2ULI Wall- Upper Large Intestine Wall. |
||||
| Target Organ | Mean (mGy/MBq) | Minimum (mGy/MBq) | Maximum (mGy/MBq) | Standard Deviation (mGy/MBq) |
| Salivary Glands | 1.499 | 0.486 | 7.957 | 1.134 |
| LLI Wall1 | 1.184 | 0.093 | 2.770 | 0.356 |
| Thyroid | 0.779 | 0.071 | 11.000 | 1.409 |
| Urinary Bladder Wall | 0.614 | 0.141 | 0.930 | 0.142 |
| ULI Wall2 | 0.514 | 0.091 | 1.120 | 0.138 |
| Liver | 0.509 | 0.180 | 7.830 | 0.862 |
| Kidneys | 0.360 | 0.085 | 0.772 | 0.163 |
| Spleen | 0.343 | 0.091 | 4.470 | 0.495 |
| Lungs | 0.323 | 0.123 | 3.170 | 0.344 |
| Heart Wall | 0.272 | 0.073 | 1.550 | 0.215 |
| Small Intestine | 0.194 | 0.085 | 0.347 | 0.042 |
| Osteogenic Cells | 0.151 | 0.085 | 0.369 | 0.044 |
| Gallbladder Wall | 0.146 | 0.083 | 0.852 | 0.094 |
| Ovaries | 0.126 | 0.000 | 0.271 | 0.046 |
| Pancreas | 0.117 | 0.068 | 0.484 | 0.054 |
| Adrenals | 0.116 | 0.067 | 0.535 | 0.059 |
| Uterus | 0.112 | 0.000 | 0.247 | 0.041 |
| Stomach Wall | 0.100 | 0.059 | 0.279 | 0.033 |
| Thymus | 0.083 | 0.049 | 0.212 | 0.027 |
| Muscle | 0.082 | 0.049 | 0.188 | 0.024 |
| Red Marrow | 0.079 | 0.048 | 0.175 | 0.022 |
| Breasts | 0.070 | 0.040 | 0.189 | 0.024 |
| Skin | 0.063 | 0.036 | 0.153 | 0.018 |
| Testes | 0.061 | 0.000 | 0.183 | 0.036 |
| Brain | 0.057 | 0.022 | 0.213 | 0.028 |
| Total Body | 0.107 | 0.064 | 0.414 | 0.045 |
3. Dosage Forms and Strengths
Injection: 555 MBq/mL (15 mCi/mL) as a clear, colorless to pale yellow solution in a single-dose vial.
5. Warnings and Precautions
5.1 Risk from Radiation Exposure
AZEDRA contributes to a patient's overall long-term radiation exposure. Long-term cumulative radiation exposure is associated with an increased risk for cancer. These risks of radiation associated with the use of AZEDRA are greater in pediatric patients than in adults [see Use in Specific Populations (8.4)].
Minimize radiation exposure to patients, medical personnel, and household contacts during and after treatment with AZEDRA consistent with institutional good radiation safety practices and patient management procedures [see Dosage and Administration (2.1)].
5.2 Myelosuppression
Severe and prolonged myelosuppression occurred during treatment with AZEDRA [see Adverse Reactions (6.1)]. Among the 88 patients who received a therapeutic dose of AZEDRA, 33% experienced Grade 4 thrombocytopenia, 16% experienced Grade 4 neutropenia, and 7% experienced Grade 4 anemia. Five percent of patients experienced febrile neutropenia. In Study IB12B following the first therapeutic dose, patients who experienced Grade 4 neutropenia reached neutrophil nadir at a median of 36 days (27 – 55 days) and remained at nadir for a median of 12 days (8 – 22 days) until recovery to less than or equal to Grade 3. Following the second dose, patients who experienced Grade 4 neutropenia reached nadir at a median of 43 days (38 – 47 days) and remained at nadir for a median of 18.5 days (8 – 31 days) until recovery to less than or equal to Grade 3.
Monitor blood cell counts weekly for up to 12 weeks or until levels return to baseline or the normal range. Withhold and dose reduce AZEDRA as recommended based on severity of the cytopenia [see Dosage and Administration (2.4)].
5.3 Secondary Myelodysplastic Syndrome, Leukemia and Other Malignancies
Myelodysplastic syndrome (MDS) or acute leukemias were reported in 6.8% of the 88 patients who received a therapeutic dose of AZEDRA [see Adverse Reactions (6.1)]. The time to development of MDS or acute leukemia ranged from 12 months to 7 years.
Two of the 88 patients developed a non-hematological malignancy. One patient developed colon cancer at 18 months and one patient developed lung adenocarcinoma at 27 months following the first therapeutic dose.
5.4 Hypothyroidism
Hypothyroidism was reported in 3.4% of the 88 patients who received a therapeutic dose of AZEDRA [see Adverse Reactions (6.1)]. The time to worsening of hypothyroidism was 4 months in one patient, and the time to development of hypothyroidism was less than one month in one patient and 18 months in one patient. Initiate thyroid-blocking medications starting at least 1 day before and continuing for 10 days after each AZEDRA dose to reduce the risk of hypothyroidism or thyroid neoplasia [see Dosage and Administration (2.3)]. Evaluate for clinical evidence of hypothyroidism and measure thyroid-stimulating hormone (TSH) levels prior to initiating AZEDRA and annually thereafter.
5.5 Elevations in Blood Pressure
Eleven percent of the 88 patients who received a therapeutic dose of AZEDRA [see Adverse Reactions (6.1)] experienced a worsening of pre-existing hypertension defined as an increase in systolic blood pressure to ≥160 mmHg with an increase of 20 mmHg or an increase in diastolic blood pressure to ≥ 100 mmHg with an increase of 10 mmHg. All changes in blood pressure occurred within the first 24 hours post infusion. Monitor blood pressure frequently during the first 24 hours after each therapeutic dose of AZEDRA.
5.6 Renal Toxicity
Of the 88 patients who received a therapeutic dose of AZEDRA [see Adverse Reactions (6.1)], 7% developed renal failure or acute kidney injury and 22% demonstrated a clinically significant decrease in glomerular filtration rate (GFR) measured at 6 or 12 months. Monitor renal function during and after treatment with AZEDRA. Patients with baseline renal impairment may be at greater risk of toxicity; perform more frequent assessments of renal function in patients with mild or moderate impairment. AZEDRA has not been studied in patients with severe renal impairment (creatinine clearance <30 mL/min).
5.7 Pneumonitis
Fatal pneumonitis occurred 9 weeks after a single dose in one patient in the expanded access program for Study IB12B (n=11). Pneumonitis was not diagnosed among the 88 patients enrolled in Study IB12 or IB12B [see Adverse Reactions (6.1)]. Monitor patients for signs and symptoms of pneumonitis and treat appropriately.
5.8 Embryo-Fetal Toxicity
Based on its mechanism of action, AZEDRA can cause fetal harm. There are no available data on the use of AZEDRA in pregnant women. No animal studies using iobenguane I 131 have been conducted to evaluate its effect on female reproduction and embryo-fetal development; however, all radiopharmaceuticals, including AZEDRA, have the potential to cause fetal harm.
Verify pregnancy status in females of reproductive potential prior to initiating AZEDRA [see Dosage and Administration (2.1)].
Advise females and males of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with AZEDRA and for 7 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment and for 4 months after the final dose [see Use in Specific Populations (8.1), (8.3)].
5.9 Risk of Infertility
Radiation exposure associated with AZEDRA may cause infertility in males and females. The recommended cumulative dose of 37 GBq of AZEDRA results in a radiation absorbed dose to the testes and ovaries within the range where temporary or permanent infertility can be expected following external beam radiotherapy [see Dosage and Administration (2.6), Use in Specific Populations (8.3)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in the labeling:
- Myelosuppression [see Warnings and Precautions (5.2)]
- Secondary Myelodysplastic Syndrome, Leukemia and Other Malignancies [see Warnings and Precautions (5.3)]
- Hypothyroidism [see Warnings and Precautions (5.4)]
- Elevations in Blood Pressure [see Warnings and Precautions (5.5)]
- Renal Toxicity [see Warnings and Precautions (5.6)]
- Pneumonitis [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in Warnings and Precautions reflect exposure to AZEDRA in 88 patients with iobenguane-scan positive recurrent or unresectable, locally advanced or metastatic pheochromocytoma or paraganglioma (PPGL) who received a therapeutic dose of AZEDRA in one of two clinical studies (IB12 or IB12B). The Warnings and Precautions also include data from 11 patients enrolled in an expanded access program for Study IB12B [see Warnings and Precautions (5)].
The safety data below was evaluated in two studies in patients with recurrent or unresectable, locally advanced or metastatic PPGL. Study IB12 was an open-label, multi-center, single-arm dose-finding study in adult patients with malignant or recurrent PPGL. The study consisted of a 12-month efficacy phase with a 1 year follow-up. Twenty-one patients received a dosimetric dose (~5 mCi), followed by one therapeutic dose (~500 mCi) of AZEDRA. Study IB12B was an open-label, multi-center, single-arm study in 68 adult and pediatric patients age 12 years and older with recurrent or unresectable, locally advanced or metastatic PPGL [see Clinical Studies (14)].
Patients with evidence of liver dysfunction (aspartate aminotransferase or alanine aminotransferase ≥ 2.5 times the upper limit of normal or total bilirubin > 1.5 times the upper limit of normal), a history of liver disease (including hepatitis and chronic alcohol abuse), or severe renal impairment (creatinine clearance < 30 mL/min) were excluded. Patients who had received external beam radiation to > 25% of bone marrow, received whole body radiotherapy, or who had received any systemic radiotherapy resulting in myelosuppression within 3 months of study entry, were also excluded. The safety data described below are based on pooled safety data from studies IB12 and IB12B. A total of 88 patients received at least one therapeutic dose of AZEDRA and 50 patients received two therapeutic doses (one patient received treatment in both studies).
Adverse reactions from studies IB12 and IB12B are presented in Table 5. The most common severe (Grade 3-4) adverse reactions were lymphopenia (78%), neutropenia (59%), thrombocytopenia (50%), fatigue (26%), anemia (24%), increased international normalized ratio (18%), nausea (16%), dizziness (13%), hypertension (11%), and vomiting (10%). Twelve percent of patients discontinued treatment due to adverse reactions (thrombocytopenia, anemia, lymphopenia, nausea and vomiting, multiple hematologic adverse reactions).
|
aNCI CTCAE version 3.0. bBased on laboratory data. cIncludes vomiting and retching. dIncludes sialoadenitis, salivary gland pain, and salivary gland enlargement. eIncludes abdominal pain, abdominal pain upper, and abdominal pain lower. fIncudes fatigue, asthenia. gIncludes upper respiratory tract infection, sinusitis, rhinorrhea, upper-airway cough syndrome, nasopharyngitis. hOnly assessed in Study IB12B (N=68). iIncludes dizziness and dizziness postural. jIncludes dysgeusia, hypogeusia and ageusia. kIncludes blood pressure increased and hypertension. |
||
| Adverse Reaction | All Gradesa, (%) | Gradesa 3 - 4, (%) |
| Hematologicb | ||
| Lymphopenia | 96 | 78 |
| Anemia | 93 | 24 |
| Thrombocytopenia | 91 | 50 |
| Neutropenia | 84 | 59 |
| Gastrointestinal | ||
| Nausea | 78 | 16 |
| Vomitingc | 58 | 10 |
| Dry mouth | 48 | 2 |
| Sialadenitisd | 39 | 1 |
| Diarrhea | 25 | 3 |
| Abdominal paine | 23 | 6 |
| Constipation | 19 | 7 |
| Oropharyngeal pain | 14 | 0 |
| Dyspepsia | 10 | 0 |
| General | ||
| Fatiguef | 71 | 26 |
| Pyrexia | 14 | 2 |
| Injection site pain | 10 | 0 |
| Hyperhidrosis | 10 | 0 |
| Alopecia | 10 | 0 |
| Infections | ||
| Upper respiratory tract infectiong | 16 | 2 |
| Urinary tract infection | 11 | 1 |
| Investigationsb | ||
| Increased international normalized ratioh | 85 | 18 |
| Increased blood alkaline phosphatase | 53 | 5 |
| Increased aspartate aminotransferase | 50 | 2 |
| Increased alanine aminotransferase | 43 | 2 |
| Metabolism and nutrition | ||
| Decreased appetite | 30 | 5 |
| Dehydration | 16 | 4 |
| Decreased weight | 16 | 1 |
| Musculoskeletal and connective tissue disorders | ||
| Back pain | 17 | 2 |
| Pain in extremity | 15 | 0 |
| Nervous system | ||
| Dizzinessi | 34 | 13 |
| Headache | 32 | 6 |
| Dysgeusiaj | 24 | 1 |
| Respiratory, thoracic, and mediastinal disorders | ||
| Cough | 18 | 0 |
| Dyspnea | 18 | 7 |
| Vascular | ||
| Hypotension | 24 | 4 |
| Hypertensionk | 20 | 11 |
| Tachycardia | 10 | 3 |
The following clinically significant adverse reactions were observed in < 10% of patients treated with AZEDRA:
Cardiac: palpitations (9%), syncope and presyncope (8%)
Endocrine: decreased TSH (5%), hypothyroidism (3%)
Gastrointestinal: dysphagia (7%), abdominal distension (6%), gastroesophageal reflux disease (6%), stomatitis (3%)
General: insomnia (9%), chills (8%), chest pain (6%)
Infections: candida infection (6%)
Investigations: prolonged prothrombin time (9%)
Musculoskeletal and connective tissue: arthralgia (8%), neck pain (8%), pain in jaw (7%), muscle spasms (6%)
Renal and urinary disorders: proteinuria (9%), renal failure (7%),
Respiratory: epistaxis (9%), nasal congestion (7%), pulmonary embolism (3%)
Skin and subcutaneous tissue: dry skin (8%), rash (8%), petechiae (7%)
Vascular: orthostatic hypotension (9%)
7. Drug Interactions
7.1 Drugs that Reduce Catecholamine Uptake or Deplete Stores
Based on the mechanism of action of iobenguane, drugs that reduce catecholamine uptake or that deplete catecholamine stores may interfere with iobenguane uptake into cells and therefore interfere with dosimetry calculations or the efficacy of AZEDRA. These drugs were not permitted in clinical trials that assessed the safety and efficacy of AZEDRA. Discontinue drugs that reduce catecholamine uptake or deplete catecholamine stores, such as those listed below, for at least 5 half-lives before administration of either the dosimetry or a therapeutic dose of AZEDRA. Do not administer these drugs until at least 7 days after each AZEDRA dose [see Dosage and Administration (2.3)].
- CNS stimulants or amphetamines (e.g. cocaine, methylphenidate, dextroamphetamine)
- Norepinephrine and dopamine reuptake inhibitors (e.g. phentermine)
- Norepinephrine and serotonin reuptake inhibitors (e.g. tramadol)
- Monoamine oxidase inhibitors (e.g. phenelzine and linezolid)
- Central monoamine depleting drugs (e.g. reserpine)
- Non-select beta adrenergic blocking drugs (e.g. labetalol)
- Alpha agonists or alpha/beta agonists (e.g. pseudoephedrine, phenylephrine, ephedrine, phenylpropanolamine, naphazoline)
- Tricyclic antidepressants or norepinephrine reuptake inhibitors (e.g. amitriptyline, bupropion, duloxetine, mirtazapine, venlafaxine)
- Botanicals that may inhibit reuptake of norepinephrine, serotonin or dopamine (e.g. ephedra, ma huang, St John's Wort, yohimbine)
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, AZEDRA can cause fetal harm [see Clinical Pharmacology (12.1)]. There are no available data on AZEDRA use in pregnant women. No animal studies using iobenguane I 131 have been conducted to evaluate its effect on female reproduction and embryo-fetal development; however, all radiopharmaceuticals, including AZEDRA, have the potential to cause fetal harm. Advise pregnant women of the risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of iobenguane I 131 in human milk or its effects on the breastfed infant or milk production. No lactation studies in animals were conducted. Because of the potential risk for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with AZEDRA and for 80 days after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating AZEDRA [see Use in Specific Populations (8.1)].
Contraception
AZEDRA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise women of reproductive potential to use effective contraception during treatment with AZEDRA and for 7 months following the final dose of AZEDRA.
Males
Based on its mechanism of action, advise males with female partners of reproductive potential to use effective contraception during treatment with AZEDRA and for 4 months following the final dose of AZEDRA [see Dosage and Administration (2.6)].
Infertility
The recommended cumulative dose of 37 GBq of AZEDRA results in a radiation absorbed dose to the testes and ovaries within the range where temporary or permanent infertility can be expected following external beam radiotherapy [see Dosage and Administration (2.6)].
8.4 Pediatric Use
The safety and effectiveness of AZEDRA have been established in patients 12 years and older with unresectable and iobenguane scan positive, locally advanced or metastatic, pheochromocytoma and paraganglioma (PPGL) which require systemic anticancer therapy. Use of AZEDRA for this indication is supported by evidence from an adequate and well-controlled study in adults and pediatric patients 12 years and older [see Adverse Reactions (6.1), Clinical Studies (14)].
The risks of radiation associated with AZEDRA is greater in pediatric patients than that in adult patients due to greater absorbed radiation doses and longer life expectancy. Ensure the therapeutic benefit of AZEDRA outweighs these greater risks prior to administration in pediatric patients.
The safety and effectiveness of AZEDRA have not been established in pediatric patients younger than 12 years old with unresectable and iobenguane scan positive, locally advanced or metastatic PPGL which require systemic anticancer therapy.
8.5 Geriatric Use
Of the patients enrolled in all clinical studies of AZEDRA, 17% were 65 years or older and 1% were 75 years or older. Clinical studies of AZEDRA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
The radiation dose to patients with renal impairment may be increased due to the delayed elimination of the drug [see Clinical Pharmacology (12)]. Adjust the therapeutic dose based on radiation exposure estimates from the dosimetry assessment [see Dosage and Administration (2.2), Clinical Pharmacology (12)]. The safety of AZEDRA in patients with severe renal impairment (CLcr < 30 mL/min) or end-stage renal disease has not been studied.
11. Azedra Injection Description
AZEDRA (iobenguane I 131) injection, for intravenous use, is a radioactive therapeutic agent. The drug substance iobenguane I 131 is a substituted benzylguanidine with I 131 in the meta position of the benzene ring.
Iobenguane I 131 is described as I 131 meta-iodobenzylguanidine. The molecular weight is 279.1 Daltons and the structural formula is as follows:
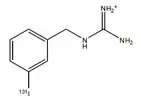
AZEDRA (iobenguane I 131) 555 MBq/mL (15 mCi/mL) injection is a sterile, clear, colorless to pale yellow solution. Each single-dose vial contains iobenguane (0.006 mg/mL), sodium ascorbate (58 mg/mL) and sodium gentisate (23 mg/mL) in Water for Injection, USP. The pH range of the solution is 4.5 to 5.5, with specific activity of ~2,500 mCi/mg (92,500 MBq/mg). Sodium hydroxide and phosphoric acid may be used for pH adjustment.
11.1 Physical Characteristics
I 131 decays with beta and gamma emissions with a physical half-life of 8.021 days. The principal beta emission has a mean energy of 191.6 keV, and the principal gamma emission has energy of 364.5 keV.
11.2 External Radiation
The specific gamma ray constant for I 131 is 2.2 R/mCi hour at 1 cm. A 2.55 cm thickness of Pb will attenuate the radiation emitted by a factor of about 1,000.
Table 6 summarizes radioactive decay properties of I 131.
|
a Calibration day. |
|
| Days | Fraction Remaining |
| 0a | 1 |
| 1 | 0.917 |
| 2 | 0.841 |
| 3 | 0.772 |
| 4 | 0.708 |
| 5 | 0.649 |
| 6 | 0.595 |
| 7 | 0.546 |
| 8 | 0.501 |
| 9 | 0.459 |
| 10 | 0.421 |
| 11 | 0.387 |
| 12 | 0.355 |
| 13 | 0.325 |
| 14 | 0.298 |
12. Azedra Injection - Clinical Pharmacology
12.1 Mechanism of Action
AZEDRA is an I 131 labeled iobenguane. Iobenguane is similar in structure to the neurotransmitter norepinephrine (NE) and is subject to the same uptake and accumulation pathways as NE. Iobenguane is taken up by the NE transporter in adrenergic nerve terminals and accumulates in adrenergically innervated tissues, such as the heart, lungs, adrenal medulla, salivary glands, liver, and spleen as well as tumors of neural crest origin. Pheochromocytoma and paraganglioma (PPGL) are tumors of neural crest origin that express high levels of the NE transporter on their cell surfaces. Following intravenous administration, AZEDRA is taken up and accumulates within pheochromocytoma and paraganglioma cells, and radiation resulting from radioactive decay of I 131 causes cell death and tumor necrosis.
12.2 Pharmacodynamics
The effect of AZEDRA on the QTc interval was evaluated in 74 patients with unresectable pheochromocytoma or paraganglioma. At the recommended therapeutic dosage, no large mean increases from baseline in the QTc interval (i.e., >20 ms) were detected.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of iobenguane I 131 following a dosimetric dose were characterized in patients with malignant PPGL and other malignancies. The mean blood area under curve (AUC) of iobenguane I 131 at the recommended dosimetric dose is 1 µCi*h/mL (CV 33%). The mean maximum concentration (Cmax) for iobenguane I 131 is 0.06 µCi/mL (CV 36%), which generally occurred at the end of the AZEDRA infusion.
Distribution
The volume of distribution (mean ± SD) of iobenguane I 131 is 2893 ± 592 mL/kg. The blood levels of radioactivity declined with a distribution half-life (mean ± SD) of 0.37 ± 0.22 hours. The non-radioactive form of iobenguane I 131 is 61% to 63% bound to human plasma proteins.
Elimination
The mean clearance is 62 ± 24 mL/hr/kg for iobenguane I 131 and the mean terminal blood half-life is 35 ± 14 hours.
Metabolism
Iobenguane I 131 does not undergo hepatic metabolism.
Excretion
Iobenguane I 131 is primarily eliminated renally with cumulative excretion of 50 ± 10% within 24 hours and 80 ± 10% within 120 hours following AZEDRA administration. Unchanged I 131 accounted for an average of 94% and 93% radioactivity excreted in urine collected at 0-6 and 6-24 hours post-dose, respectively. Minor metabolites detected in some patients included free I 131, quantifiable in 55% of 11 patients in Study IB11, as well as meta-iodohippuric acid (MIHA) and meta-iodobenzyl bisguanidine (MMIBG) quantifiable in one patient each.
Specific Populations
Eight of 42 patients (19%) with mild or moderate renal impairment (CLcr ≥ 30-89 mL/min by Cockcroft-Gault) required therapeutic dose reductions based on radiation dose estimates to critical organs exceeding Emami limits (absorbed renal dose exceeding 23 Gy). The pharmacokinetics of iobenguane I 131 has not been studied in patients with severe renal impairment (CLcr < 30 mL/min) or end-stage renal disease [see Use in Specific Populations (8.6)].
Drug Interaction Studies
In Vitro Studies
The non-radioactive form of iobenguane does not inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A. It does not induce CYP1A, 2B6, 2C9, 2C19, or 3A. It is not a substrate or inhibitor of P-glycoprotein.
14. Clinical Studies
The efficacy of AZEDRA in patients with iobenguane scan-positive, unresectable, locally advanced or metastatic pheochromocytoma or paraganglioma (PPGL) which require systemic anticancer therapy was established in Study IB12B, an open-label, single-arm, multicenter clinical trial (NCT00874614). Patients were at least 12 years of age and were ineligible for curative therapy. Patients also progressed on prior therapy for PPGL or were not candidates for chemotherapy. Other eligibility criteria required patients' tumors to have definitive iobenguane avidity; at least one tumor site identified by computed tomography (CT), magnetic resonance imaging (MRI), or iobenguane I 131 scan; Karnofsky performance status ≥60; absence of active central nervous system lesions, and no changes to their antihypertensive regimen in the 30 days prior to the first therapeutic dose.
The major efficacy outcome measure was the proportion of patients who experienced a 50% or greater reduction of all antihypertensive medication(s) lasting for at least six months (28 days per month). Overall tumor response measured by RECIST (Response Evaluation Criteria in Solid Tumors version 1.0) was also evaluated. After the final 12-month assessment, patients entered into long-term follow-up for up to 4 additional years.
A total of 74 patients received the dosimetric dose of AZEDRA. Following dosimetry, 68 patients received at least one therapeutic dose and 50 patients received two therapeutic doses administered at least 90 days apart. The dosimetric dose was 185 mBq to 222 MBq (5 mCi to 6 mCi) for patients weighing > 50 kg and 3.7 MBq/kg (0.1 mCi/kg) for patients weighing ≤ 50 kg. The therapeutic dose was 18,500 MBq (500 mCi) for patients weighing > 62.5 kg and 296 MBq/kg (8 mCi/kg) for patients weighing ≤ 62.5 kg. Among the 68 patients, the median age was 55 years (16 to 72 years), 57% were male, 75% were White, 21% were Black and 4% were Asian. For the primary tumor diagnosis, 78% had pheochromocytoma, 21% had paraganglioma, and 1% had both. Fifty percent (50%) of patients with evaluable imaging studies had lung or liver metastases and 61% had bone metastases at baseline. Eighty-eight percent (88%) underwent prior surgery, 50% received prior external radiation, 31% received prior I 131 MIBG, 31% received prior chemotherapy, 15% received prior kinase inhibitors and 4% received other prior systemic therapies. The median (range) of prior therapies per patient is 2 (0, 7).
The efficacy results are summarized in Table 7. All confirmed responses per RECIST were partial responses.
|
a Calculated using the Agresti-Coull method. b Exact Confidence Interval. |
|
| At least the first therapeutic dose N=68 |
|
| Reduction of all antihypertensive medications by at least 50% maintained for at least 6 months, n (%) | |
| Number of patients | 17 |
| Proportion of patients (95% CIa) | 25% (16%, 37%) |
| Best confirmed overall tumor response per RECIST | |
| Number of patients | 15 |
| Overall response rate (95%CIb) | 22% (14%, 33%) |
| % Responders with Duration of Response ≥ 6 months | 53% |
16. How is Azedra Injection supplied
AZEDRA injection, containing 555 MBq/mL (15 mCi/mL) of I-131 (as iobenguane I 131) and 0.006 mg/mL of iobenguane, is a sterile, clear, colorless to pale yellow solution for intravenous use supplied in a colorless Type 1 borosilicate glass 30 mL single-dose vial. AZEDRA is supplied in dosimetric (2 mL) and therapeutic (22.5 mL) presentations:
- Dosimetric: 1,110 MBq (30 mCi) of iobenguane I 131 at calibration time (NDC 71258-015-02).
- Therapeutic: 12,488 MBq (337.5 mCi) of iobenguane I 131 at calibration time (NDC 71258-015-22).
The product vial is in a lead shielded container placed in a re-sealable plastic bag. The product is shipped on dry ice in a USA DOT Type A Radioactive package.
Store at -70°C (-94°F).
The shelf life is 6 days post calibration time. Discard appropriately at 144 hours.
17. Patient Counseling Information
Hydration
Advise patients to drink at least 2 liters of liquid a day before and for one week following each dose of AZEDRA to minimize irradiation of the bladder [see Dosage and Administration (2.3)].
Radiation Risks
Advise patients to minimize radiation exposure to household contacts consistent with institutional good radiation safety practices and patient management procedures [see Dosage and Administration (2.3), Warnings and Precautions(5.1)].
Myelosuppression
Advise patients to contact their health care provider for any signs or symptoms of neutropenia, thrombocytopenia, or anemia [see Warnings and Precautions (5.2)].
Secondary Myelodysplastic Syndrome, Leukemia and Other Malignancies
Advise patients of the potential for secondary cancers, including myelodysplastic syndrome, acute leukemia, and other malignancies [see Warnings and Precautions (5.3)].
Hypothyroidism
Advise patients to take thyroid-blocking agents as prescribed. Advise patients of the need for life-long monitoring for hypothyroidism [see Warnings and Precautions (5.4)].
Elevations in Blood Pressure
Advise patients to contact their health care provider for signs or symptoms that may occur following tumor-hormone catecholamines release and possible risk of increased blood pressure during or 24 hours following each therapeutic AZEDRA dose [see Warnings and Precautions (5.5)].
Pneumonitis
Advise patients to contact their health care provider for signs or symptoms of pneumonitis [see Warnings and Precautions (5.7)].
Drug Interactions
Advise patients that some medicines interact with AZEDRA and to contact their health care provider before starting any over the counter medicines or herbal or dietary supplements.
Embryo-Fetal Toxicity
Advise pregnant women and males and females of reproductive potential of the potential risk to a fetus. Advise females to inform their health care provider of a known or suspected pregnancy [see Warnings and Precautions (5.8), Use in Specific Populations (8.1), (8.3)].
Advise females of reproductive potential to use effective contraception during treatment with AZEDRA and for 7 months after the final dose [see Use in Specific Populations (8.1), (8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with AZEDRA and for 4 months after the final dose [see Warnings and Precautions (5.8), Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with AZEDRA and for 80 days after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males patients that AZEDRA may impair fertility [see Warnings and Precautions (5.9), Use in Specific Populations (8.3)].
Manufactured for:
Progenics Pharmaceuticals, Inc., a Lantheus company
331 Treble Cove Road
N. Billerica, Ma 01862
AZEDRA® is a registered trademark of Progenics Pharmaceuticals Inc.
516104-0521
| AZEDRA®
iobenguane I 131 injection for intravenous use |
|||||
| Package Handling Instructions | |||||
| Always handle dry ice with care and wear personal protective equipment | Figure 1: TempTale® Ultra Dry Ice Probe Monitor
|
||||
IMPORTANT: Make sure to save the following items for courier to pick up (see directions below):
| |||||
| – Temperature monitor | – Aluminum tubes | – Shipping box | |||
| |||||
1. Reading the Temperature Monitor
| |||||
| Figure 2: Temperature monitor location
|
||||
|
Figure 3: Stopped temperature monitor with
Figure 4: Stopped temperature monitor with
|
||||
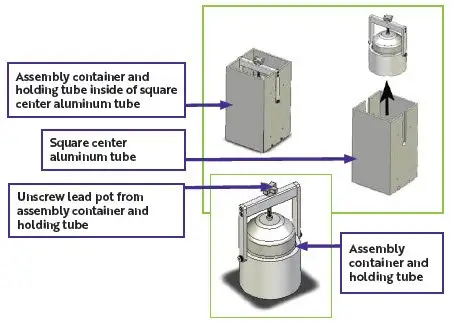
2. Unpacking Procedures
| Figure 5: Unpacking for lead pot containing AZEDRA
|
||||
 in the upper left corner of the screen.
in the upper left corner of the screen. will appear in the upper right corner of the screen next to the
will appear in the upper right corner of the screen next to the  icon (
icon ( icon (
icon (
AZEDRA® is a registered trademark of
Progenics Pharmaceuticals, Inc., a Lantheus company
516124-0422

| AZEDRA
iobenguane i-131 injection, solution |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - Progenics Pharmaceuticals, Inc. (195551247) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Progenics Pharmaceuticals, Inc. | 116991706 | MANUFACTURE(71258-015) , LABEL(71258-015) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Almac Sciences Limited | 232665666 | API MANUFACTURE(71258-015) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Catalent Pharma Solutions, LLC | 014904112 | LABEL(71258-015) | |
